Introduction
Ecto-5′-nucleotidase (CD73, eN) is a
glycosylphosphatidylinositol-linked sialylglycoprotein anchored to
the outer surface of the plasma membrane and is expressed in a
large number of cell types. CD73 is able to hydrolyze extracellular
nucleoside monophosphates to nucleosides. As it is highly specific
for 5′-AMP, it can generate bioactive adenosine during the end
stage of the ectonucleotide breakdown cascade where, along with
CD39 (nucleoside triphosphate diphosphohydrolase-1; NTPDase1), the
sequential hydrolysis of ATP to AMP occurs (1,2).
According to the hypoxic hypothesis, such nucleotide breakdown and
adenosine accumulation occur in solid tumors with no adequate blood
supply (3,4). Hypoxic conditions can stimulate the
process of tumor angiogenesis via direct mitogenic effects on
vasculature and the production of pro-angiogenic factors (5,6).
Adenosine can signal via any of four membrane
G-coupled receptors (ARs): A1, A2A,
A2B and A3. Each of these ARs functions via a
different internal effector mechanism and exhibits a distinct
pattern of tissue distribution. Different ARs were found to be
upregulated in cancer cells (7). To
date, there is no universal hypothesis regarding the role of
different ARs in tumor growth. For instance, activation of the
A1AR can impair glioblastoma growth, but it enhances
breast cancer cell proliferation (4,7,8).
A2AAR is key in mediating the adenosine-induced
anti-inflammatory response to tumor cells via inhibition of T-cell
function, which leads to a decrease in the proliferation of
hormone-dependent breast cancer cells, but paradoxically leads to
an increase in the viability of human melanoma cells (4,7,9).
Stimulation of A3AR in normal cells can induce the
production of growth factors; however, in tumor cells,
A3AR expression is frequently upregulated and apoptosis
is induced or growth is inhibited (4,7).
Finally, adenosine receptors influence the development of the tumor
vascular bed through inhibition of endothelial cell growth via
A1AR or stimulation of angiogenesis through both
A2AAR and A2BAR (7,8).
Evidence that CD73 plays a direct role in tumor
progression via regulation of cell adhesion and T-cell signaling
was outlined by several investigators (10–12).
Other mechanisms were also proposed; for example, CD73 interacts
with tenascin-C and leads to CD73 inhibition, which results in a
decrease in extracellular adenosine and weakly interferes with the
anti-adhesive properties of tenascin-C in the context of other ECM
proteins (13,14). The indication of a crucial function
for CD73 in the regulation of the invasive potential of tumor cells
is frequently raised.
Recently, it was reported that both CD73-deficiency
in transgenic mice and pharmacological inhibition of CD73 in
wild-type mice significantly slows the development of subcutaneous
tumors, experimental lung metastases (9,15,16)
and human xenograft tumors (17,18).
CD73 was further suggested to be a potential negative prognostic
marker in certain cancer types, such as human colorectal cancer
(19). CD73 overexpression in some
human breast cancer cell lines increased their ability to invade,
migrate and adhere to ECM proteins, thus increasing their
metastatic potential. Furthermore, inhibition of CD73 activity
reversed those effects (18,20).
Nonetheless, some clinical studies have shown that CD73 exhibits
low activity in breast cancer cells when compared to the adjacent
non-involved tissue (21,22) and that elevated CD73 expression in
tumor cells was a positive prognostic marker for disease-free
survival in stage I–III breast cancer patients (23). Therefore, the precise involvement of
CD73 in tumor progression requires further clarification.
The murine B16F10 melanoma cell line is frequently
used as the model of choice for studies regarding tumor progression
as it is highly metastatic and syngenic with the C57BL/6 mouse
strain which is commonly used for transgenesis. It has been
previously reported that the B16F10 cell line shows low, although
significant, activity of ecto-5′-nucleotidase (CD73). Specifically,
CD73 activity was 50% lower in the B16F10 line when compared to two
low-metastatic variants: B16-F1 and B16-F10Lr
(lymphocyte resistant) (24,25).
In the present study, we analyzed the influence of
CD73 activity on B16F10 cell migration and invasion potential in
vitro, its effects on subcutaneous tumor growth and the
development of the vascular bed in CD73-deficient mice.
Materials and methods
Reagents
Anti-CD73 mAb (clone 2B6), anti-CD39 mAb (clone
H-85), HRP-conjugated mAb and A375, MDA-MB-231 and HEK293T whole
cell lysates were purchased from Santa Cruz Biotechnology (Santa
Cruz, CA, USA). PE-conjugated anti-CD73 mAb (clone TY/23) and its
isotype control were purchased from BD Pharmingen (San Jose, CA,
USA). Adenosine 5′-α,β-methylene diphosphate (AOPCP), adenosine
receptor agonists [2-chloro-N6-cyclopentyladenosine
(CCPA) (towards A1),
2-p-(2-carboxyethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine
hydrochloride hydrate (CGS-21680) (towards A2A) and
N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide
(IB-MECA) (towards A3)], cell culture reagents, heparin,
hemoglobin and Drabkin’s solution were purchased from Sigma-Aldrich
(St. Louis, MO, USA). bFGF was purchased from Calbiochem
(Darmstadt, Germany) and Matrigel (Basement Membrane Matrix) was
purchased from BD Biosciences (Albany, NY, USA).
Mice
Wild-type and CD73-deficient C57BL/6 mice (26) were bred and maintained at the
Tri-City Central Animal Laboratory of the Medical University of
Gdańsk (Poland) under SPF conditions. Animal studies were performed
on male 4- to 8-week-old mice according to EU and national
guidelines with the approval of the Local Independent Ethics
Committee.
Cell cultures
The B16F10 murine melanoma cell line was kindly
provided by Professor J. Konopa (Gdańsk University of Technology,
Poland). The HEK293T cell line was purchased from Open Biosystems
(Lafayette, CO, USA). Both cell lines were cultured in DMEM
supplemented with 10% FBS in a humidified atmosphere containing 5%
CO2 at 37°C.
Cell adhesion assay
The wells of a 96-well plate were pre-coated with
100 μl of 16 μg/ml Matrigel, 15 μg/ml fibronectin or 50 μg/ml type
I collagen and non-specific binding sites were blocked with 10
mg/ml denatured BSA. B16F10 cells were grown to ~80% confluence in
DMEM containing 10% FBS, pre-incubated in serum-free DMEM for 2 h,
trypsinized and suspended at a concentration of 3×105
cells/ml. Cell suspension (100 μl) in serum-free DMEM was applied
to each well for 15 min (Matrigel) or 30 min (fibronectin or type I
collagen). When indicated, medium was supplemented with 200 μM
AOPCP and/or 10 μM adenosine receptor agonists. Non-adherent cells
were subsequently removed by gentle washing with PBS. The relative
number of adherent cells was calculated using an MTT metabolic
assay. The optical density of wells containing cells incubated only
with a serum-free medium was assumed to be 100% of cell
adhesion.
Wound healing assay
B16F10 cells were seeded in a 96-well plate
(2×105 cells/well) and cultured until 90% confluent.
Cells were subsequently serum-starved overnight, and a linear wound
was applied to the monolayer using a 200-μl pipette tip. Loose
cells were washed away with PBS. When indicated, serum-free DMEM
was supplemented with 200 μM AOPCP and/or 10 μM adenosine receptor
agonist. Images were captured immediately after wounding and again
after 24 h. The wound width was calculated using arbitrary units
with the use of ImageJ software (NIH). The cell migration distance
was determined by subtracting the width of the wound after 24 h
from its initial width at time 0 (immediately after wounding). The
values were plotted as the percentage of the wound closure, with
the initial width set to 0%.
Matrigel invasion assay
The thin coating Matrigel method in a 24-well plate
Transwell system was used (27).
Transwell membrane inserts (BD Biosciences, Franklin Lakes, NJ,
USA) with a pore size of 8 μm were pre-coated with 5 μg of Matrigel
per filter and dried overnight. B16F10 cells were serum-starved
overnight and added (2.5×104/filter) to the upper
compartment in serum-free DMEM supplemented with 200 μM AOPCP
and/or 10 μM adenosine receptor agonist, when indicated.
Conditioned medium from the NIH 3T3 cell culture was added to the
lower chamber as a chemoattractant. After 22 h of incubation, the
Matrigel coating on the upper surface of the filter was wiped off
using a cotton swab. Cells that migrated through the filter were
fixed, stained with Giemsa stain and counted under the microscope
(magnification, ×400) from eight randomly selected fields for each
filter isolated from triplicate chambers.
Western blot analysis
Cells (8×106) were lysed in 100 μl of
lysis buffer (1% glycerol, 5 mM EDTA, 0.1% NP40 in PBS and pH 7.4)
containing complete protease inhibitor cocktail (Roche Applied
Sciences, Indianapolis, IN, USA) and centrifuged for 10 min at
17,000 × g. Proteins in the supernatant were denatured by boiling
in 5X Laemmli buffer, subjected to SDS-PAGE electrophoresis and
transferred to a PVDF membrane (Immobilon; Millipore, Etten-Leur,
The Netherlands). The next steps were performed according to the BM
Chemiluminescence Blotting Substrate instructions (Roche Applied
Sciences), with an overnight incubation in the primary antibody
(mouse anti-CD73 or anti-CD39 mAb) and a 30-min incubation in a
secondary goat anti-mouse HRP-conjugated mAb (Santa Cruz
Biotechnology).
Immunofluorescence microscopy
B16F10 cells were plated on glass coverslips and
fixed with 4% paraformaldehyde after 48 h. For detection of
internal antigens, cells were permeabilized with 0.1% Triton X-100
for 3 min. Cells were subsequently blocked for 15 min with 0.5% BSA
in PBS, incubated overnight with a primary mouse anti-CD73 mAb and
with the goat anti-mouse secondary Cy3-conjugated antibody (Jackson
ImmunoResearch, West Grove, PA, USA) for 30 min. Permeabilized
cells were counterstained with 0.05% DAPI (Sigma) for 5 min. The
stained cells were mounted on glass slides in n-propyl-gallate, and
images were obtained using an Axiovert Zeiss microscope and
AxioVision software (Zeiss, Oberkochen, Germany).
Analysis of cells by flow cytometry
Cells (1×105) were collected, suspended
in a staining solution (2% FBS, 0.09% NaN3 in PBS) and
incubated for 5 min at 4°C. Relevant PE-conjugated antibodies were
added to the final concentration of 1 μg per 106 cells
and incubated for 30 min at 4°C. Cells were subsequently washed
twice with a staining solution and suspended in staining solution
for analysis. When the visualization of the intracellular antigen
was required, cells were fixed with 4% paraformaldehyde and treated
with permeabilization-wash buffer (BioLegend, San Diego, CA, USA)
as an additional step prior to incubation with antibodies. The
analysis was performed using an LSR II (Becton-Dickinson, Franklin
Lakes, NJ, USA) flow cytometer equipped with an argon ion laser
(488 nm). All the measurements were performed for 1×104
cells by gating the cells that exhibited the typical forward and
side scatter features of non-disintegrated cells. The percentage of
positive cells was measured from a cut-off set using an
isotype-matched non-specific control antibody. Data were analyzed
offline using the BD FACSDiva (BD Biosciences) and Cyflogic (CyFlo
Ltd., Turku, Finland).
Tumor inoculation
B16F10 tumor cells were injected subcutaneously into
the left flank of syngenic C57BL/6 wild-type or CD73-deficient mice
(2.5×106 tumor cells/animal). When indicated, the s.c.
injection was supplemented with 200 μM AOPCP, and AOPCP was
subsequently administered via intraperitoneal injection at a dose
of 20 mg/kg on days 5, 7, 9 and 12. Control animals were injected
with vehicle only. Tumor volume was monitored with calipers and
calculated using the formula: V= 4/3 π × (D/2) × (d/2)2,
where V is the volume (mm3), D is the long diameter (mm)
and d is the short diameter (mm). Mice were euthanized after 14
days and tumors were fixed in formalin for subsequent histological
analysis.
Matrigel plug angiogenesis assay
Mice were inoculated s.c. on the right side of the
linea alba with 600 μl of Matrigel (10 mg/ml) mixed with heparin
(12.5 U) and bFGF (200 ng), supplemented with 200 μM AOPCP or
saline (control and CD73−/−plugs). When indicated, the
animals were injected every second day around the plug (four
injection sites) with AOPCP (100 μl of 200 μM solution) or saline.
In the cancer-induced angiogenesis assay, the inoculation contained
600 μl of Matrigel mixed with B16F10 cells (4×105 cells)
supplemented with 200 μM AOPCP or saline (control plugs). Plugs
were removed from euthanized mice after 8 days. Matrigel plug
vascularization was quantified by measuring hemoglobin
concentration in homogenized plugs using Drabkin’s solution
according to the manufacturer’s instructions.
Histological analysis
Excised tumors were fixed in formalin and embedded
in paraffin. Paraffin sections were stained with hematoxylin and
eosin using the method of May-Grünwald-Giemsa or were stained with
Heidenhain’s AZAN modification of Mallory’s trichrome stain. Images
from microscope slides were acquired with an iPOLiS SNB-7000
digital camera (Samsung, Daegu, China) connected to an Eclipse E800
microscope (Nikon, Tokyo, Japan).
Statistical analysis
Mean values were obtained from at least three
separate experiments and reported as the mean (± SD). For
statistical analysis, the Mann-Whitney test for two unpaired groups
of a non-Gaussian population was used. Advanced hemorrhagic
necrosis incidence in tumors was evaluated using Fisher’s
non-parametric test. P-values <0.05 were considered to indicate
statistically significant differences.
Results
B16F10 cells possess significant amounts
of CD73 and CD39
To verify the benefits of the chosen cell model for
an in vitro analysis of CD73 influence on invasive
potential, we evaluated the B16F10 cell line for the presence of
both CD39 (NTPDase1), an ectonucleotidase that catalyzes the
sequential hydrolysis of ATP to AMP and CD73, which converts AMP to
adenosine.
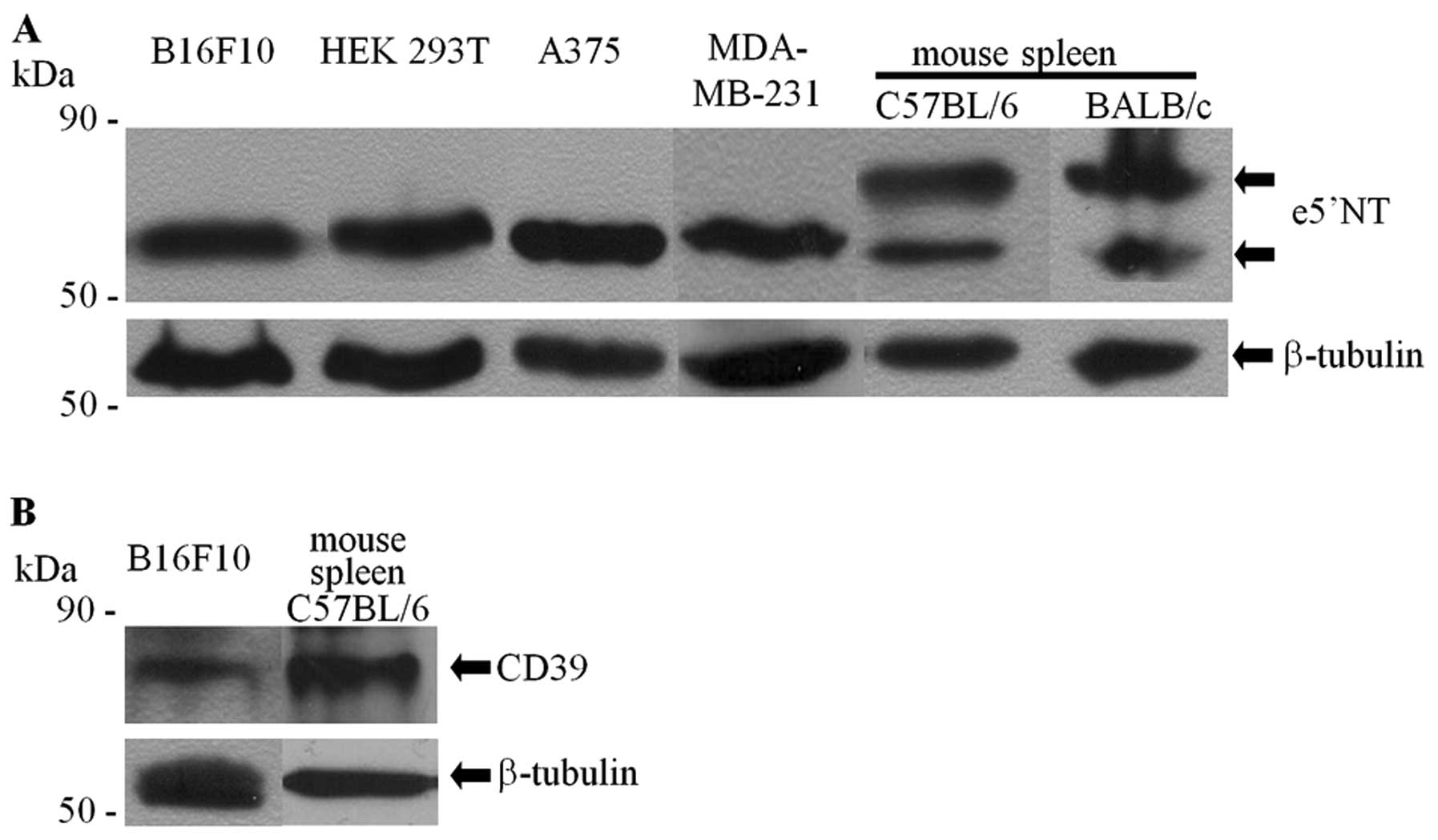
The CD73 content in the whole cell lysate of B16F10
cells (Fig. 1A) was examined by
western blot analysis and compared with other tissues and cells
exhibiting diverse levels of ecto-5′-nucleotidase enzymatic
activity: human melanoma A375 cell line (high level) (12), human breast adenocarcinoma
MDA-MB-231 cell line (high level) (17) and human embryonic kidney cell line
HEK293T (low level) (28,29). Judging by the intensity and
localization of specific bands, the levels of CD73 protein in
B16F10 cells appeared to be quantitatively and qualitatively
comparable to the levels found in other cell lines. The molecular
mass of the detected protein was similar to the non-glycosylated
form of CD73 (58 kDa) (30). Only
lysates prepared from the spleen of mouse strains syngenic
(C57BL/6) or non-syngenic (BALB/c) to B16F10 exhibited the
additional major band with a higher molecular mass, a
characteristic of highly glycosylated CD73 present on lymphocytes
(10).
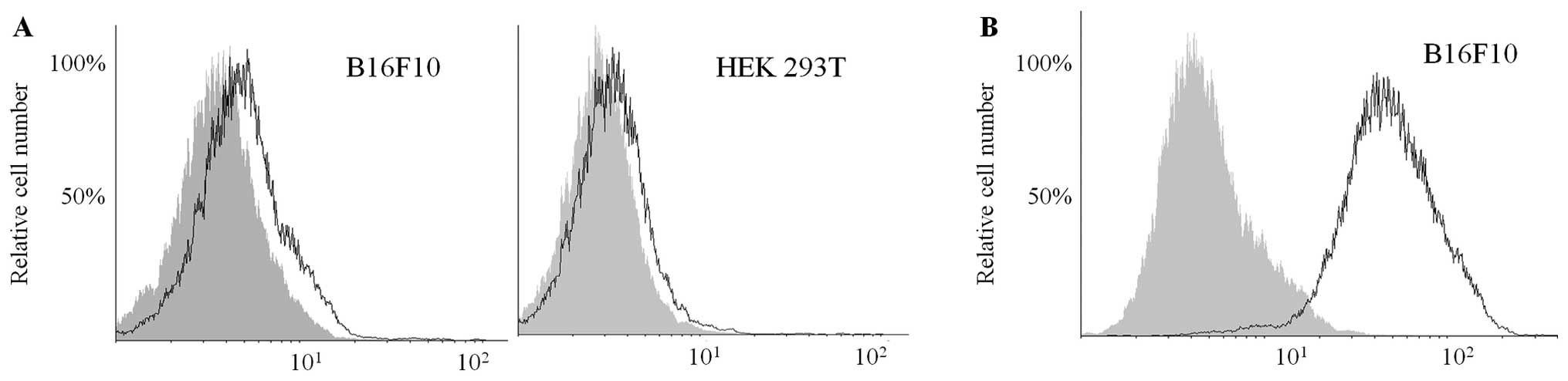
Immunofluorescence staining (Fig. 1C and D) revealed that CD73 was less
densely distributed (but still significantly expressed) on the
B16F10 cell surface when compared to HEK293T. The use of flow
cytometry to detect CD73 with a PE-conjugated anti-CD73 mAb (clone
TY/23) did not deliver conclusive results, as cell-surface CD73 was
not detectable in non-fixed B16F10 or HEK293T cells, although the
intracellular antigen was apparent in the fixed cells (Fig. 2). Targeting either CD73 with AOPCP
or various adenosine receptors with specific agonists (IB-MECA,
CGS-2180 and CCPA) did not change the total CD73 protein levels or
its distribution in B16F10 cells (data not shown).
CD39 content was significantly high and comparable
among the whole cell lysates obtained from B16F10 cells or the
spleen of a syngenic mouse (Fig.
1B).
Targeting CD73 with AOPCP modulates
B16F10 melanoma cell ability to adhere to the ECM, migrate and
invade in vitro
We analyzed the ability of melanoma cells to adhere
to ECM proteins, migrate and invade. The specific competitive CD73
inhibitor AOPCP, a hydrolytically stable analog of ADP, was used
for this purpose. Selective adenosine receptor agonists CCPA
(A1R), CGS-21680 (A2AR) and IB-MECA
(A3R) were used in the presence of AOPCP to determine
which of the adenosine receptors was involved in mediating these
processes. Although all the agonists, AOPCP and serum-starvation
were tested, none of these treatments influenced the viability of
B16F10 cells under the experimental conditions described (data not
shown).
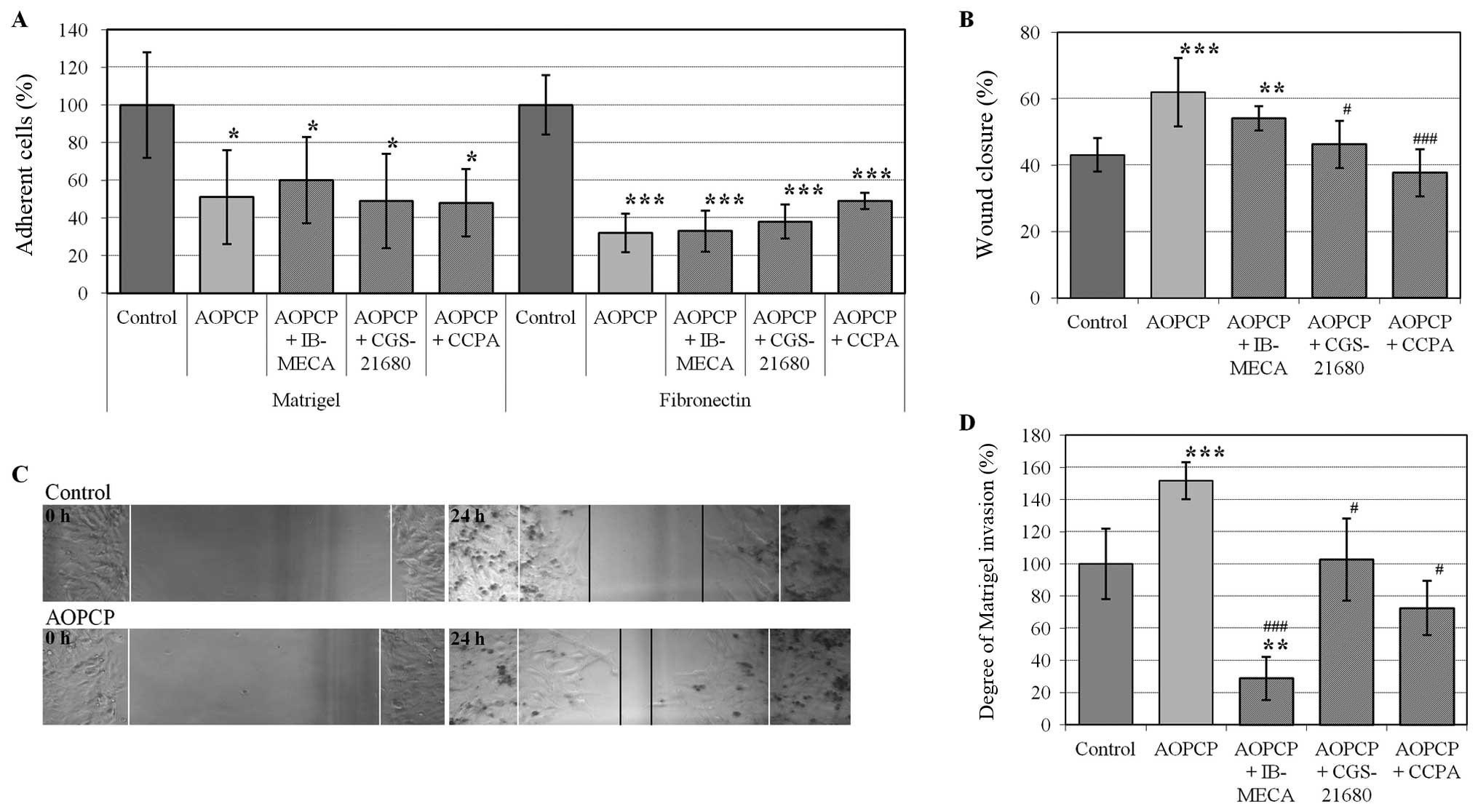
B16F10 cell adherence to Matrigel and fibronectin
was measured (Fig. 3A). The major
component of the Matrigel basement membrane matrix is laminin, but
Matrigel also contains type IV collagen, heparan sulfate
proteoglycans and entactin/nidogen. Attachment of B16F10 cells to
the matrix was significantly inhibited by AOPCP and was ~51% of the
control (P<0.05). Melanoma attachment to fibronectin displayed a
larger decrease of ~32% of the control (P<0.005). Blocking CD73
by AOPCP was ~20% (±2%) stronger (P<0.01) when compared to the
blocking ability of monoclonal anti-CD73 antibodies (data not
shown). No significant changes in the ability of the melanoma cells
to attach to ECM proteins were observed upon the simultaneous
addition of adenosine receptor agonists and AOPCP.
The wound healing assay was used to investigate
directional cell migration after breaking the continuity of a
monolayer. Addition of AOPCP to the medium significantly increased
the motility of B16F10 cells, as demonstrated by an increase in the
wound closure from 43% in control cells to 62% upon AOPCP treatment
(P<0.005) 24 h after wound formation (Fig. 3B and C). Addition of adenosine
A1 or A2A receptor agonists reversed the
effects of AOPCP (P<0.005 or P<0.05, respectively). Upon
stimulation of the A3 receptor, the observed effects
were not statistically significant (P=0.11) (Fig. 3B).
The rate of Matrigel invasion was quantified via the
use of a Transwell chamber (Boyden) (Fig. 3D). The number of cells migrating
through a membrane coated with Matrigel towards a chemotactic agent
was increased by ~50% in the presence of AOPCP (P<0.005). This
effect was completely reversed by simultaneous stimulation of
adenosine receptors of type A1 or A2A
(P<0.05) and significantly diminished below the control level by
stimulation of A3R (P<0.01 compared to the
control).
B16F10 melanoma growth in vivo is
inhibited in CD73-deficient mice
We compared the melanoma growth rates in the
wild-type and CD73-deficient mice to discriminate between the
influence of the host cell- and cancer cell-derived CD73 on tumor
growth. B16F10 tumor cells were injected subcutaneously and the
tumor growth was monitored during the early phase of its growth (up
to 14 days after inoculation). As shown in Fig. 4A, the tumor growth rate was
significantly reduced in CD73-deficient mice (P<0.05, days 9 and
14 after inoculation). Simultaneous targeting of CD73 on tumor
cells with AOPCP further reduced the tumor growth rate when
compared to both the wild-type mice (P<0.01, day 9; P<0.05,
days 12 and 14) and the CD73-deficient mice not treated with AOPCP
(P<0.05, day 12). No lung metastases were observed within 14
days of the subcutaneous injection.
The development of local blood vessels is commonly
recognized as a growth-limiting mechanism for neoplasms and the
process of angiogenic switch has previously been found to be
adenosine-dependent (31). In view
of these reports, we assessed whether the decrease in the tumor
growth rate in CD73-deficient mice with CD73-inhibited cancer cells
was directly related to a decrease in angiogenic response. This
hypothesis was supported by our observation of advanced hemorrhagic
necrosis (manifested as a tumor perforation and a blood clot on its
surface). Incidence of such necrosis was significantly higher
(P<0.05) 14 days after tumor inoculation in CD73-deficient hosts
(38% of untreated and 50% of AOPCP-treated mice) when compared to
the wild-type animals (no animals exhibited necrosis on day 14)
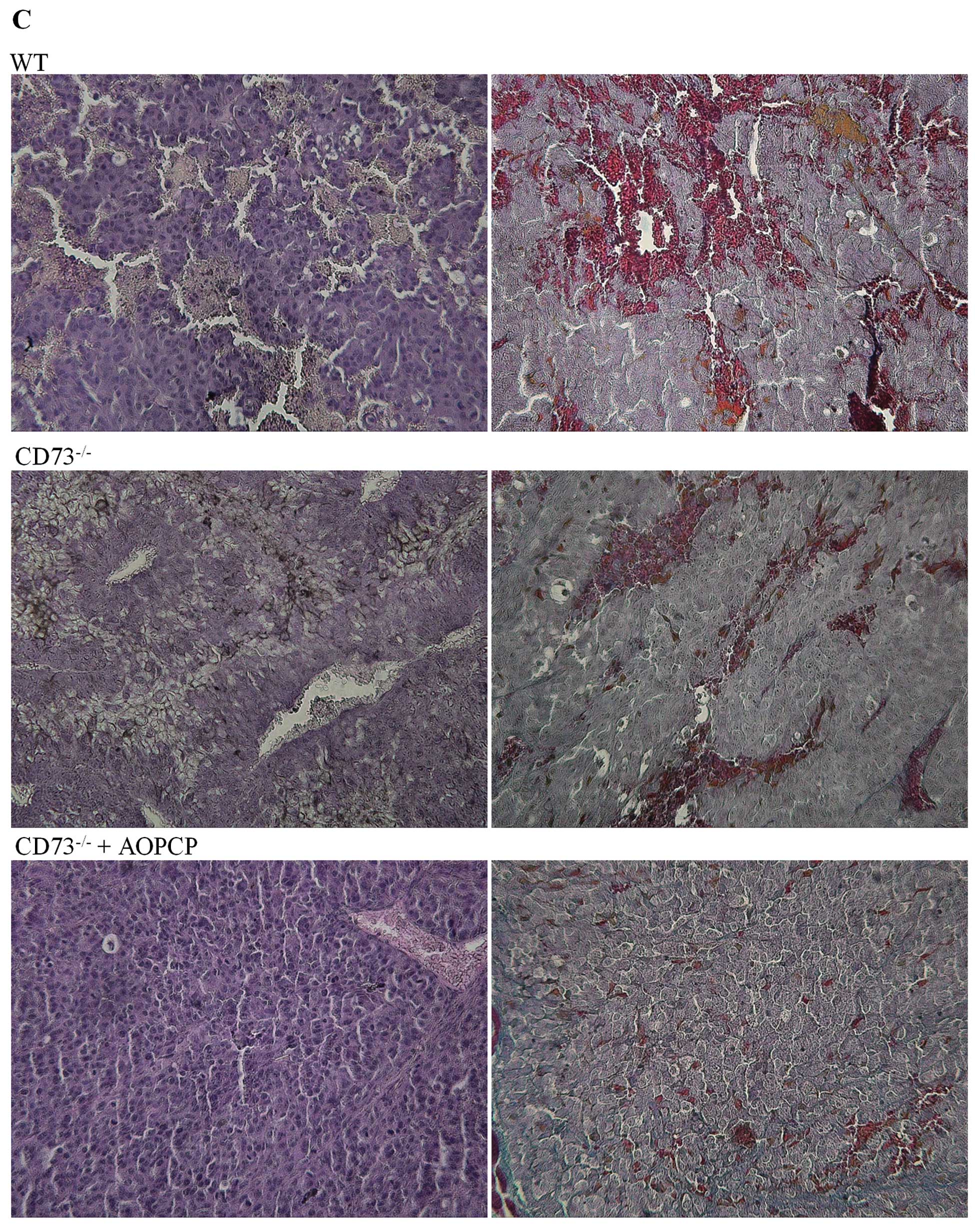
(Fig. 4B). This hypothesis was also
supported by histological analysis of tumors (Fig. 4C) by H&E or Azan-Mallory
staining. We observed a visible decrease in the vascular density of
tumors isolated from AOPCP-treated CD73-deficient mice when
compared to the highly vascularized tumors observed in both
wild-type controls and CD73-deficient mice not treated with AOPCP
(no significant difference was observed between the two latter
groups).
The angiogenic response of the host and
B16F10 melanoma-induced angiogenesis are reduced upon CD73
inhibition
To analyze a putative role of CD73 in in vivo
angiogenesis, the Matrigel plug angiogenesis assay was used. The
assay was performed by adding CD73 inhibitor (AOPCP) locally to the
plug in a wild-type animal or by using CD73-deficient hosts.
Neovascularization of the plug was analyzed 8 days after injection
when the blood vessel formation was robust enough for hemoglobin
measurements and the total volume of the angiogenic blood vessels
in a Matrigel plug could be quantified.
Substantial ingrowth of new blood vessels into the
Matrigel plug was observed in wild-type mice in the presence of
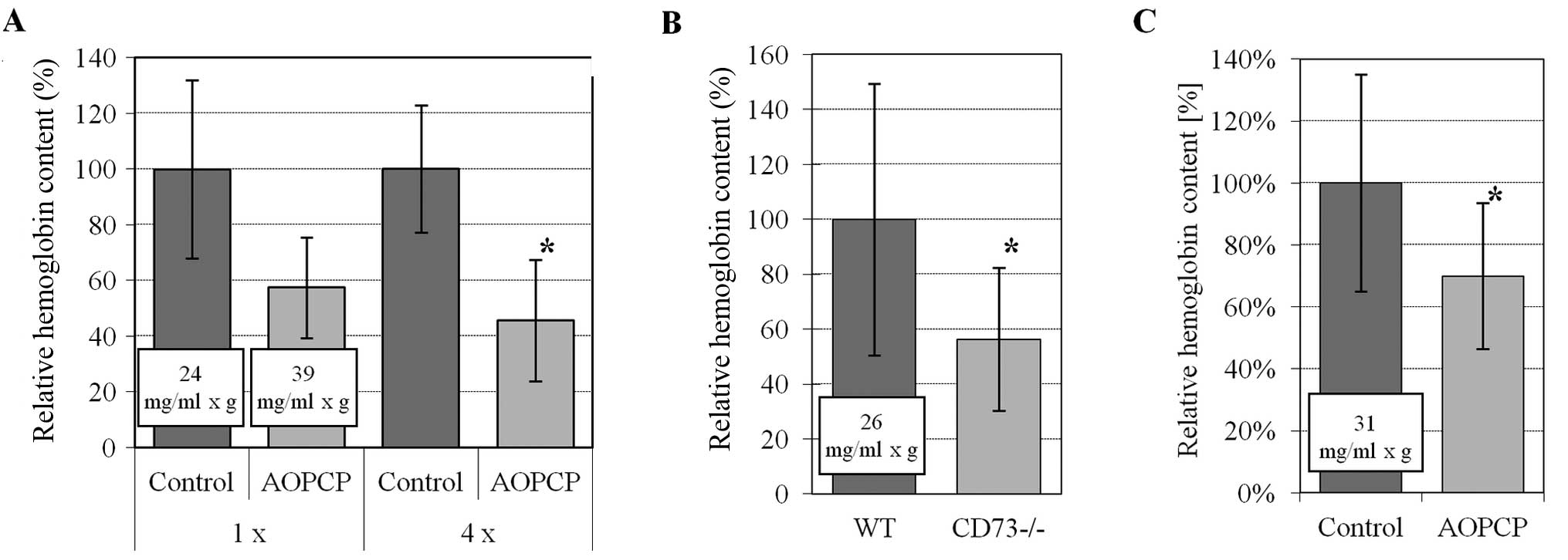
proangiogenic bFGF. A single application of AOPCP decreased the
volume of blood in new vessels; however, this result was not
statistically significant (P=0.09). Statistical significance was
achieved after four consecutive local injections of AOPCP around
the plug, which decreased the total volume of blood vessels to ~46%
of the control (P<0.05) (Fig.
5A). Moreover, we observed that neoangiogenesis was compromised
in CD73-deficient mice when compared to wild-type mice, with the
total blood volume in plugs exhibiting a significant decrease to
~56% of wild-type mice (P<0.05) (Fig. 5B).
When B16F10 melanoma cells were introduced into a
Matrigel plug in wild-type animals, the angiogenic response was
strong (~31 mg/ml × g) and the ingrowth of tumor blood vessels was
significantly decreased to ~60% of the control values after a
single application of AOPCP (Fig.
5C).
Thus, we found that formation of new vessels in
C57BL/6 mice and tumor-induced angiogenesis in Matrigel plugs is
inhibited by decreasing CD73 enzymatic activity.
Discussion
Recently published reports confirmed the association
between CD73 and tumor progression in melanoma (9,12,15,16).
The authors claim that either CD73-deficiency in transgenic mice or
a pharmacological inhibition of CD73 in wild-type mice can protect
against the development of subcutaneous tumors and experimental
lung metastases (9,15,16).
However, murine B16F10 melanoma was characterized by some authors
as a CD73-negative cell line despite earlier reports that low, but
significant, cell surface 5′-nucleotidase activity was present
(24,25). In the present study, we analyzed
CD73 expression in B16F10 cells using various techniques: western
blotting, immunofluorescence microscopy and flow cytometry with two
different antibodies (non-conjugated and PE-conjugated mAb). These
results convincingly showed that B16F10 cells contain
ecto-5′-nucleotidase levels comparable to those found in high
CD73-expressing human melanoma A375 (12) or breast adenocarcinoma MDA-MB-231
cell lines (17). The protein
displayed a similar molecular mass in all analyzed cell lines,
which was smaller than the more heavily glycosylated form present
on T lymphocytes (10). A
significant amount of CD73 was detected on the B16F10 cell surface
by immunofluorescence; however, its main localization pattern
appeared to be intracellular. This is in agreement with an earlier
hypothesis stating that an extensive intracellular distribution of
CD73 is present in a membrane-bound pool (i.e., lysosomes, Golgi
apparatus and transcytotic vesicles) and that CD73 undergoes
continual exchange between the plasmatic and internal membranes
(1,32,33).
We encountered a problem detecting CD73 on the surface of non-fixed
B16F10 and HEK293T cells by flow cytometry (although the
intracellular antigen was apparent in the fixed cells), but this
might be due to ecto-domain shedding after binding of the anti-CD73
antibody. Such an effect was demonstrated by Airas et al
(34) when CD73 expression was
examined on the surface of lymphocytes.
The paradigm for the role of CD73 in tumor
progression has not yet been established. In the literature,
examples suggesting that a high level of CD73 expression on tumor
cells can be used as either a negative (19) or a positive (23) prognostic marker can be found.
However, an explicitly positive correlation between CD73 expression
and invasive potential was shown in some cancer cells in
vitro. Overexpression of CD73 in human breast cancer cells
increased their ability to invade, migrate and adhere to the ECM,
thus upregulating their metastatic potential (17,20).
For the highly invasive human melanoma cell lines, it was shown
that CD73 was upregulated and correlated with a number of
metastasis-related markers (12).
On the contrary, some other clinical research studies showed
reduced CD73 activity in breast cancer cells when compared to the
adjacent non-involved tissue (21,22).
Additionally, the reverse correlation between CD73 activity and
metastatic potential was occasionally indicated for rat breast
cancer cell lines (35) and B16F10
cells (24,25). In the present study, we showed that
the highly invasive murine B16F10 cells contained levels of CD73
protein comparable to those found in highly invasive human melanoma
A375 (12) and human breast
adenocarcinoma MDA-MB-231 cell lines (17). Moreover, the targeting of CD73 in
B16F10 cells with a selective inhibitor led to a significant
increase in their invasive potential in vitro.
It should be noted that blocking CD73 with AOPCP
decreased B16F10 cell adherence to fibronectin and to the basement
membrane matrix (Matrigel) and that this adherence was not
modulated via adenosine receptor signaling. This is particularly
significant with regards to the decisive role of cell adherence to
ECM components in the escape and invasion of ectopic environments
(36). It has been shown that
isolated CD73 binds to fibronectin in a specific and saturable
manner (11,37). Such specificity suggests a direct
role of CD73 in the binding of B16F10 cells to fibronectin. It is
possible that the conformation of ecto-5′-nucleotidase changed in
the presence of inhibitory AOPCP, thus interfering with ECM
binding, similar to previously indicated results for eukaryotic
cells and the 5′-nucleotidase from E. coli (38). The difference between its effect on
B16F10 adhesion to Matrigel and fibronectin might be explained by
the presence of laminin in Matrigel and its ability to direct more
CD73 to the cell surface (39). The
adherence-dependent processes of migration and invasion were
upregulated in AOPCP-treated B16F10 cells and downregulated by a
set of adenosine receptor agonists. Therefore, the involvement of
CD73 in melanoma cell invasion was confirmed and probably occurs
via direct CD73-mediated interactions with ECM constituents.
Nevertheless, the exact molecular mechanisms of this phenomenon
require further study.
By contrast, in vivo experiments showed that
CD73-deficiency in the host caused impaired tumor growth in mice.
Additionally, pharmacological inhibition of CD73 activity on the
grafted B16F10 melanoma in CD73-deficient hosts displayed even
further decreases in tumor growth. Earlier reports suggest that
B16F10 melanoma growth is slower in CD73-deficient mice when
compared to a wild-type mouse and that this effect was attributable
to the immunomodulatory role of the host-derived CD73 on
lymphocytes (9,15,16).
We propose here a similar role for the angiogenic switch, a
critical step in melanoma tumor growth correlating with the
transition to a metastatic phase (31); it also correlates with an increase
in tumor thickness and resultant hypoxia (40). Adenosine is regarded as an important
hypoxia-counteracting mediator (41,42)
which exerts a mitogenic effect on the endothelium and regulates
the synthesis of pro- and anti-angiogenic factors (such as VEGF
through the A3 receptor in melanoma cells) (6,43).
Experiments performed on human breast cancer xenografts in a nude
mouse model have demonstrated that both the chemical inhibition of
CD73 and systemic CD73-deficiency can decrease angiogenesis not
only in vitro (in isolated pulmonary microvascular
endothelial cells) but also in vivo by contributing to an
increase in the necrotic area of the tumor (17,18).
The regulatory role of adenosine produced extracellularly by the
grafted B16F10 cells could be variable. Therefore, we suggested
that full inhibition of the adenosine producing enzyme may lead to
decreased neoangiogenesis. The histological analysis of the B16F10
tumors have shown that the complete lack of active CD73 in host and
grafted cells induced quite significant changes in the vascular
density, similar to the results obtained from breast cancer
xenografts (17,18), and also led to an increase in
necrosis of the tumor tissue (18).
However, even the low level expression of CD73 in B16F10 cells in
CD73-deficient mice was enough to abrogate this effect, similar to
the results described by Yegutkin et al (15). Nevertheless, the increase in the
visible hemorrhagic necrosis in this experimental group suggests
some less visible changes in tumor vasculature as the lower
vascular density correlates with the lower perfusion rate of the
tumor tissue, which generally increases the incidence of necrosis
(44). These changes remain under
investigation.
The Matrigel plug angiogenesis assay is one of the
most frequently used angiogenesis assays in current practice
(45), and further analysis
demonstrated a significant decrease in the total volume of new
blood vessels due to both CD73 inhibition in wild-type mice (also
in the presence of B16F10 cells) or as a result of CD73-deficiency
in animals. Therefore, we propose that inhibition of CD73 in
vivo does interfere with the development of the melanoma
vascular bed, possibly by influencing either the number or the
maturation status of the developing blood vessels. The mechanism
has yet to be fully clarified, but aside from a direct influence on
vessels, these effects may be mediated through the modulation of
some known B16F10 function (e.g., VEGF production), such as those
shown by Pötgens et al for the human melanoma xenografts
(46).
In conclusion, the present study strongly supports a
long-standing hypothesis that CD73 is one of the most important
molecules regulating the progression of melanoma. However, a more
complex set of interactions was identified in the present study.
These results suggest that the membrane enzyme itself and its
product, adenosine, can influence the progression by various
mechanisms, some of which are contradictory. The ecto-enzyme can
function as an adhesion protein and by itself influence melanoma
progression. The adenosine product alone can signal to inhibit
migration and invasion of melanoma cells, while simultaneously
contributing to the development of tumor vasculature. Therefore,
final outcomes regarding CD73 influence on tumor progression may be
the result of maintaining a critical balance between its enzymatic
and non-enzymatic activities regulating many aspects of the
invasive potential of tumor cells. Further in-depth studies of CD73
and adenosine receptor signaling will provide us with solid
premises regarding the functions of CD73 and which stages of tumor
development should be targeted in cancer therapy.
Acknowledgements
The present study was supported by the W-732
intramural grant from the Medical University of Gdansk (2007–2008),
the core fund of the Intercollegiate Faculty of Biotechnology
(2008–2010) and the grant N N401 006938 from the National Science
Centre (2010–2013). This research was co-funded by the European
Social Fund POKL.04.01.01-00-017/10-00 (to M.G.) ‘We educate the
best - a comprehensive program of development of graduate students,
young doctors and academic teaching staff of the University of
Gdańsk’.
References
|
1
|
Zimmermann B: 5′-Nucleotidase: molecular
structure and functional aspects. Biochem J. 285:345–365. 1992.
|
|
2
|
Bianchi V and Spychala J: Mammalian
5′-nucleotidases. J Biol Chem. 278:46195–46198. 2003.
|
|
3
|
Bly J, White TD and Hoskin DW: The
extracellular fluid of solid carcinomas contains immunosuppressive
concentrations of adenosine. Cancer Res. 57:2602–2605.
1997.PubMed/NCBI
|
|
4
|
Merighi S, Baraldi PG, Gessi S, Iannotta
V, Klotz KN, Leung E, Mirandola P, Tabrizi MA, Varani K and Borea
PA: Adenosine receptors and human melanoma. Drug Dev Res.
58:377–385. 2003. View Article : Google Scholar
|
|
5
|
Bergers G and Benjamin LE: Tumorigenesis
and the angiogenic switch. Nat Rev Cancer. 3:401–410. 2003.
View Article : Google Scholar
|
|
6
|
Auchampach JA: Adenosine receptors and
angiogenesis. Circ Res. 101:1075–1077. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fishman P, Bar-Yehuda S, Synowitz M,
Powell JD, Klotz KN, Gessi S and Borea PA: Adenosine receptors and
cancer. Adenosine Receptors in Health and Disease, Handbook of
Experimental Pharmacology. Wilson CN and Mustafa SJ:
Springer-Verlag; Berlin: pp. 399–441. 2009, View Article : Google Scholar
|
|
8
|
Stagg J and Smyth MJ: Extracellular
adenosine triphosphate and adenosine in cancer. Oncogene.
29:5346–5358. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stagg J, Divisekera U, Duret H, Sparwasser
T, Teng MW, Darcy PK and Smyth MJ: CD73-deficient mice have
increased anti-tumor immunity and are resistant to experimental
metastasis. Cancer Res. 71:2892–2900. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Resta R, Yamashita Y and Thompson LF:
Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol
Rev. 161:95–109. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stochaj U, Dieckhoff J, Mollenhauer J,
Cramer M and Mannherz HG: Evidence for the direct interaction of
chicken gizzard 5′-nucleotidase with laminin and fibronectin.
Biochim Biophys Acta. 992:385–392. 1989.
|
|
12
|
Sadej R, Spychala J and Skladanowski AC:
Expression of ecto-5′-nucleotidase (eN, CD73) in cell lines from
various stages of human melanoma. Melanoma Res. 16:213–222.
2006.
|
|
13
|
Orend G and Chiquet-Ehrismann R: Adhesion
modulation by antiadhesive molecules of the extracellular matrix.
Exp Cell Res. 261:104–110. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sadej R, Inai K, Rajfur Z, Ostapkowicz A,
Kohler J, Skladanowski AC, Mitchell BS and Spychala J: Tenascin C
interacts with Ecto-5′-nucleotidase (eN) and regulates adenosine
generation in cancer cells. Biochim Biophys Acta. 1782:35–40.
2008.PubMed/NCBI
|
|
15
|
Yegutkin GG, Marttila-Ichihara F,
Karikoski M, Niemelä J, Laurila JP, Elima K, Jalkanen S and Salmi
M: Altered purinergic signaling in CD73-deficient mice inhibits
tumor progression. Eur J Immunol. 41:1231–1241. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Forte G, Sorrentino R, Montinaro A,
Luciano A, Adcock IM, Maiolino P, Arra C, Cicala C, Pinto A and
Morello S: Inhibition of CD73 improves B cell-mediated anti-tumor
immunity in a mouse model of melanoma. J Immunol. 189:2226–2233.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou X, Zhi X, Zhou P, Chen S, Zhao F,
Shao Z, Ou Z and Yin L: Effects of ecto-5′-nucleotidase on human
breast cancer cell growth in vitro and in vivo. Oncol
Rep. 17:1341–1346. 2007.
|
|
18
|
Wang L, Tang S, Wang Y, Xu S, Yu J, Zhi X,
Ou Z, Yang J, Zhou P and Shao Z: Ecto-5′-nucleotidase (CD73)
promotes tumor angiogenesis. Clin Exp Metastasis. 30:671–680.
2013.
|
|
19
|
Eroglu A, Canbolat O, Demirci S, Kocaoglu
H, Eryavuz Y and Akgül H: Activities of adenosine deaminase and
5′-nucleotidase in cancerous and noncancerous human colorectal
tissues. Med Oncol. 17:319–324. 2000.
|
|
20
|
Wang L, Zhou X, Zhou T, Ma D, Chen S, Zhi
X, Yin L, Shao Z, Ou Z and Zhou P: Ecto-5′-nucleotidase promotes
invasion, migration and adhesion of human breast cancer cells. J
Cancer Res Clin Oncol. 134:365–372. 2008.
|
|
21
|
Pohl AL, Reiner G, Kolb R, Sauermann G,
Moser KV and Spona J: Enzyme activities in human breast tumor cells
and sera. Cancer Detect Prev. 8:57–66. 1985.PubMed/NCBI
|
|
22
|
Krüger KH, Thompson LF, Kaufmann M and
Möller P: Expression of ecto-5′-nucleotidase (CD73) in normal
mammary gland and in breast carcinoma. Br J Cancer. 63:114–118.
1991.
|
|
23
|
Supernat A, Markiewicz A,
Welnicka-Jaskiewicz M, Seroczynska B, Skokowski J, Sejda A, Szade
J, Czapiewski P, Biernat W and Zaczek A: CD73 expression as a
potential marker of good prognosis in breast carcinoma. Appl
Immunohistochem Mol Morphol. 20:103–107. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Raz A, McLella WL, Hart IR, Bucana CD,
Hoyer LC, Sela BA, Dragsten P and Fidler IJ: Cell surface
properties of B16 melanoma variants with differing metastatic
potential. Cancer Res. 40:1645–1651. 1980.PubMed/NCBI
|
|
25
|
Schroeder F and Gardiner JM: Membrane
lipids and enzymes of cultured high-and low-metastatic B16 melanoma
variants. Cancer Res. 44:3262–3269. 1984.PubMed/NCBI
|
|
26
|
Koszalka P, Özüyaman B, Huo Y, Zernecke A,
Flögel U, Braun N, Buchheiser A, Decking UKM, Smith ML, Sévigny J,
Gear A, Weber A, Molojavyi A, Ding Z, Weber C, Ley K, Zimmermann H,
Gödecke A and Schrader J: Targeted disruption of
cd73/ecto-5′-nucleotidase alters thromboregulation and augments
vascular inflammatory response. Circ Res. 95:814–821. 2004.
|
|
27
|
Shaw LM: Tumor cell invasion assays:
methods in molecular biology. Cell Migration: Developmental Methods
and Protocols. Guan JL: 294. Humana Press Inc; New York: pp.
97–105. 2005, View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Morabito L, Montesinos MC, Schreibman DM,
Balter L, Thompson LF, Resta R, Carlin G, Huie MA and Cronstein BN:
Methotrexate and sulfasalazine promote adenosine release by a
mechanism that requires ecto-5′-nucleotidase-mediated conversion of
adenine nucleotides. J Clin Invest. 101:295–300. 1998.PubMed/NCBI
|
|
29
|
St Hilaire C, Ziegler SG, Markello TC, et
al: NT5E mutations and arterial calcifications. N Engl J Med.
364:432–442. 2011.PubMed/NCBI
|
|
30
|
Flocke K and Mannherz HG: Isolation and
characterization of 5′-nucleotidase of a human pancreatic tumor
cell line. Biochim Biophys Acta. 1076:273–281. 1991.
|
|
31
|
Ria R, Reale A, Castrovilli A, Mangialardi
G, Dammacco F, Ribatti D and Vacca A: Angiogenesis and progression
in human melanoma. Dermatol Res Pract. Article ID 185687.
View Article : Google Scholar : 2010.
|
|
32
|
Widnell CC, Schneider YJ, Pierre B,
Baudhuin P and Trout A: Evidence for a continual exchange of
5′-nucleotidase between the cell surface and cytoplasmic membranes
in cultured rat fibroblasts. Cell. 28:61–70. 1982.
|
|
33
|
van den Bosch R, Geuze HJ, du Maine APM
and Strous GJ: Transport and metabolism of 5′-nucleotidase in a rat
hepatoma cell line. Eur J Biochem. 160:49–54. 1986.
|
|
34
|
Airas L, Niemelä J, Salmi M, Puurunen T,
Smith DJ and Jalkanen S: Differential regulation and function of
CD73, a glycosyl-phosphatidylinositol-linked 70-kD adhesion
molecule, on lymphocytes and endothelial cells. J Cell Biol.
136:421–431. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chatterjee SK, Kim U and Bielat K: Plasma
membrane associated enzymes of mammary tumors as the biochemical
indicators of metastasizing capacity. Analyses of enriched plasma
membrane preparations. Br J Cancer. 33:15–26. 1976. View Article : Google Scholar
|
|
36
|
Lugassy C, Vernon SE, Busam K, Engbring
JA, Welch DR, Poulos EG, Kleinman HK and Barnhill RL: Angiotropism
of human melanoma: studies involving in transit and other cutaneous
metastases and the chicken chorioallantoic membrane: implications
for extravascular melanoma invasion and metastasis. Am J
Dermatopathol. 28:187–193. 2006. View Article : Google Scholar
|
|
37
|
Stochaj U, Richter H and Mannherz HG:
Chicken gizzard 5′-nucleotidase is a receptor for the extracellular
matrix component fibronectin. Eur J Cell Biol. 51:335–338.
1990.
|
|
38
|
Sträter N: Ecto-5′-nucleotidase: Structure
function relationships. Purinergic Signal. 2:343–350. 2006.
|
|
39
|
Méhul B, Aubery M, Mannherz HG and Codogno
P: Dual mechanism of laminin modulation of ecto-5′-nucleotidase
activity. J Cell Biochem. 52:266–274. 1993.PubMed/NCBI
|
|
40
|
Murphy BJ: Regulation of malignant
progression by the hypoxia-sensitive transcription factors HIF-1a
and MTF-1. Comp Biochem Physiol B Biochem Mol Biol. 139:495–507.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ohta A and Sitkovsky M: Role of
G-protein-coupled adenosine receptors in downregulation of
inflammation and protection from tissue damage. Nature.
414:916–920. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Spychala J: Tumor-promoting functions of
adenosine. Pharmacol Ther. 87:161–173. 2000. View Article : Google Scholar
|
|
43
|
Merighi S, Simioni C, Gessi S, Varani K,
Mirandola P, Tabrizi MA, Baraldi PG and Borea PA: A2B
and A3 adenosine receptors modulate vascular endothelial
growth factor and interleukin-8 expression in human melanoma cells
treated with etoposide and doxorubicin. Neoplasia. 11:1064–1073.
2009.
|
|
44
|
Tufto I, Lyng H and Rofstad EK: Vascular
density in human melanoma xenografts: relationship to angiogenesis,
perfusion and necrosis. Cancer Lett. 123:159–165. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Norrby K: In vivo models of angiogenesis.
J Cell Mol Med. 10:588–612. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pötgens AJ, Lubsen NH, van Altena MC,
Schoenmakers JG, Ruiter DJ and de Waal RM: Vascular permeability
factor expression influences tumor angiogenesis in human melanoma
lines xenografted to nude mice. Am J Pathol. 146197–146209.
1995.PubMed/NCBI
|