Introduction
Death-associated protein kinase (DAPK), a 160-kDa
calcium/calmodulin-dependent Ser/Thr kinase protein, is an
essential mediator for apoptotic cell death induced by various
stimuli, including interferon-γ (IFN-γ), tumor necrosis factor-α
(TNF-α), transforming growth factor-β (TGF-β) and Fas ligand
(1–3). DAPK is a well-known pro-apoptotic
protein as well as a tumor suppressor and is involved in a wide
variety of cellular apoptotic signaling cascades (3–7). It is
also triggered by cell death-regulator genes and deregulated in
various tumor types, including head, neck, non-small cell lung and
pancreatic cancer (8–10). In addition, the downregulation of
DAPK expression is observed in various types of tumors, including
B-cell lymphoma (11,12). The death domain of DAPK controls its
interactions with tumor necrosis factor receptor-1 (1), FADD (13), RSK (14), the mitogen-activated protein kinase
extracellular signal-regulated kinase (ERK), Src (15) and the UNC5H2-dependent receptor
(16). Of note, clinical studies
have significantly related the loss of DAPK expression with a more
malignant type of cancer, while upregulating the metastatic ability
of human cancer. In non-small cell lung tumors, DAPK promoter
methylation was closely associated with aggressive disease types
and poor survival (9,17–19).
However, how the cellular activities of DAPK are regulated in
vitro remains poorly understood.
The proliferation, migration, blood vessel invasion
and expansion of the primary capillary-like tubular network of the
endothelial cells during embryogenesis, angiogenesis and
vasculogenesis are mostly operated by the vascular endothelial
growth factor (VEGF) signaling cascade via the VEGF receptor-2
(VEGFR-2) (20). In tumor
angiogenesis and pathogenesis, the tumor microenvironment is one of
the most important factors for development, in which the
environment is often hypoxic. Tumor hypoxia is a condition of
oxygen deprivation in carcinoma cells, where hypoxic tumor cells
are generally resistant to chemo- and radio-therapy (21). The hypoxic environment leads to
genetic instability, which is beneficially related to tumor
progression. In addition, hypoxia promotes of hypoxia-inducible
factor-1α (HIF-1α) protein expression, which stimulates
angiogenesis and is correlated with poorer prognosis with the
activation of several genes such as VEGF, erythropoietin (EPO) and
nitric oxide synthases that are related to metastasis (22). The upregulation of VEGF occurs in
most aggressive solid cancers, including ovarian, breast, lung,
colon and uterus tumors, and is also closely connected to cancer
progression and poor prognosis (23–26).
In addition, VEGF and VEGFR-2 overexpression is also closely
related to a reduced disease-free survival rate (26) and overall survival with ovarian
tumors (27,28). Thus, the precise molecular mechanism
for ovarian carcinoma cells remains to be fully elucidated.
For the objective of the present study, we analyzed
ovarian carcinoma cells SKOV-3 as a model system in order to
examine the functional mechanism of the anti-angiogenic properties
of DAPK. We observed that DAPK markedly suppressed the expression
of VEGF and HIF-1α in carcinoma cells. Thus, we investigated a
possible molecular mechanism by which DAPK reduced VEGF production
in an in vitro model system.
Materials and methods
Cell lines, culture, chemicals and
antibodies
Human epithelial ovarian cancer cell lines SKOV-3
were grown in Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with 10% fetal bovine serum (FBS). Primary human
umbilical vein endothelial cells (HUVECs) were cultured on 0.3%
gelatin-coated dishes (Sigma, St. Louis, MO, USA) using EGM-2
BulletKit medium (Clonetics). Two cell lines were purchased from
the American Type Culture Collection (ATCC, Manassas, VA, USA). All
cells were cultured under 5% CO2 at 37°C. Rapamycin was
obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA)
and wortmannin and LY294002 were from Sigma. The primary antibodies
used in the present study were: anti-DAPK, anti-phospho-specific
PI3K, anti-PI3K, anti-phospho-specific Akt, anti-Akt, anti-HIF-1α,
anti-phospho-specific PDK-1, anti-PDK-1, anti-phospho-specific
TSC-2, anti-TSC-2, anti-VEGFR-2 (Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA), anti-VEGF121 (Ab-1; Oncogene,
Cambridge, MA, USA), anti-phospho-specific mTOR, anti-mTOR,
anti-phospho-specific 4E-BP1, anti-4E-BP1, anti-phospho-specific
p70S6K, anti-p70S6K (Cell Signaling Technology, Inc.) and
anti-β-actin (Sigma).
[3H]thymidine incorporation
assay
Cell proliferation was calculated by the
incorporation of radio-labeled thymidine in trichloroacetic acid as
previously described (29,30). HUVECs were plated in 96-well culture
plates at densities of 1.5×104 cells/well and were
labeled with [methyl-3H]thymidine for the last 4
h at 0.5 μCi/ml. The cells were filtered by absorption onto paper
and were rinsed twice with 5% TCA and twice with 95% ethanol. The
zone of dried paper corresponding to each well was eliminated with
a round hole-punch, and [3H]thymidine incorporation was
estimated by the liquid scintillation counter (Beckman
Instruments).
Cell invasion assay
Transwell cell chambers (8.0-μm pore size; Costar),
with 24 wells, were used to assay the cell invasion (29,31).
For the invasion assay, the lower surface of a filter was coated
with 10 μg/ml of gelatin overnight. M199 containing 1% FBS with
VEGF (10 ng/ml) was placed in the lower wells. The cells were
collected by trypsinization, washed, and re-suspended to
1×105 cells in 100 μl of fresh DMEM. Subsequently, the
cells were inoculated into the upper chamber and incubated for 6 h
at 37°C. The chamber was fixed with methanol and stained with 10
mg/ml H&E. The level of cell invasion was then quantified by
calculating the stained cells in five random areas per
membrane.
Capillary tube formation assay
The tube formation assay was carried out as
previously described using growth factor-reduced Matrigel (30,32).
In brief, HUVECs (3.5×105) were cultured on the surface
of the Matrigel. Seeded cells were then incubated with or without
10 ng/ml of VEGF for 48 h in M199 containing 1% FBS. After rinsing,
images were captured at ×40 magnification. Tube formation lengths
were estimated using an inverted microscope equipped with a digital
CCD camera (Zeiss), and quantification was measured using Image Lab
imaging software (MCM Design). The experiments were performed in
triplicate.
Western blotting
After transfections, cultured cells were collected,
rinsed twice with ice-cold PBS and lysed by adding ice-cold RIPA
lysis buffer containing a protease inhibitor cocktail (Sigma) at
4°C for 1 h. The extract was then transferred to a micro-tube and
centrifuged at 15,000 × g for 10 min. Subsequently, equal amounts
of protein were separated with 8–12% SDS-PAGE, and transferred onto
a Hybond-ECL nitrocellulose membrane (GE Healthcare, Little
Chalfont, Buckinghamshire, UK). After blocking, the membranes were
incubated with the indicated specific primary antibodies at 4°C
overnight. The membranes were rinsed thrice with TBST buffer and
incubated in either goat anti-rabbit or anti-mouse secondary
antibodies. Protein bands were developed using an ECL detection
system (GE Healthcare).
PI3K activity analysis
PI3 kinase activity was carried out as previously
reported (33,34). In brief, cells were grown at a
density of 2.6×106 cells. Following overnight
incubation, cells were transfected according to various
concentrations of the indicated specific expression plasmid. The
cells were then lysed with ice-cold buffer containing a protease
inhibitor cocktail. The lysates were centrifuged at 20,000 × g for
15 min at 4°C, and the supernatants were used as the cell lysate.
In order to immunoprecipitate PI3K, total proteins were incubated
with anti-p85 antibody, followed by incubation with protein
A-agarose beads for an additional 1 h at 4°C. Immunoprecipitates
were mixed with a kinase reaction buffer containing 200 μg/ml of
phosphatidylinositol and 2 μCi of [32P] ATP/assay
mixture at 37°C for 15 min. The reaction materials were detected
using auto-radiography and the radioactive compounds were evaluated
using a liquid scintillation counter.
Yeast two-hybrid system
cDNA, which encodes full-length human DAPK, was
introduced into the EcoRI and XhoI restriction enzyme
sites of a pGilda-LexA expression shuttle vector. A human
full-length VEGFR-1 and VEGFR-2 cDNA was subcloned into the pJG4–5
activation vector in order to generate B42 fusion proteins in the
EcoRI and XhoI sites, respectively. Positive
interactions were attested by the formation of black colonies on
the X-gal-containing medium as previously described (35). The binding activity of the
interaction was compared by estimating the relative expression
level of o-nitrophenyl β-D-galactopyranoside
(ONPG) β-galactosidase (35,36).
Luciferase reporter-gene assay
In vitro VEGFR-2 promoter activity was
carried out as previously reported (36). Briefly, cells at 85% confluency were
transfected with a VEGFR-2 reporter expression plasmid. After lysis
with RIPA buffer, total cell lysates were cleared with
centrifugation at 14,000 rpm for 15 min and cell extracts were
incubated with the luciferase substrate reagent at room temperature
for 30 min according to the manufacturer’s protocols. Then, a 5 μl
aliquot of each sample was quantitated using a MicroLumat Plus
LB96V luminometer.
Statistical analysis
Statistical analysis of the results was performed
using the Student’s t-test when compared between two groups. Data
are presented as the means ± SD with the error of the mean given
for triplicate experiments. The index for statistical significance
was P<0.05. The values with 95% confidence (P<0.05) are
depicted with an asterisk (*) on each graph. Each
experiment was repeated three times with similar results.
Results
Suppressor DAPK protein inhibits
VEGF-induced endothelial cell proliferation, migration and tube
formation ability
VEGF, as a potent multifunctional cytokine, is a
crucial regulator of the pathological and physiological
angiogenesis in tumors. The upregulation of VEGF appears in most
solid tumors involved in ovarian, breast, lung, colon and
aggressive uterus tumors, and is very closely related to tumor
progression and poor prognosis (24–26).
Therefore, the suppression of VEGF expression has been shown to
suppress tumor growth as well as migration, invasion and
metastasis. As a result of these observations, it may be stated
that VEGF enhances cell proliferation and migration via multiple
signaling pathways. To investigate whether DAPK may inhibit
angiogenesis, we first evaluated the effect of DAPK on endothelial
cell proliferation by a [3H]thymidine incorporation
assay. Generally, VEGF-stimulated DNA synthesis was calculated with
both untransfected cells and empty vector control-transfected cells
and then compared with the uninduced cells. DAPK significantly
decreased cell proliferation to 55–65% that of the expression
vector only (control) in these cells. In contrast, the loss of
function of DAPK by siRNA-mediated knockdown did not suppress cell
proliferation (Fig. 1A).
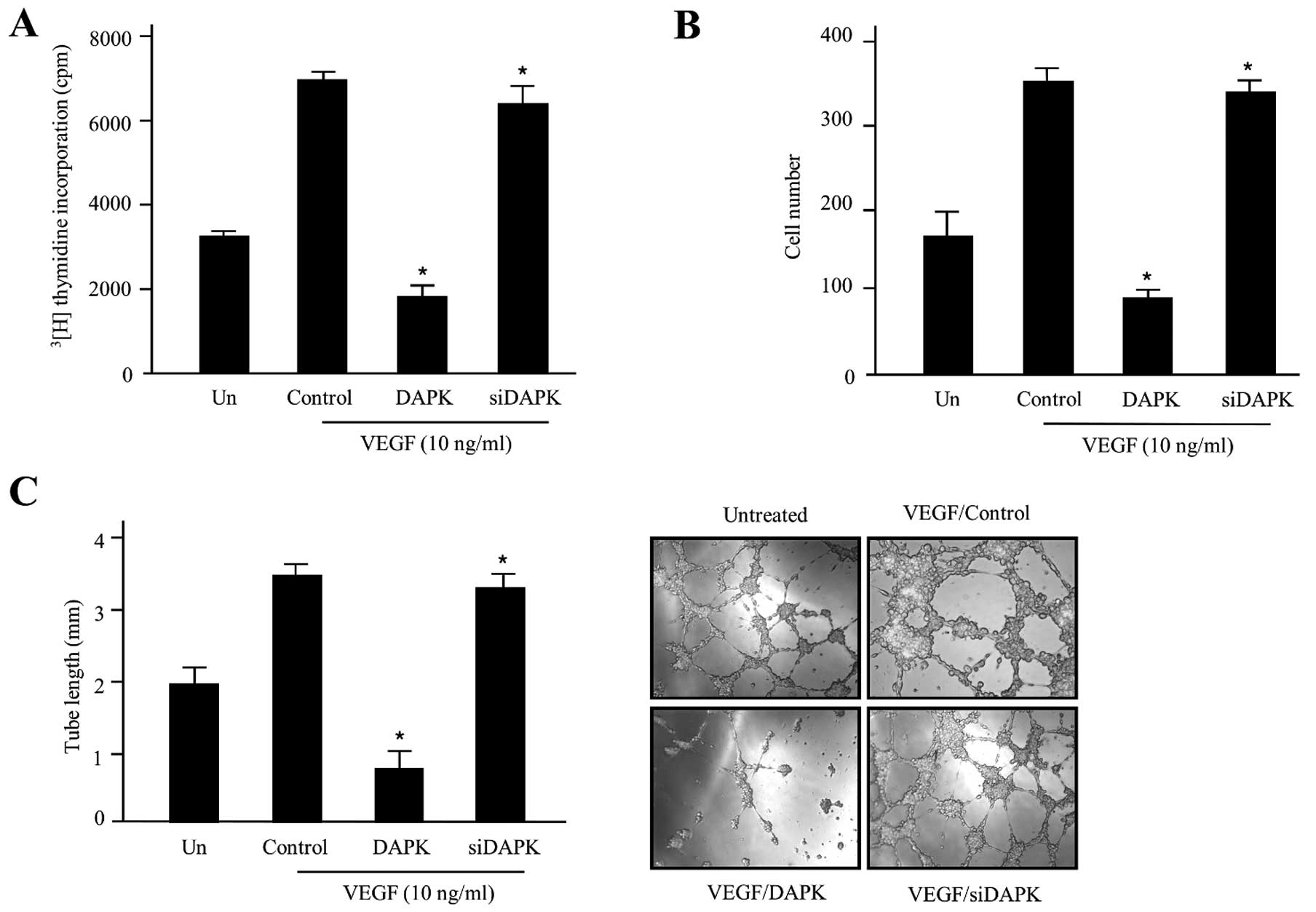 | Figure 1DAPK promotes anti-angiogenic
activity in HUVECs. (A) The inhibitory effect of DAPK on HUVEC
proliferation. Cells were incubated with 10 ng/ml stimulator VEGF
and then transfected with the control (expression vector only),
DAPK, or DAPK-siRNA, respectively. The cpm value of
[3H]thymidine was calculated using a liquid
scintillation counter. Data are shown as the means ± SD of three
separate experiments. (B) Invasion of HUVECs was evaluated using
the Transwell Boyden chamber. In the absence or presence of VEGF,
HUVECs (4.5×104 cells) were transfected with the control
(expression vector only), DAPK, or DAPK-siRNA, respectively. After
48 h incubation at 37°C, invading cells were counted under a
microscope and the mean values were estimated. The data are
expressed as the means ± SD of four separate experiments. (C)
Inhibitory effect of DAPK on HUVEC tube formation. HUVECs
(9.5×104) were cultured in 24-well plates containing
growth factor reduced Matrigel, and pre-treated with/without VEGF,
followed by transfection with the control, DAPK, or DAPK-siRNA.
Capillary-like tubular structures were photographed with a digital
camera attached to an inverted microscope. The tube lengths were
estimated and the data are presented as the means ± SD. Three
independent experiments were performed in triplicate.
*P<0.05 compared to the control. DAPK,
death-associated protein kinase; HUVECs, human umbilical vein
endothelial cells; VEGF, vascular endothelial growth factor. |
To address the potential function for DAPK
upregulation in decreasing cell proliferation and VEGF-modulated
angiogenesis, we additionally observed the invasive cell levels of
DAPK in HUVECs by the Boyden chamber Transwell assay. Cell invasion
is also an important step required for angiogenesis. As presented
in Fig. 1B, DAPK-siRNA did not
inhibit cell invasion of HUVECs, but DAPK markedly suppressed
VEGF-induced cell invasion without killing the cells. These data
indicate that DAPK may suppress the invasion of endothelial cells
by carcinoma cells. In the latter process, endothelial cell
capillary-like structures are an important event to become
elongated into tubes to form new blood vessels for angiogenesis. We
then examined the anti-angiogenic activities of DAPK on
VEGF-induced tube formation using an in vitro angiogenesis
model system. In the presence of DAPK-siRNA, endothelial cells were
almost none affected. In the presence of DAPK, the linear
structures of the capillary networks were disrupted (Fig. 1C). Collectively, these results
suggest that DAPK suppressed HUVEC invasion and network formation
may occur possibly via interruption of VEGF-mediated signaling
pathways.
DAPK downregulates PI3K and Akt
phosphorylation in a dose-dependent manner
The phosphorylation of PI3K/Akt is an important
process for signaling cascade in tumor angiogenesis. Akt plays a
pivotal role as a downstream regulator of PI3K and is also
regulated by various growth factors, such as epidermal growth
factor (EGF), insulin-like growth factor-1 (IGF-1) and transforming
growth factor-β1 (TGF-β1). Therefore, we assessed whether DAPK
suppressed PI3K and Akt phosphorylation in carcinoma cells. Total
cell lysates from the control and transfected cells of various
concentrations (0.2–1.0 μg) were subjected to immunoblot analysis,
which showed that the VEGF-stimulated phosphorylation of PI3K and
Akt played a pivotal role in VEGF-induced angiogenesis. As
presented in Fig. 2A, the activity
of PI3K transfected with various concentrations of DAPK was
suppressed in a concentration-dependent manner, with the maximum
effect at 1.0 μg. Following transfection with 0.4–0.6 μg of DAPK,
cells showed significantly suppressed phosphorylation of PI3K. We
also evaluated the effect of DAPK on the inhibition of Akt, which
is one of the major downstream components of PI3K. As expected,
DAPK gradually decreased the phosphorylation of Akt in a
dose-dependent manner (Fig. 2B).
This substrate was similarly able to suppress phosphorylation in
HUVECs (data not shown). Taken together, our results strongly
suggest that the PI3K/Akt-dependent signal cascades are critically
contained in the biological function of DAPK-regulated endothelial
cell. This inhibitory effect was comparable to that of some
well-known PI3K inhibitors, such as wortmannin and rapamycin, which
are well-known for targeting the mammalian target of rapamycin
(mTOR) signaling pathways. As indicated in Fig. 2C, DAPK markedly reduced the
phosphorylation of Akt (Ser473) and the phosphorylation of
eukaryotic translation initiation factor 4E-binding protein 1
(4E-BP1) (Tyr37/46), as well as the phosphorylation of
phosphoinositide-dependent protein kinase-1 (PDK1) (Ser241)
(Fig. 3C), one of the best
characterized targets of the mTOR complex. Subsequently, when DAPK
was co-treated with rapamycin, an inhibitor of mTOR, DAPK
significantly displayed the effects of deactivation in p-4E-BP1.
These results provide evidence for our former theory that DAPK is a
powerful inhibitor of PI3K/Akt activity.
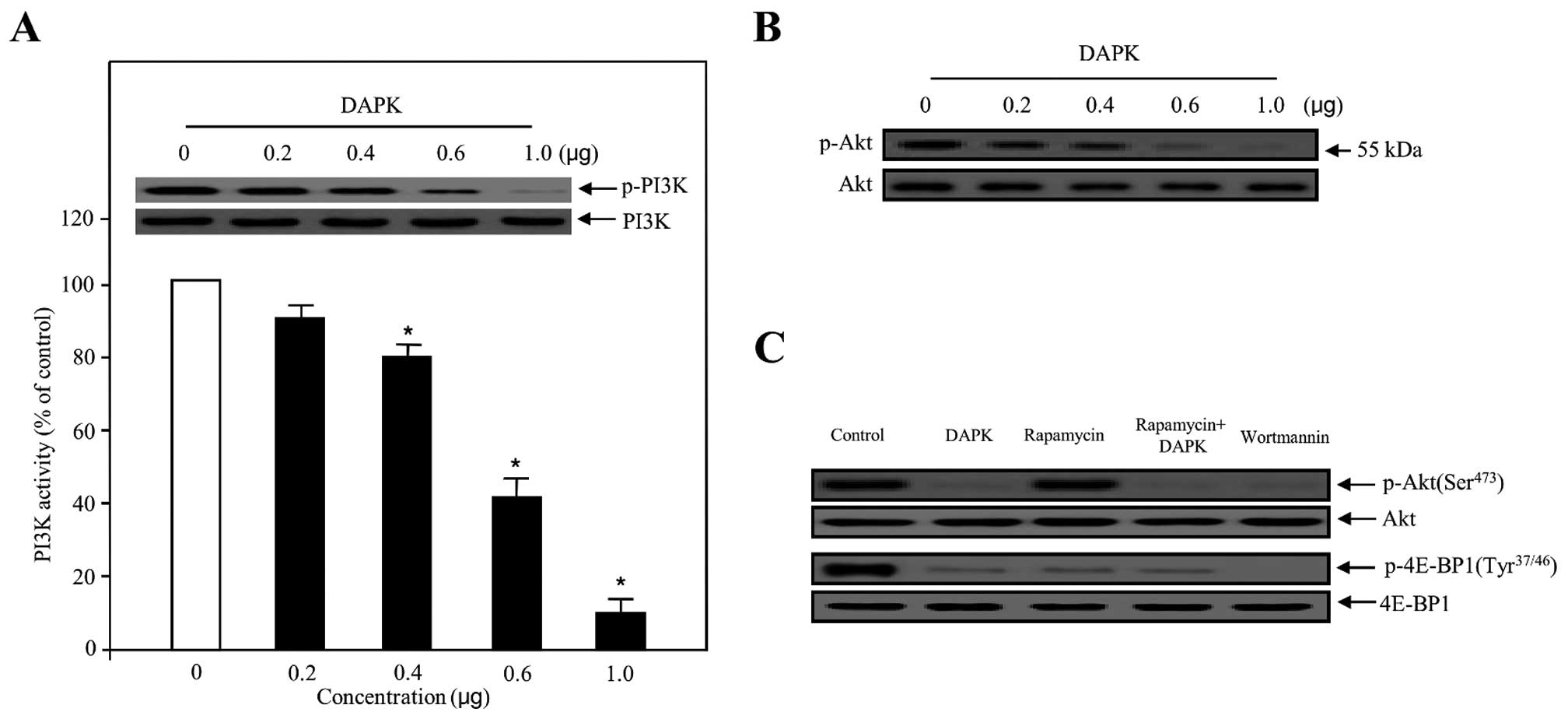 | Figure 2Effect of DAPK on PI3K activity and
PI3K/Akt phosphorylation in carcinoma cells. (A) The inhibitory
effect on PI3K activity, in the presence of DAPK, was evaluated
using the in vitro PI3 kinase assay system (bottom panel).
Experiments were performed in triplicate and error bars are shown
as means ± SD. *P<0.05. PI3K was transfected with
increasing concentrations of DAPK, collected and introduced to
western blotting for the indicated dose proteins (upper panel).
PI3K was used to verify equal loading of the samples. (B) Ovarian
carcinoma cells were transfected with various concentrations of
DAPK. The phosphorylation of Akt was determined using western blot
analysis. (C) Cells were transfected with control (expression
vector only), DAPK, rapamycin as an mTOR inhibitor, rapamycin plus
DAPK and wortmannin as a PI3K inhibitor, respectively. After 24 h,
the cells were collected, lysed on ice-cold RIPA buffer and western
blotting was carried out. Western blotting for unphosphorylated Akt
and 4E-BP1 was used as a loading control. DAPK, death-associated
protein kinase. |
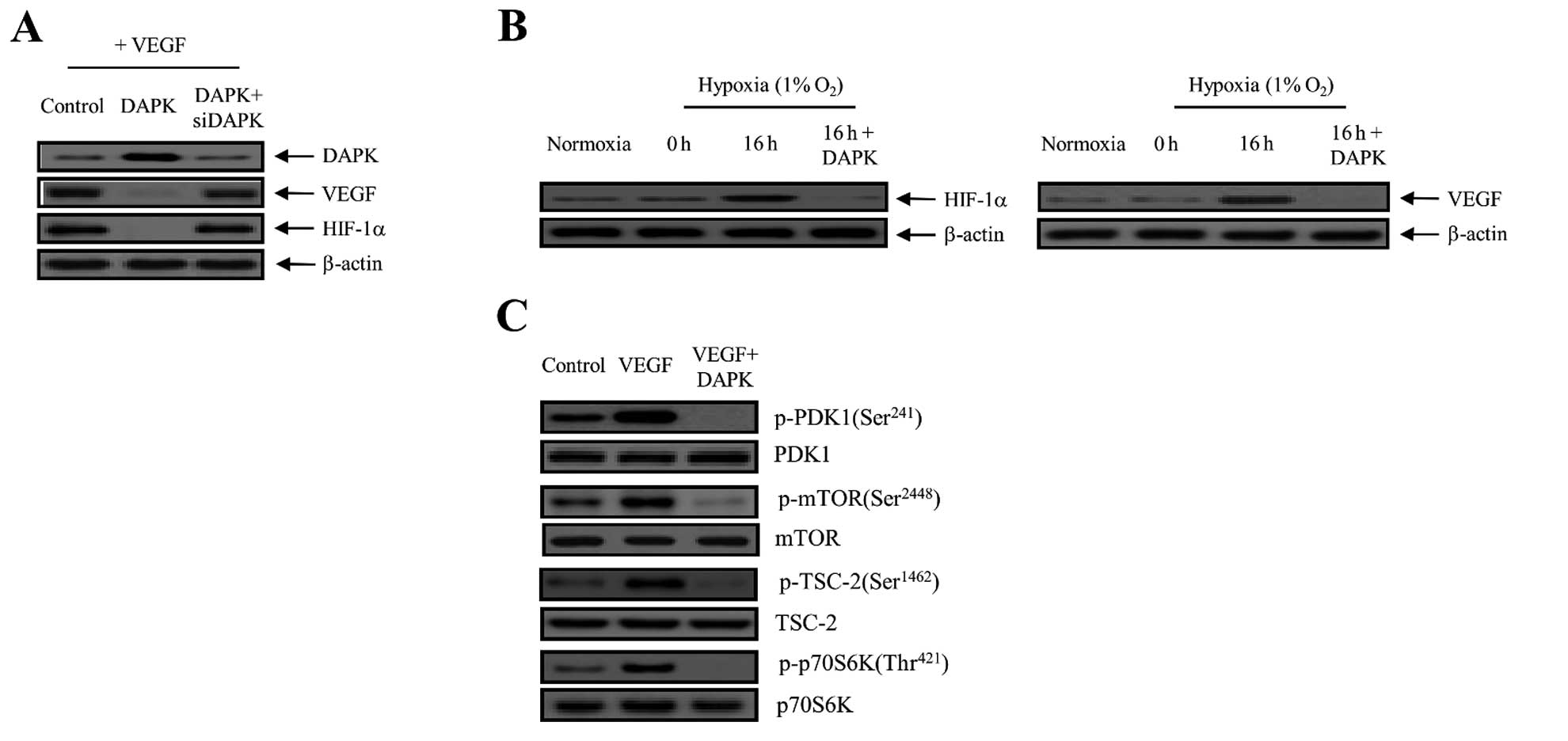 | Figure 3DAPK suppresses the expression of
VEGF and HIF-1α in carcinoma cells. (A) Cells were incubated with
10 ng/ml VEGF and then control (expression vector only)-transfected
or transfected with DAPK or DAPK plus DAPK-siRNA. VEGF and HIF-1α
expression was then detected by immunoblotting. Three independent
experiments were performed in triplicate. (B) DAPK inhibits
hypoxia-induced VEGF and HIF-1α activation. Cells were transfected
with DAPK for 16 h under either normoxic or hypoxic conditions.
Equal protein amounts were introduced to SDS-PAGE, blotted, and
incubated with either specific primary VEGF, HIF-1α, or β-actin
antibody. (C) After transfection with DAPK, cells were collected
for western blot analysis with primary antibodies specific for the
phosphorylated and non-phosphorylated proteins, which is indicative
of PDK1 phosphorylation, mTOR phosphorylation, TSC-2
phosphorylation and the phosphorylation of downstream modulators,
including p70S6K. The results shown are representative of three
independent experiments. DAPK, death-associated protein kinase;
VEGF, vascular endothelial growth factor; HIF-1α, hypoxia-inducible
factor-1α; PDK1, phosphoinositide-dependent protein kinase-1;
TSC-2, tuberous sclerosis complex-2. |
DAPK inhibits the expression of VEGF and
HIF-1α in ovarian cancer cells
In rapidly growing cancer, hypoxic conditions
strongly activate the expression of the transcription factor
HIF-1α, which in turn stimulates the expression of VEGF proteins in
carcinoma cells. VEGF expression levels modulate the effects of
other angiogenic regulators and therefore play key roles in the
regulation of tumor angiogenesis. In order to disrupt new blood
vessel formation in cancer, it is key to suppress the expression of
the VEGF and HIF-1α proteins in carcinoma cells. To address whether
DAPK inhibits the level of VEGF and HIF-1α protein expression via
the PI3K/Akt signaling pathway, we first evaluated the inhibitory
effect of DAPK on VEGF expression in SKOV-3 ovarian carcinoma
cells. As presented in Fig. 3A,
overexpression of DAPK clearly decreased the VEGF expression level,
whereas DAPK-siRNA had no effect. We also measured the effect of
DAPK on HIF-1α protein expression. HIF-1α is a key regulator for
VEGF expression as a transcription factor. As indicated in Fig. 3A, DAPK markedly decreased the
expression of the HIF-1α protein. On the other hand, the inhibitory
effect of DAPK was completely restored by DAPK-siRNA transfection.
Subsequently, we measured the levels of HIF-1α and VEGF protein
expression in SKOV-3 cells exposed to normoxia or 1% O2
hypoxia and DAPK transfection. After a 16-h treatment, levels of
HIF-1α and VEGF protein expression indicated that they were fully
activated. In contrast, HIF-1α and VEGF expression levels were
rapidly reduced after DAPK transfection (Fig. 3B). Therefore, it may be suggested
that hypoxia significantly promoted the levels of HIF-1α and VEGF
expression, whereas DAPK suppressed their activation. Next, we
examined the effects of DAPK on the downstream regulators in the
PI3K/Akt signaling pathway. DAPK significantly suppressed the
phosphorylation of mTOR on the Ser2448 residue position,
tuberous sclerosis complex-2 (TSC-2) on the Ser1462
residue position, and p70 ribosomal protein S6 kinase (p70S6K) on
the Thr421 residue position, which are downstream
factors essential to mTOR (Fig.
3C). These results indicate that DAPK inhibits the autocrine
effect of VEGF in endothelial cells and, therefore, has a direct
anti-angiogenic effect, which leads to the inhibition of tumor
angiogenesis, metastasis and tumorigenesis.
DAPK inhibits VEGF-induced VEGFR-2
phosphorylation through the interaction with VEGFR-2 protein
VEGFR-2 is a key modulator of the VEGF-induced
endothelial cell involved cellular based-physiological/pathological
function. To explore the biological/functional relevance of the
association between VEGFR-2 and DAPK, we evaluated the effect of
DAPK on VEGF-stimulated VEGFR-2 phosphorylation in HUVECs. As shown
in Fig. 4A, overexpression of DAPK
markedly decreased VEGF-stimulated VEGFR-2 phosphorylation, while
DAPK-siRNA had no effect. These new findings indicated that DAPK
strongly suppressed protein levels of VEGFR-2 phosphorylation in
vitro in HUVECs. Direct interaction between the two proteins is
crucial for the majority of cellular biological/physiological
mechanisms. For example, signal transductions from the exterior of
the cell are specifically mediated to the inside of the cell by
protein-protein interactions of the signaling components. This
process plays a crucial role in the cellular biological mechanisms
and in various diseases involving aggressive solid cancer.
Protein-protein interactions are central to almost every process in
the living cell. Thus, we also demonstrated the interaction between
DAPK and VEGFR-2 protein in an in vivo and in vitro
system. VEGFR-1 and expression vector only were used as the
negative control. As shown in Fig.
4B, DAPK with VEGFR-2, but not with VEGFR-1 and expression
vector only, permitted yeast cell growth on a leucine-depleted
plate, and β-galactosidase activity between DAPK and VEGFR-2 was
fully activated (84.27±1.15). However, control (vector only)
(1.64±0.58) and VEGFR-1 (1.88±0.54) expression activity failed,
suggesting a specific interaction between DAPK and VEGFR-2.
Consistent with these results, direct interactions between DAPK and
VEGFR-2 were confirmed using co-immunoprecipitation in vitro
assays. Recombinant plasmids of DAPK (pcDNA3.1/Flag-DAPK) and
VEGFR-2 (pcDNA3.1-VEGFR-2), or pcDNA3.1/Flag-DAPK and expression
vector only (pcDNA3.1) were co-transfected into the HEK293T cells.
Subsequently, an immunoprecipitation was incubated using anti-Flag
specific primary antibody with total cell lysates from both
transfected cells. After immunoprecipitation, the precipitated
proteins were detected with a specific anti-DAPK or anti-VEGFR-2
primary antibody. As indicated in Fig.
4C, pcDNA3.1-VEGFR-2 was co-immunoprecipitated with
pcDNA3.1/Flag-DAPK (lane 2 in upper panel), whereas it failed with
pcDNA3.1 plasmid (expression vector only) (lane 1 in upper panel).
At the same time, in order to confirm the direct interaction
between endogenous DAPK and the VEGFR-2 protein in cellular
physiological conditions, we subjected this construct to
immunoprecipitation with the proteins endogenously expressed in the
HEK293T cells using a specific anti-DAPK or anti-VEGFR-2 primary
antibody. The endogenous DAPK protein was then directly
co-immunoprecipitated with the VEGFR-2 protein (Fig. 4D). Our results strongly indicate the
binding of endogenous VEGFR-2 to the DAPK protein in the cells.
Next, to address whether DAPK regulates VEGFR-2, the effect of DAPK
on VEGFR-2 transcription activity was also calculated by a
luciferase reporter assay, using a construct integrating a VEGFR-2
promoter fused to the luciferase gene. The luciferase activity was
gradually inhibited by transient transfection of DAPK in a
concentration-dependent manner (Fig.
4E), providing further evidence of the importance of DAPK for
the regulation of VEGFR-2 activity. These results strongly suggest
that the overexpression of DAPK downregulates its transcriptional
activity. The results validated that DAPK obstructs the VEGFR-2
transcriptional activation level by inhibiting VEGFR-2
phosphorylation via the PI3K/Akt signaling cascade.
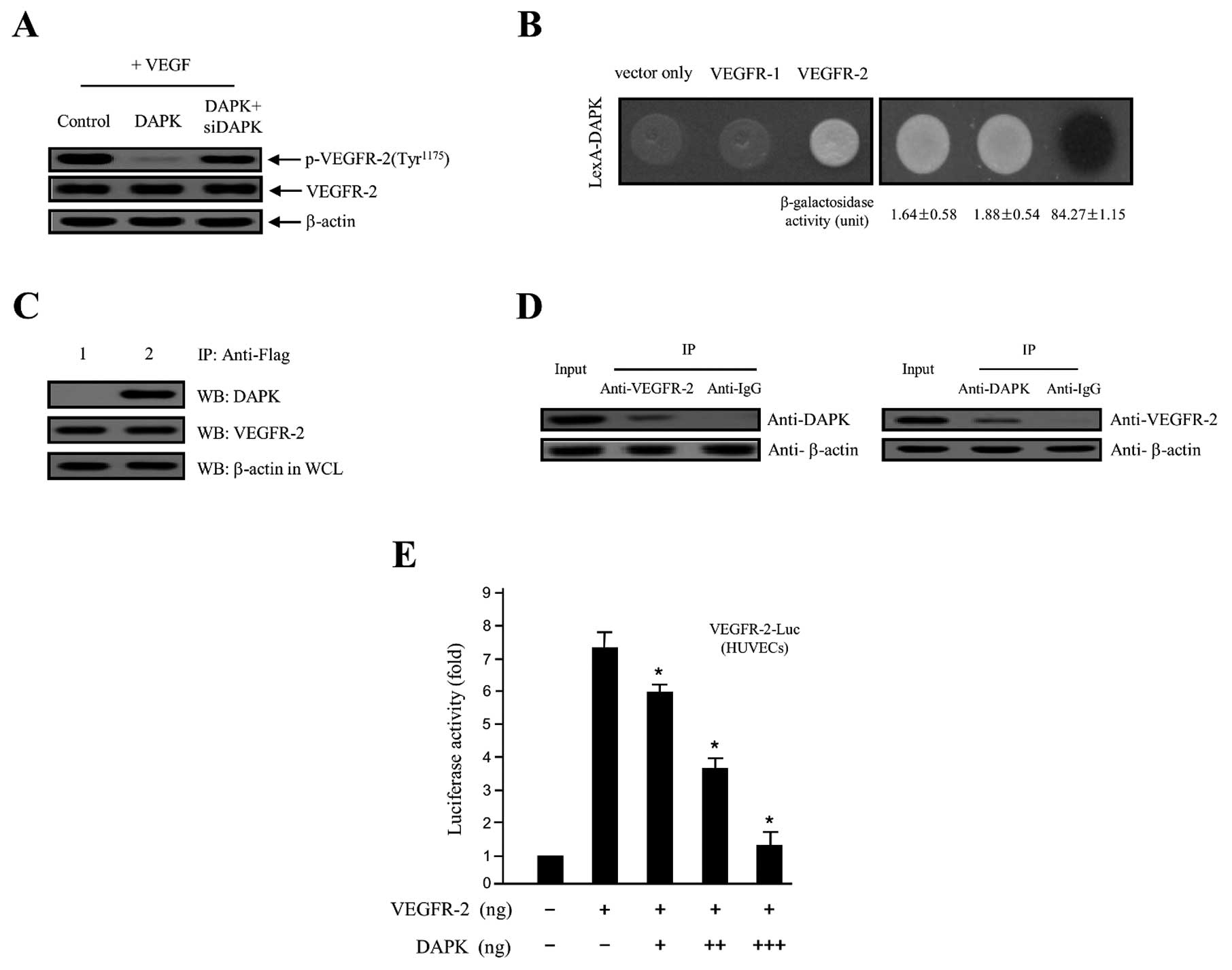 | Figure 4DAPK inhibits VEGF-induced VEGFR-2
phosphorylation through the interaction with VEGFR-2 protein. (A)
HUVECs were treated with VEGF angiogenesis inducer and then
transfected with the control (expression vector only), DAPK, or
DAPK plus DAPK-siRNA, respectively. Phosphorylation of VEGFR-2 was
detected using the indicated specific primary antibody.
Unphosphorylated VEGFR-2 and β-actin were used as the loading
control. (B) Physical interaction between GABARBP and VEGFR-2.
Direct interactions were observed by monitoring cell growth on
deplete-leucine amino acid plate, and through the formation of a
black colony on the cultured plate containing the X-gal reagent
(37,38). The values of the β-galactosidase
activity (unit), calculated by adding o-nitrophenyl
β-D-galactopyranoside (ONPG) assays, are indicated below
the corresponding lanes (37,38).
(C) Co-immunoprecipitation of DAPK with VEGFR-2.
Immunoprecipitation began by using primary anti-Flag antibody with
lysates from both transfected HEK293T cells. After
immunoprecipitation, precipitated proteins were visualized using
the primary anti-DAPK and anti-VEGFR-2 antibody. Lane 1, pcDNA3.1
(expression vector only) and pcDNA3.1/Flag-DAPK transfectant; lane
2, pcDNA3.1/Flag-DAPK and pcDNA3.1-VEGFR-2 transfectant. (D) In
vitro co-immunoprecipitation between the endogenous VEGFR-2 and
DAPK shows the interactions of the two proteins. (E) Inhibition of
the VEGFR-2-dependent transcriptional activity by DAPK. HUVECs were
co-transfected with 500 ng of VEGFR-2-Luc, 500 ng of a VEGFR-2
inserted expression vector (pcDNA3.1/VEGFR-2) and increasing
concentrations of gene-encoding DAPK (pcDNA3.1/Flag-DAPK) (100, 250
and 500 ng). The results shown are representative of at least three
independent experiments. The values are expressed as the means ±
SD. *P<0.05 compared to the control. DAPK,
death-associated protein kinase; VEGF, vascular endothelial growth
factor; HUVECs, human umbilical vein endothelial cells; VEGFR-2,
VEGF receptor-2. |
Discussion
In the present study, we demonstrated that the
possible intracellular mechanisms of DAPK have an inhibitory effect
on the roles of VEGF-induced angiogenesis in the endothelial cell
system. In angiogenesis and vasculogenesis, VEGF is a key regulator
serving as a potent cytokine in endothelial cell proliferation,
migration and survival (37).
Generally, angiogenic signaling cascades are a route mediated by
VEGF and their receptors (VEGFRs) (38). VEGF expression is closely correlated
with the regulation of tumor growth, metastasis and aggression, as
well as poor survival (27,39,40).
Notably, most human solid tumors greatly increase VEGF and HIF-1α
expression, which promote tumor angiogenesis and tumor growth.
Thus, inhibition of tumor angiogenesis by the interruption of VEGF
and HIF-1α expression is becoming a crucial approach for cancer
treatment (41). One of many
studies reported that the maximal suppression of tumor angiogenesis
and growth can be accomplished by completely disturbing the
circulation of VEGF (42). Herein,
we established a new cellular molecular mechanism for DAPK as a
novel potent angiogenic factor that may target through the
disturbance of VEGF and HIF-1α expression in the PI3K/Akt signaling
cascade using a tumor model system. As presented in Fig. 1, ectopically expressed DAPK markedly
inhibited major events in VEGF-induced angiogenesis in
vitro, which involved endothelial cell proliferation and cell
migration. Meanwhile, overexpression of DAPK completely abrogated
the VEGF-induced capillary-like tubular structure network. In the
presence of DAPK-siRNA, the endothelial cells were nearly none
affected. Collectively, these results strongly indicate that DAPK
specifically mediates VEGF-induced HUVEC migration and tube
formation.
HIF-1 increases the transcriptional levels of
various genes, including VEGF in tumor progression (43). HIF-1α is frequently overexpressed in
several human solid cancers (44),
and its activation is closely associated with tumor angiogenesis
and tumorigenesis progression in cells (45). Additionally, HIF-1 upregulates the
transcription of the VEGF gene by binding to the hypoxia-response
element (HRE) in the VEGF promoter region (46). Fang et al (47) reported that apigenin significantly
suppresses the expression of HIF-1α and VEGF in ovarian carcinoma
cells. As mentioned above, HIF-1α is a major modulator for VEGF
expression as a transcription factor. As shown in Fig. 3A, DAPK strongly downregulated the
expression of HIF-1α protein. In contrast, the inhibitory effect of
DAPK was completely restored by DAPK-siRNA transfection.
Subsequently, we assessed the levels of HIF-1α and VEGF protein
expression in SKOV-3 ovarian carcinoma cells exposed to normoxia or
1% O2 hypoxia and DAPK transfection. After a 16-h
treatment, the levels of HIF-1α and VEGF protein expression
indicated that they were fully activated. On the other hand, levels
of HIF-1α and VEGF protein expression rapidly decreased after DAPK
transfection (Fig. 3B).
Collectively, conditions of hypoxia were significantly upregulated
in the levels of HIF-1α and VEGF protein expression, whereas DAPK
inhibited its activation.
VEGF phosphorylates through the interaction with
VEGFR-2 and its downstream signaling component fully activates
PI3K/Akt phosphorylation, which is a potent cytokine in the
cellular control of endothelial cell growth and survival of various
types of tumor, including ovarian cancer (48–51).
Akt activates mTOR and is mediated by mTOR via a negative- and
positive-feedback biological system (52), while also acting as a key regulator
for cell proliferation, enhancing cell survival through various
biological mechanisms. Furthermore, Akt is involved in that Akt1
and Akt3, two downstream modulators of the PI3K signaling pathway,
have their crucial roles in ovarian tumorigenesis played via
control of VEGF secretion and angiogenesis (53,54).
In spite of these reports, the functional mechanisms of ovarian
tumor angiogenesis are still not understood. In the present study,
we found that DAPK can inhibit the phosphorylation of VEGF-induced
PI3K and Akt in a dose-dependent manner in vitro (Fig. 2). In contrast, DAPK-siRNA
transfection fully restored DAPK-reduced phosphorylation of both
PI3K and Akt (data not shown). This inhibitory effect was
comparable to that of other well-known PI3K inhibitors, such as
wortmannin and rapamycin, which are well-known mTOR signaling
pathways. As presented in Fig. 2C,
DAPK clearly downregulated the phosphorylation of Akt (Ser473) and
the phosphorylation of 4E-BP1 (Tyr37/46), as well as the
phosphorylation of PDK1 (Ser241) (Fig.
3C), one of the best characterized targets of the mTOR complex.
Subsequently, ectopic expression of DAPK significantly reduced
VEGF-induced VEGFR-2 phosphorylation, whereas DAPK-siRNA did not
have any effect. This new finding suggests that DAPK strongly
inhibits the levels of VEGFR-2 phosphorylation in vitro in
HUVECs. The direct interaction between two proteins is important
for the majority of the cellular biological/functional mechanisms
(Fig. 4). Collectively, the results
indicate that DAPK can obstruct VEGFR-2 transcriptional activity by
inhibiting VEGFR-2 phosphorylation through the PI3K/Akt signaling
cascade.
In summary, we propose that the phosphorylation of
VEGFR-2 has an influence on the cellular biological function of
tumor angiogenesis. In the present study, we further validated a
new biological/physiological function and cellular molecular
mechanism for DAPK accurately, which serves as a novel potential
anti-angiogenic factor that can mediate the phosphorylation of
PI3K/mTOR/4E-BP1 via the interruption of the VEGF and HIF-1α
expression levels. Therefore, a combination therapy with DAPK and
traditional drugs may be a useful approach for more advanced
ovarian tumor, recurrent and certain malignant patients.
Acknowledgements
This study was supported by grants from the National
Cancer Center (NCC-1210470-2).
Abbreviations:
|
DAPK
|
death-associated protein kinase
|
|
VEGF
|
vascular endothelial growth factor
|
|
HIF-1α
|
hypoxia-inducible factor-1α
|
|
PDK1
|
phosphoinositide-dependent protein
kinase-1
|
|
mTOR
|
mammalian target of rapamycin
|
|
TSC-2
|
tuberous sclerosis complex-2
|
|
4E-BP1
|
eukaryotic translation initiation
factor 4E binding protein 1
|
|
HUVECs
|
human umbilical vein endothelial
cells
|
References
|
1
|
Cohen O, Inbal B, Kissil JL, et al:
DAP-kinase participates in TNF-α- and Fas-induced apoptosis and its
function requires the death domain. J Cell Biol. 146:141–148.
1999.
|
|
2
|
Jang CW, Chen CH, Chen CC, et al: TGF-β
induces apoptosis through Smad-mediated expression of DAP-kinase.
Nat Cell Biol. 4:51–58. 2002.
|
|
3
|
Bialik S and Kimchi A: The
death-associated protein kinases: structure, function, and beyond.
Annu Rev Biochem. 75:189–210. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Deiss LP, Feinstein E, Berissi H, Cohen O
and Kimchi A: Identification of a novel serine/threonine kinase and
a novel 15-kD protein as potential mediators of the gamma
interferon-induced cell death. Genes Dev. 9:15–30. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cohen O, Feinstein E and Kimchi A:
DAP-kinase is a Ca2+/calmodulin-dependent,
cytoskeletal-associated protein kinase, with cell death-inducing
functions that depend on its catalytic activity. EMBO J.
16:998–1008. 1997.
|
|
6
|
Raveh T, Droguett G, Horwitz MS, DePinho
RA and Kimchi A: DAP kinase activates a p19ARF/p53-mediated
apoptotic checkpoint to suppress oncogenic transformation. Nat Cell
Biol. 3:1–7. 2001.PubMed/NCBI
|
|
7
|
Michie AM, McCaig AM, Nakagawa R and
Vukovic M: Death-associated protein kinase (DAPK) and signal
transduction: regulation in cancer. FEBS J. 277:74–80. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sanchez-Cespedes M, Esteller M, Wu L, et
al: Gene promoter hypermethylation in tumors and serum of head and
neck cancer patients. Cancer Res. 60:892–895. 2000.PubMed/NCBI
|
|
9
|
Kim DH, Nelson HH, Wiencke JK, et al:
Promoter methylation of DAP-kinase: association with advanced stage
in non-small cell lung cancer. Oncogene. 20:1765–1770. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dansranjavin T, Möbius C, Tannapfel A, et
al: E-cadherin and DAP kinase in pancreatic adenocarcinoma and
corresponding lymph node metastases. Oncol Rep. 15:1125–1131.
2006.PubMed/NCBI
|
|
11
|
Kissil JL, Feinstein E, Cohen O, et al:
DAP-kinase loss of expression in various carcinoma and B-cell
lymphoma cell lines: possible implications for role as tumor
suppressor gene. Oncogene. 15:403–407. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Katzenellenbogen RA, Baylin SB and Herman
JG: Hypermethylation of the DAP-kinase CpG island is a common
alteration in B-cell malignancies. Blood. 93:4347–4353.
1999.PubMed/NCBI
|
|
13
|
Henshall DC, Araki T, Schindler CK, et al:
Expression of death-associated protein kinase and recruitment to
the tumor necrosis factor signaling pathway following brief
seizures. J Neurochem. 86:1260–1270. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Anjum R, Roux PP, Ballif BA, Gygi SP and
Blenis J: The tumor suppressor DAP kinase is a target of
RSK-mediated survival signaling. Curr Biol. 15:1762–1767. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen CH, Wang WJ, Kuo JC, et al:
Bidirectional signals transduced by DAPK-ERK interaction promote
the apoptotic effect of DAPK. EMBO J. 24:294–304. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liambi F, Lourenço FC, Gozuacik D, et al:
The dependence receptor UNC5H2 mediates apoptosis through
DAP-kinase. EMBO J. 24:1192–1201. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tang X, Khuri FR, Lee JJ, et al:
Hypermethylation of the death-associated protein (DAP) kinase
promoter and aggressiveness in stage I non-small-cell lung cancer.
J Natl Cancer Inst. 92:1511–1516. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Harden SV, Tokumaru Y, Westra WH, et al:
Gene promoter hypermethylation in tumors and lymph nodes of stage I
lung cancer patients. Clin Cancer Res. 9:1370–1375. 2003.PubMed/NCBI
|
|
19
|
Lu C, Soria JC, Tang X, et al: Prognostic
factors in resected stage I non-small-cell lung cancer: A
multivariate analysis of six molecular markers. J Clin Oncol.
22:4575–4583. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Risau W: Mechanisms of angiogenesis.
Nature. 386:671–674. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Denny WA: Prodrug strategies in cancer
therapy. Eur J Med Chem. 36:577–595. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Blagosklonny MV: Antiangiogenic therapy
and tumor progression. Cancer Cell. 5:13–17. 2004. View Article : Google Scholar
|
|
23
|
Olson TA, Mohanraj D, Carson LF and
Ramakrishnan S: Vascular permeability factor gene expression in
normal and neoplastic human ovaries. Cancer Res. 54:276–280.
1994.PubMed/NCBI
|
|
24
|
Paley PJ, Staskus KA, Gebhard K, et al:
Vascular endothelial growth factor expression in early stage
ovarian carcinoma. Cancer. 80:98–106. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ohta Y, Tomita Y, Oda M, et al: Tumor
angiogenesis and recurrence in stage I non-small cell lung cancer.
Ann Thorac Surg. 68:1034–1038. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ishigami SI, Arii S, Furutani M, et al:
Predictive value of vascular endothelial growth factor (VEGF) in
metastasis and prognosis of human colorectal cancer. Br J Cancer.
78:1379–1384. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yamamoto S, Konishi I, Mandai M, et al:
Expression of vascular endothelial growth factor (VEGF) in
epithelial ovarian neoplasms: correlation with clinicopathology and
patient survival, and analysis of serum VEGF levels. Br J Cancer.
76:1221–1227. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Spannuth WA, Nick AM, Jennings NB, et al:
Functional significance of VEGFR-2 on ovarian cancer cells. Int J
Cancer. 124:1045–1053. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee OH, Kim YM, Lee YM, et al: Sphingosine
1-phosphate induces angiogenesis: its angiogenic action and
signaling mechanism in human umbilical vein endothelial cells.
Biochem Biophys Res Commun. 264:743–750. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Byun HJ, Lee JH, Kim BR, et al:
Anti-angiogenic effects of thioridazine involving the FAK-mTOR
pathway. Microvasc Res. 84:227–234. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Senger DR, Ledbetter SR, Claffey KP, et
al: Stimulation of endothelial cell migration by vascular
permeability factor/vascular endothelial growth factor through
cooperative mechanisms involving the alphavbeta3 integrin,
osteopontin, and thrombin. Am J Pathol. 149:293–305. 1996.
|
|
32
|
Benelli R and Albini A: In vitro models of
angiogenesis: the use of Matrigel. Int J Biol Markers. 14:243–246.
1999.PubMed/NCBI
|
|
33
|
Fruman DA, Mauvais-Jarvis F, Pollard DA,
et al: Hypoglycaemia, liver necrosis and perinatal death in mice
lacking all isoforms of phosphoinositide 3-kinase p85α. Nat Genet.
26:379–382. 2000.PubMed/NCBI
|
|
34
|
Lee KB, Byun HJ, Park SH, et al: CYR61
controls p53 and NF-κB expression through PI3K/Akt/mTOR pathways in
carboplatin-induced ovarian cancer cells. Cancer Lett. 315:86–95.
2012.PubMed/NCBI
|
|
35
|
Rho SB, Kim MJ, Lee JS, et al: Genetic
dissection of protein-protein interactions in multi-tRNA synthetase
complex. Proc Natl Acad Sci USA. 96:4488–4493. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rho SB, Song YJ, Lim MC, et al: Programmed
cell death 6 (PDCD6) inhibits angiogenesis through PI3K/mTOR/p70S6K
pathway by interacting of VEGFR-2. Cell Signal. 24:131–139. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Plate KH: Control of tumor growth via
inhibition of tumor angiogenesis. Adv Exp Med Biol. 451:57–61.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ferrara N: Role of vascular endothelial
growth factor in physiologic and pathologic angiogenesis:
therapeutic implications. Semin Oncol. 29(Suppl 16): S10–S14. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mu J, Abe Y, Tsutsui T, et al: Inhibition
of growth and metastasis of ovarian carcinoma by administering a
drug capable of interfering with vascular endothelial growth factor
activity. Jpn J Cancer Res. 87:963–971. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hartenbach EM, Olson TA, Goswitz JJ, et
al: Vascular endothelial growth factor (VEGF) expression and
survival in human epithelial ovarian carcinomas. Cancer Lett.
121:169–175. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ferrara N and Davis-Smyth T: The biology
of vascular endothelial growth factor. Endocr Rev. 18:4–25. 1997.
View Article : Google Scholar
|
|
42
|
Gerber HP, Kowalski J, Sherman D, Eberhard
DA and Ferrara N: Complete inhibition of rhabdomyosarcoma xenograft
growth and neovascularisation requires blockade of both tumor and
host vascular endothelial growth factor. Cancer Res. 60:6253–6258.
2000.
|
|
43
|
Semenza GL: Hypoxia, clonal selection, and
the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol.
35:71–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhong H, De Marzo AM, Laughner E, et al:
Overexpression of hypoxia-inducible factor 1α in common human
cancers and their metastases. Cancer Res. 59:5830–5835. 1999.
|
|
45
|
Maxwell PH, Dachs GU, Gleadle JM, et al:
Hypoxia-inducible factor-1 modulates gene expression in solid
tumors and influences both angiogenesis and tumor growth. Proc Natl
Acad Sci USA. 94:8104–8109. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Forsythe JA, Jiang BH, Iyer NV, et al:
Activation of vascular endothelial growth factor gene transcription
by hypoxia-inducible factor 1. Mol Cell Biol. 16:4604–4613.
1996.PubMed/NCBI
|
|
47
|
Fang J, Xia C, Cao Z, et al: Apigenin
inhibits VEGF and HIF-1 expression via PI3K/AKT/p70S6K1 and
HDM2/p53 pathways. FASEB J. 19:342–353. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Altomare DA, Wang HQ, Skele KL, et al: AKT
and mTOR phosphorylation is frequently detected in ovarian cancer
and can be targeted to disrupt ovarian tumor cell growth. Oncogene.
23:5853–5857. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Olsson AK, Dimberg A, Kreuger J and
Caesson-Welsh L: VEGF receptor signaling - in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Engelman JA: Targeting PI3K signalling in
cancer: opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hanrahan AJ, Schultz N, Westfal ML, et al:
Genomic complexity and AKT dependence in serous ovarian cancer.
Cancer Discov. 2:56–67. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Guertin DA and Sabatini DM: An expanding
role for mTOR in cancer. Trends Mol Med. 11:353–361. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Xia C, Meng Q, Cao Z, Shi X and Jiang BH:
Regulation of angiogenesis and tumor growth by p110 alpha and AKT1
via VEGF expression. J Cell Physiol. 209:56–66. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Liby TA, Spyropoulos P, Buff Lindner H, et
al: Akt3 controls vascular endothelial growth factor secretion and
angiogenesis in ovarian cancer cells. Int J Cancer. 130:532–543.
2012. View Article : Google Scholar : PubMed/NCBI
|














