Introduction
Synthetic sickness/lethality interaction is a highly
attractive strategy for cancer therapy (1–4). For
example, in cancer cells with a KRAS gene mutation, the
inhibition of polo-like kinase 1 (PLK1) resulted in cell death
(5). Similarly, cancer cells with
the KRAS mutation were sensitive to the suppression of the
serine/threonine kinase STK33 (6).
Moreover, dysfunction of DNA double-strand break repair caused by
mutations in BRCA1 or BRCA2 gene sensitized cells to
the inhibition of poly-ADP ribose polymerase (PARP) enzymatic
activity, resulting in chromosomal instability, cell cycle arrest,
and subsequent apoptosis (7). This
concept had been proved by a phase II trial where olaparib, a PARP
inhibitor, provided objective antitumor activity in patients with a
BRCA1 or BRCA2 mutation (8).
TP53 is the most commonly mutated tumor
suppressor gene in several different types of human cancer
(9). TP53 encodes the 393
amino acid p53 protein, which binds to specific DNA sequences in
the regulatory region of downstream genes (10). A variety of cellular stressors
including ultraviolet rays, ionizing radiation, chemotherapeutic
drugs, and hypoxia stabilize the p53 protein, and
post-translational modifications activate it; this results in
various cellular responses including cell cycle arrest, DNA repair
and apoptosis (11,12).
According to the TP53 mutation databases,
~75% of the mutations are missense mutations (13,14);
to date, >1,200 distinct missense mutations have been reported.
Among them, those at residues Arg175(R175),
Gly245(G245), Arg248(R248),
Arg249(R249), Arg273(R273) and
Arg282(R282) have been reported most frequently
(15). The most common p53 mutant
proteins caused by TP53 hot-spot mutations are R175H, G245S,
R248W, R248Q, R249S and R273H; these mutations cause a loss of the
trans-activation function of downstream genes (16). However, some p53 mutants gain new
functions that are not observed in wild-type p53 (so called
gain-of-function mutations). For example, mice with the knock-in
mutant p53 R172H and R270H, which correspond to human p53 R175H and
R273H mutations, develop a variety of novel tumors such as lung
adenocarcinoma, renal cancer, hepatocellular carcinoma, and
intestinal carcinoma which are not generally observed in
TP53-null mice (17). In
addition, embryonic fibroblasts derived from p53 R172H knock-in
mice gained activities of cell proliferation, DNA synthesis and
retroviral transformation (18).
Moreover, human p53 R273H or R248W interacted with Mre11 and
suppressed the binding of the Mre11-Ras50-NBS1 (MRN) complex to DNA
double-strand breaks, resulting in the chromosomal translocation
and abrogation of the G2/M check point (19). According to these results, it has
been hypothesized that some p53 mutant proteins, such as the
activated K-ras protein, are oncogenic and contribute to
carcinogenesis and cancer progression.
In the present study, we conducted high-throughput
RNAi screening by a lentiviral gene suppression system to identify
synthetic sick/lethal genes in the presence of p53 R175H, which
accounts for ~6% of the missense mutations identified in human
cancer (20). As a result, we
identified that inhibitor of differentiation 1 (ID1) is the
first gene that causes synthetic sickness when paired with p53
R175H mutant protein.
Materials and methods
Cell lines and culture
The stable SF126 cell line expressing the
doxycycline (Dox)-inducible p53 R175H mutant (SF126-tet-R175H) was
constructed according to the protocol described previously
(21). In addition, SF126-tet-TON,
which does not express p53, was used as a control (21). Mutant p53 was induced with 10 ng/ml
doxycycline (Sigma-Aldrich, St. Louis, MO, USA). Five human cell
lines including SKBr3 and HCC1395 (both derived from breast
cancer), VMRC-LCD (derived from lung cancer), Detroit 562 (derived
from head and neck cancer), and LS123 (derived from colon cancer)
express p53 R175H endogenously. Colon cancer cell lines HT-29 and
SW480 express p53 R273H endogenously. HCT116 (derived from colon
cancer) expressed wild-type p53 endogenously. Four cell lines
including PC3 (derived from prostate cancer), H1299 and Calu-1
(both derived from lung cancer), and SK-N-MC (derived from
neuroblastomas) are TP53-null. PC3 was purchased from Cell
Research Center for Biomedical Research, Institute of Development,
Aging and Cancer, Tohoku University (Sendai, Japan). SKBr3,
HCC1395, LS123, H1299, Calu-1, SK-N-MC, HCT116 and SW480 were
purchased from American Type Culture Collection (ATCC; Manassas,
VA, USA). Detroit 562 and VMRC-LCD were purchased from DS Farma
Biomedical (Osaka, Japan) and Human Science Research Resources Bank
(Tokyo, Japan), respectively. HT-29 was a gift from Dr John M.
Mariadason. SKBr3, HCC1395, HT-29, SW480, H1299 and PC3 were
cultured in RPMI-1640, and LS123, Calu-1, SK-N-MC, Detroit 562 and
VMRC-LCD were cultured in minimum essential medium with 10% FBS at
37°C. The identity of SF126, PC3, HCC1395, LS123, H1299, Detroit
562, VMRC-LCD and HT-29 cells was tested using a set of 10 short
tandem repeats produced by BEX Co., Ltd (Tokyo, Japan) in 2011.
SKBr3, Calu-1, SK-N-MC, SW480, and HCT116 were passaged for <6
months.
Mammalian p53 expression vectors
To express p53 R175H mutant, the p53 expression
vectors pCR259-R175H and pCR259 were used (16,22).
Each plasmid was transfected into TP53-null cells using
Effectene Transfection Reagent (Qiagen, Hercules, CA, USA),
following the manufacturer’s recommendations. Expression of p53
R175H was confirmed by western blot analysis.
RNAi screening
One million SF126-tet-R175H and SF126-tet-TON cells
were seeded in 10-cm culture plates for 24 h. The medium was
removed from the plates and the Decode RNAi Viral Screening Library
(Thermo Scientific Open Biosystems, Huntsville, AL, USA) was added
to the plate at the multiplicity of infection (MOI) of 0.3 with
serum-free medium. After 6 h, the medium was replaced with
virus-free medium. After 48 h, puromycin was added at a final
concentration of 2 mg/ml to select the infected cells. Finally,
7×106 of lentivirus-infected SF126-tet-R175H and
SF126-tet-TON cells were obtained. These cells theoretically
contain 70,000 distinct shRNAs, and each cell should express a
single shRNA product. These cells were divided into 2 groups, and
each group was cultured with or without doxycycline for 10 days.
Genomic DNA was extracted from each group using the Blood &
Cell Culture DNA Mini kit (Qiagen), according to the manufacturer’s
recommendation. Barcode sequences corresponding to specific shRNAs
were amplified by the following primers located outside the barcode
sequence: forward, 5′-caaggggctactttaggagcaattatcttg-3′ and
reverse, 5′-ggttgattgttccagacgcgt-3′.
Amplified PCR products were separated in 1.5% TAE
agarose gel and extracted using Wizard SV Gel and PCR Clean-Up
system (Promega Corporation, Madison WI, USA). Each purified PCR
product was labeled with Cy5 (doxycycline-on group) or Cy3
(doxycycline-off group) using Agilent’s Genomic DNA Labeling kit
(Agilent Technologies, Inc., Santa Clara, CA, USA) and was
hybridized on the barcode microarray in the hybridization oven at
65°C for 17 h. After hybridization, the arrays were scanned with
the Agilent DNA microarray scanner to quantify log2
Cy5/Cy3. The ‘log2 Cy5/Cy3’ indicates increase and decrease of
cells in the primary screening and negative value of ‘log2 Cy5/Cy3’
shows that the counts of R175H expressing cells (dyed with Cy5) is
smaller than the counts of R175H unexpressed cells (dyed with Cy3).
We conducted 2 independent experiments, and obtained 3 independent
values of log2 Cy5/Cy3 were analyzed by Student’s t-test. Candidate
genes were identified after analyzing raw data for each shRNA using
the Gene Spring software (Agilent Technologies). Microarray data
were deposited in GEO (accession no. 33362).
Knockdown analysis of candidate genes
using siRNA
The siRNAs of 50 candidate genes, identified from
primary screening, were synthesized by Hokkaido System Science
(Hokkaido, Japan). The sequences of synthesized siRNA for candidate
genes are listed in Table I.
ID1-2 siRNA was synthesized as described previously
(23). TP53 siRNA was
purchased from Applied Biosystems (Foster City, CA, USA), and
TP53-2 siRNA was purchased from Cell Signaling Technology,
Inc. (Boston, MA, USA). A total of 3.5–5.0×103
cells/well were seeded and incubated in a 96-well plate for 24 h.
Each candidate siRNA and negative control siRNA was added to the
cells to make a final concentration of 30 nM or 100 nM using
DharmaFECT 1 (Dharmacon, Lafayette, CO, USA). Cell proliferation
assays were performed using Cell Counting kit-8 (Dojin
Laboratories, Kumamoto, Japan), as previously described (21).
 | Table ISequences of synthesized siRNA for 50
candidate genes. |
Table I
Sequences of synthesized siRNA for 50
candidate genes.
| Gene symbol | siRNA sense
sequence | siRNA antisense
sequence |
|---|
| Smallest
p-value |
| UROS |
CCTCTGTGGAAGCCAGCTTAA |
TTAAGCTGGCTTCCACAGAGG |
| GYPC |
GCTCAGAACGATTGGAAATAA |
TTATTTCCAATCGTTCTGAGC |
|
PRO1596 |
CGATGAATATCTCTGTGAATA |
TATTCACAGAGATATTCATCG |
| CD69 |
GGAGCATTTATAAATGGACAA |
TTGTCCATTTATAAATGCTCC |
| PDXP |
GGTACCAGTTTAGGTTCCTAA |
TTAGGAACCTAAACTGGTACC |
| THADA |
CGCTTACAGATGATTCTGAAT |
ATTCAGAATCATCTGTAAGCG |
| KCNJ10 |
GCAGATATCTTGGCCTGGTTA |
TAACCAGGCCAAGATATCTGC |
| ABCA12 |
CCAAATTTCCTCCAACTGCAA |
TTGCAGTTGGAGGAAATTTGG |
| UBA6 |
GCATTGTTACTTGCCTTGAAA |
TTTCAAGGCAAGTAACAATGC |
| CDC7 |
GCACTTTCAGCTCTGTTTATT |
AATAAACAGAGCTGAAAGTGC |
| ID1 |
GGAAATTGCTTTGTATTGTAT |
ATACAATACAAAGCAATTTCC |
| CTBS |
GGCTCCTTATTATAACTATAA |
TTATAGTTATAATAAGGAGCC |
| EIF2B3 |
CGGAGTGAACTGATTCCATAT |
ATATGGAATCAGTTCACTCCG |
| UFM1 |
GGTAGCAAAGTGTTACAGAAA |
TTTCTGTAACACTTTGCTACC |
| PTCD1 |
CCTCGATGTGTTCAAGGAAAT |
ATTTCCTTGAACACATCGAGG |
| TPCN2 |
GGAGCTCCTGTTCAGGGATAT |
ATATCCCTGAACAGGAGCTCC |
| NEURL |
GGGTAACAACTTCTCCAGTAT |
ATACTGGAGAAGTTGTTACCC |
| STEAP4 |
GCACTATATTAGGTTAAGTAT |
ATACTTAACCTAATATAGTGC |
|
C19orf40 |
GCATATTGTGGCCAATGAGAA |
TTCTCATTGGCCACAATATGC |
|
C19orf38 |
CCACCTTGGATGATCACTCAG |
CTGAGTGATCATCCAAGGTGG |
|
C14orf37 |
GGAACTCCTTACAAGCACTAA |
TTAGTGCTTGTAAGGAGTTCC |
| Largest
fold-change |
|
MGC42105 |
CCAGCTGACGCCCTTCGAGAA |
TTCTCGAAGGGCGTCAGCTGG |
| GP6 |
GGGCTCCAGACGGATCTCTAA |
TTAGAGATCCGTCTGGAGCCC |
|
BCL2L14 |
GCCTGTAGCTTCAAGTTCTAA |
TTAGAACTTGAAGCTACAGGC |
| HILS1 |
GCCAAGTGCCACTGCAATTAA |
TTAATTGCAGTGGCACTTGGC |
| CLDND2 |
GAAGAATGCGTGGAAGAACAA |
TTGTTCTTCCACGCATTCTTC |
| UMOD |
CCACTGACACCTCAGAAGCAA |
TTGCTTCTGAGGTGTCAGTGG |
| RHAG |
GGGCATATTCTTTGAGTTATA |
TATAACTCAAAGAATATGCCC |
| GFPT2 |
GCTACGAGTTTGAGTCAGAAA |
TTTCTGACTCAAACTCGTAGC |
| SATL1 |
AAGCATGGATTACTTTCAAAT |
ATTTGAAAGTAATCCATGCTT |
|
RTN4IP1 |
GCTGCCAGTGTAAATCCTATA |
TATAGGATTTACACTGGCAGC |
| DOK7 |
CCAAGCGGATTCATCTTTGAA |
TTCAAAGATGAATCCGCTTGG |
| NUP98 |
GGCCAAAGGCTTTACAAACAA |
TTGTTTGTAAAGCCTTTGGCC |
| PWWP2A |
GCCATGCCGCTCCAAAGTAAT |
ATTACTTTGGAGCGGCATGGC |
| MEP1A |
GCCTATAAGGCCATCATAGAA |
TTCTATGATGGCCTTATAGGC |
| CCT6B |
TGGCTGAAGCTCTTGTTACAT |
ATGTAACAAGAGCTTCAGCCA |
| PDXP |
GGTACCAGTTTAGGTTCCTAA |
TTAGGAACCTAAACTGGTACC |
| ZNF300 |
GCTAATATTAGCTTGTCATAA |
TTATGACAAGCTAATATTAGC |
|
DEFB125 |
AGAGGATATAACATTGGATTA |
TAATCCAATGTTATATCCTCT |
| GJA5 |
CGTTGCTCATTAATTCTAGAA |
TTCTAGAATTAATGAGCAACG |
| EFNA4 |
CCATGTTCAATTCTCAGAGAA |
TTCTCTGAGAATTGAACATGG |
| Double entry |
| NMNAT1 |
TCATCTGAAGTGTCACGTAAA |
TTTACGTGACACTTCAGATGA |
| KLHL10 |
GCTGAGTACTTCATGAACAAT |
ATTGTTCATGAAGTACTCAGC |
| LMLN |
CCACAGTGAAACATGAGGTTA |
TAACCTCATGTTTCACTGTGG |
| FBXO22 |
TCCCTCAAATTGAAGGAATAA |
TTATTCCTTCAATTTGAGGGA |
| ITGB7 |
GGACAGTAATCCTCTCTACAA |
TTGTAGAGAGGATTACTGTCC |
| CPN1 |
GGAATGCAAGACTTTAATTAT |
ATAATTAAAGTCTTGCATTCC |
| COLQ |
GGCTACAATGCTCTTCCTCTT |
AAGAGGAAGAGCATTGTAGCC |
| AP3B2 |
GGATTGCACCTGATGTCTTAA |
TTAAGACATCAGGTGCAATCC |
| ANXA11 |
CGTGGTGAAATGTCTCAAGAA |
TTCTTGAGACATTTCACCACG |
Western blot analysis
Cells were harvested and resuspended in lysis buffer
containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 5 mM EDTA, and 1%
protease inhibitor cocktail (Sigma-Aldrich). The lysate was
subjected to western blot analysis, as previously described
(24). Anti-p53 (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), anti-β-actin
(Sigma-Aldrich), anti-Id1, and anti-GAPDH (Applied Biosystems)
antibodies were used.
Cell cycle analysis by FACS
A total of 1.5×104 cells/plate were
seeded and incubated in 6-cm culture plates for 24 h. The cells
were further incubated in the presence of drugs for 48 h. These
cells were collected, and FACS analysis was performed, as
previously described (24).
Results
Screening of synthetic lethal genes that
interact with p53 R175H mutant
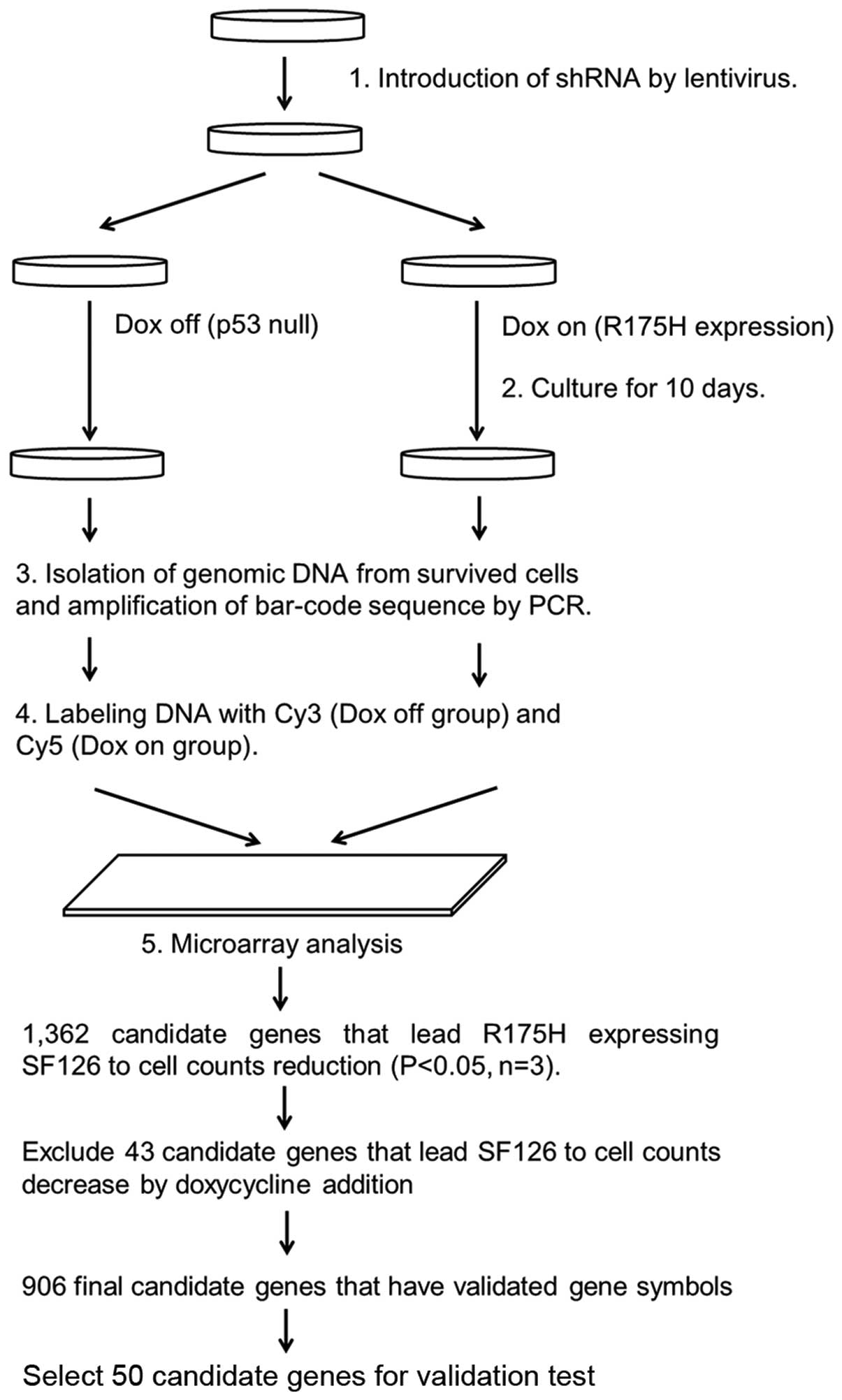
A flow chart of the high-throughput screening of
synthetic lethal genes interacting with p53 R175H is shown in
Fig. 1. By comparative analysis,
1,362 candidate genes were identified for synthetic lethality with
p53 R175H expression in the SF126-tet-R175H cell line (p<0.05
according to t-test, n=3). Among these, 43 were excluded as
suppression of these genes also resulted in decreasing cell numbers
in SF126-tet-TON cells after doxycycline treatments (no R175H
expression). In the remaining 1,319 genes, 906 genes have validated
gene symbols, which have p-value <0.05. Among these, we selected
50 genes (21 genes from the group with the smallest p-values, 20
genes from the group with the largest fold-change, and 9 genes
reproduced by different siRNA sequences) for further validation
testing (Table I).
Suppression of candidate genes by siRNA
in p53 R175H expressing cell lines and TP53-null cell lines
To investigate whether the suppression of candidate
genes by siRNA resulted in p53 R175H-dependent inhibition of cell
growth, candidate gene siRNAs were transfected into cell lines
expressing endogenous p53 R175H (SKBr3, LS123, HCC1395, Detroit 562
and VMRC-LCD) and TP53-null cells (PC3, H1299, SK-N-MC and
Calu-1). We obtained the ratio of cell growth inhibition of
candidate gene siRNA transfected cells for negative control siRNA
transfected cells on day 4. In 50 candidate genes, suppression of
GYPC, NUP98, GP6, EFNA4 and ID1 by siRNA
significantly decreased the number of p53 R175H expressing cells
compared with TP53-null cells (t-test) (Table II).
| Table IITop 5 candidate genes for synthetic
lethal interaction with R175H on t-test. |
Table II
Top 5 candidate genes for synthetic
lethal interaction with R175H on t-test.
| Gene symbol | p-value |
|---|
| GYPC | 0.002446 |
| NUP98 | 0.029324 |
| GP-6 | 0.043416 |
| EFNA4 | 0.055412 |
| ID1 | 0.061552 |
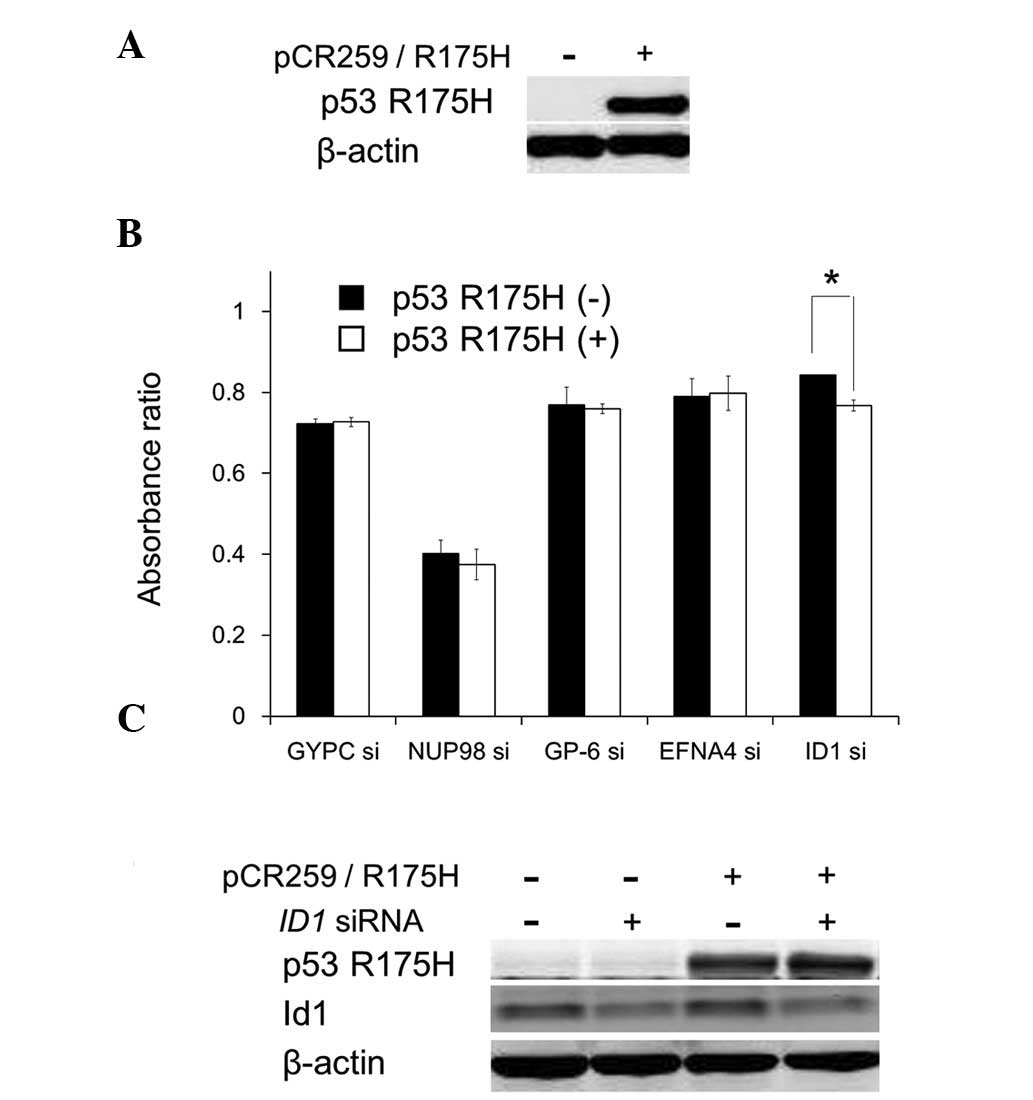
To examine whether the cell growth inhibition
resulting from suppression of the candidate genes depends on p53
R175H expression, GYPC, NUP98, GP6, EFNA4 and ID1
were suppressed by specific siRNAs in PC3 cells transiently
expressing p53 R175H. Suppression of these genes inhibited cell
growth; however, among the candidate genes, ID1 suppression
significantly accelerated the cell growth inhibition under
transient p53 R175H expression (Fig. 2A
and B). ID1 suppression and p53 R175H overexpression did
not influence the other protein expression level (Fig. 2C). These results suggest that p53
R175H expression and ID1 suppression cooperate to cause cell
growth inhibition.
Cell growth inhibition by ID1 and/or TP53
suppression in endogenously expressing p53 R175H, wt p53 cells and
in TP53-null cells
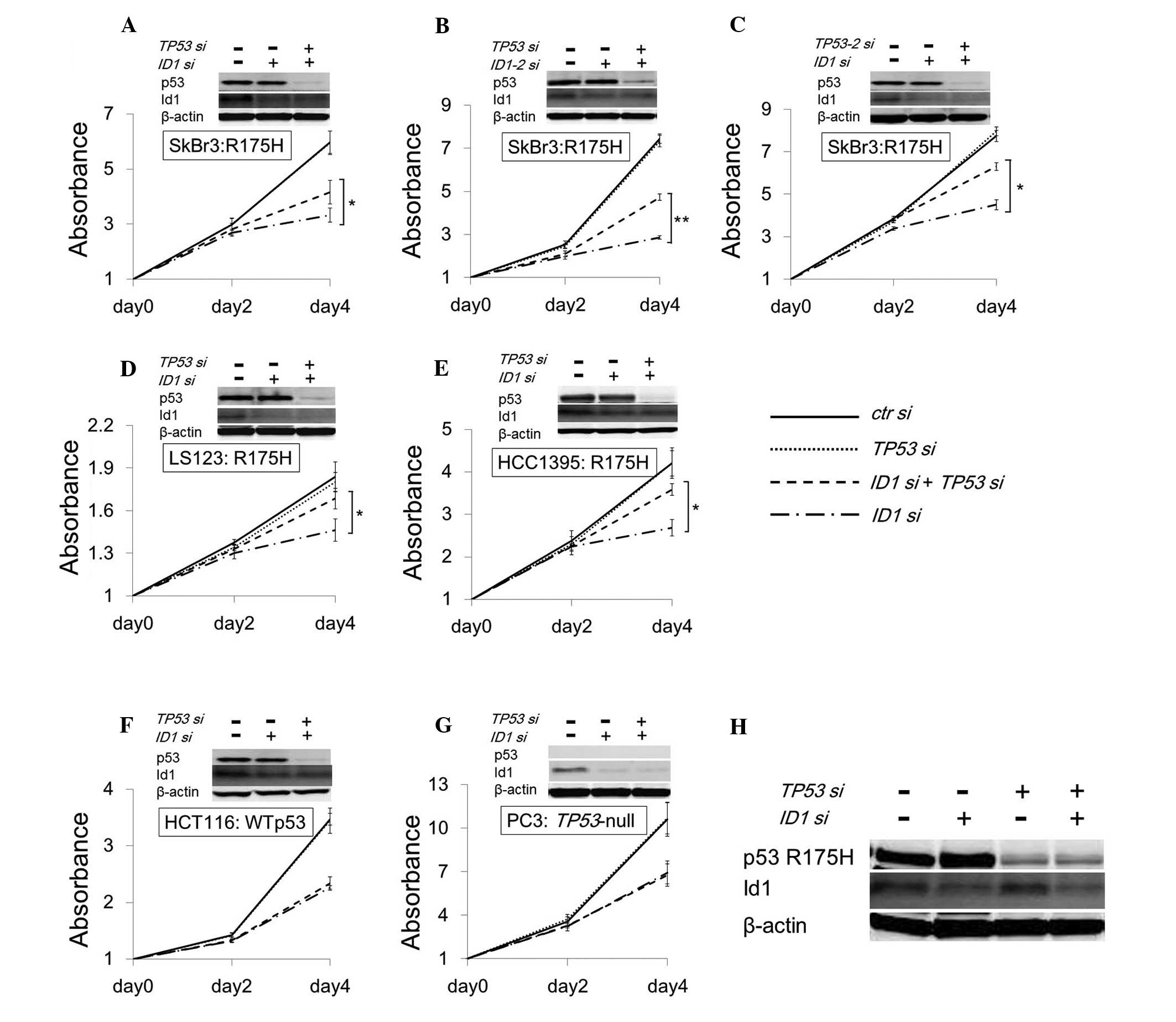
To determine whether cell growth inhibition is
rescued by the suppression of both candidate genes and p53 R175H,
siRNAs of the targeting candidate genes and TP53 were
transfected into SKBr3, a p53 R175H expressing cell line.
Downregulation of p53 R175H rescued cell growth inhibition caused
by ID1 suppression (Fig.
3A), but not by GYPC, NUP98, GP6 and EFNA4
suppression (data not shown). To exclude the off-target effect of
siRNA, other siRNA for ID1 and TP53 targeting
different sites (ID1-2 and TP53-2) were transfected
into SKBr3, and we observed reproducible results to the original
siRNAs (Fig. 3B and C). Moreover,
similar results were observed only in cell lines expressing p53
R175H (LS123, Fig. 3D and HCC1395,
Fig. 3E), but not in wt p53
(HCT116, Fig. 3F), and
TP53-null (PC3, Fig. 3G).
The quantity of the Id1 protein in SKBr3 was not altered by p53
R175H suppression (Fig. 3H), same
as p53 R175H transient expression. These results support the
finding that cell growth inhibition by ID1 suppression is
accelerated by p53 R175H.
Suppression of ID1 in cell lines
expressing another common mutant p53 (R273H)
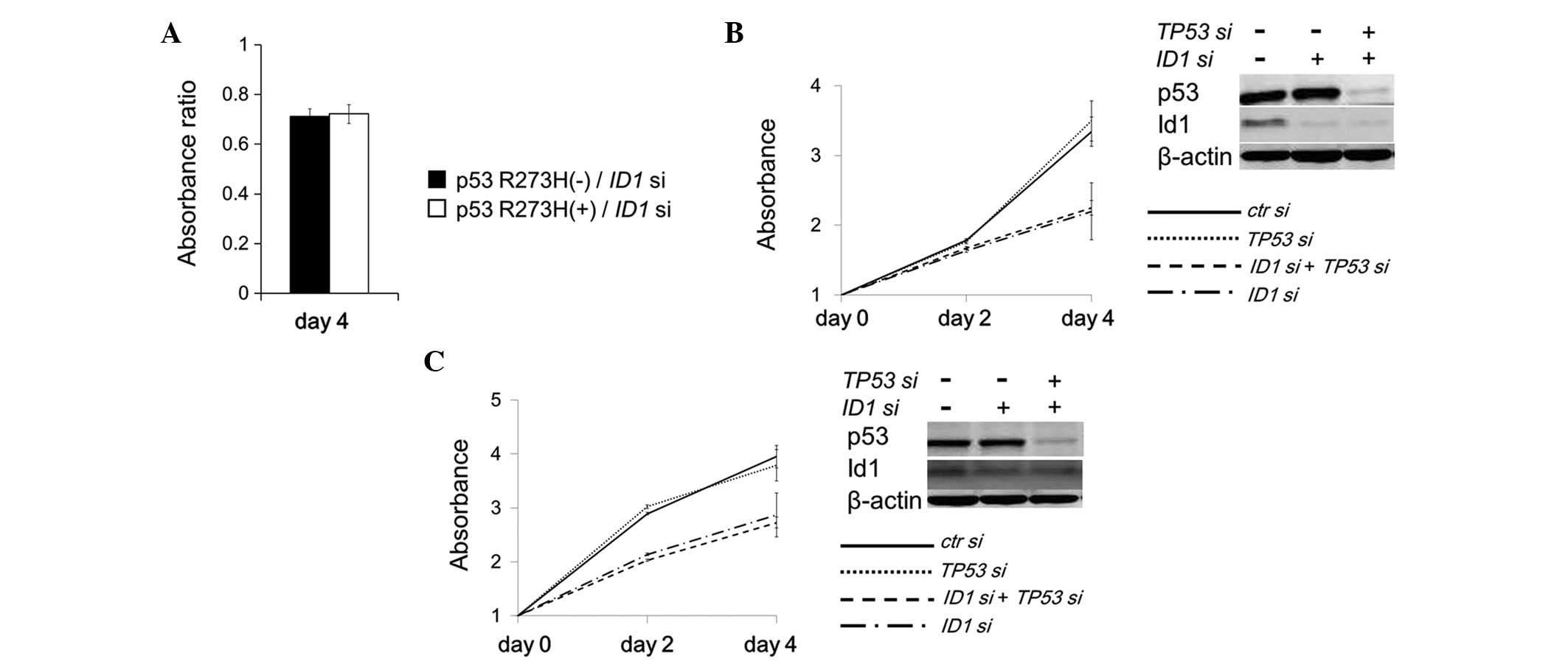
To examine whether cell growth inhibition caused by
ID1 suppression is accelerated specifically by p53 R175H
expression, another common p53 mutant (R273H) was expressed in a
PC3 cell line (TP53-null). Unlike p53 R175H expression, p53
R273H expression did not accelerate the cell growth inhibition
caused by ID1 suppression (Fig.
4A). Furthermore, the cell growth inhibition caused by
ID1 suppression was not restored by simultaneous suppression
of TP53 in HT-29 cells expressing endogenous p53 R273H
(Fig. 4B). Similar results were
observed in SW480 cells expressing endogenous p53 R273H/P309S
double mutants (Fig. 4C). These
results indicated that the growth inhibition induced by ID1
suppression may be accelerated by p53 R175H expression in a
specific manner.
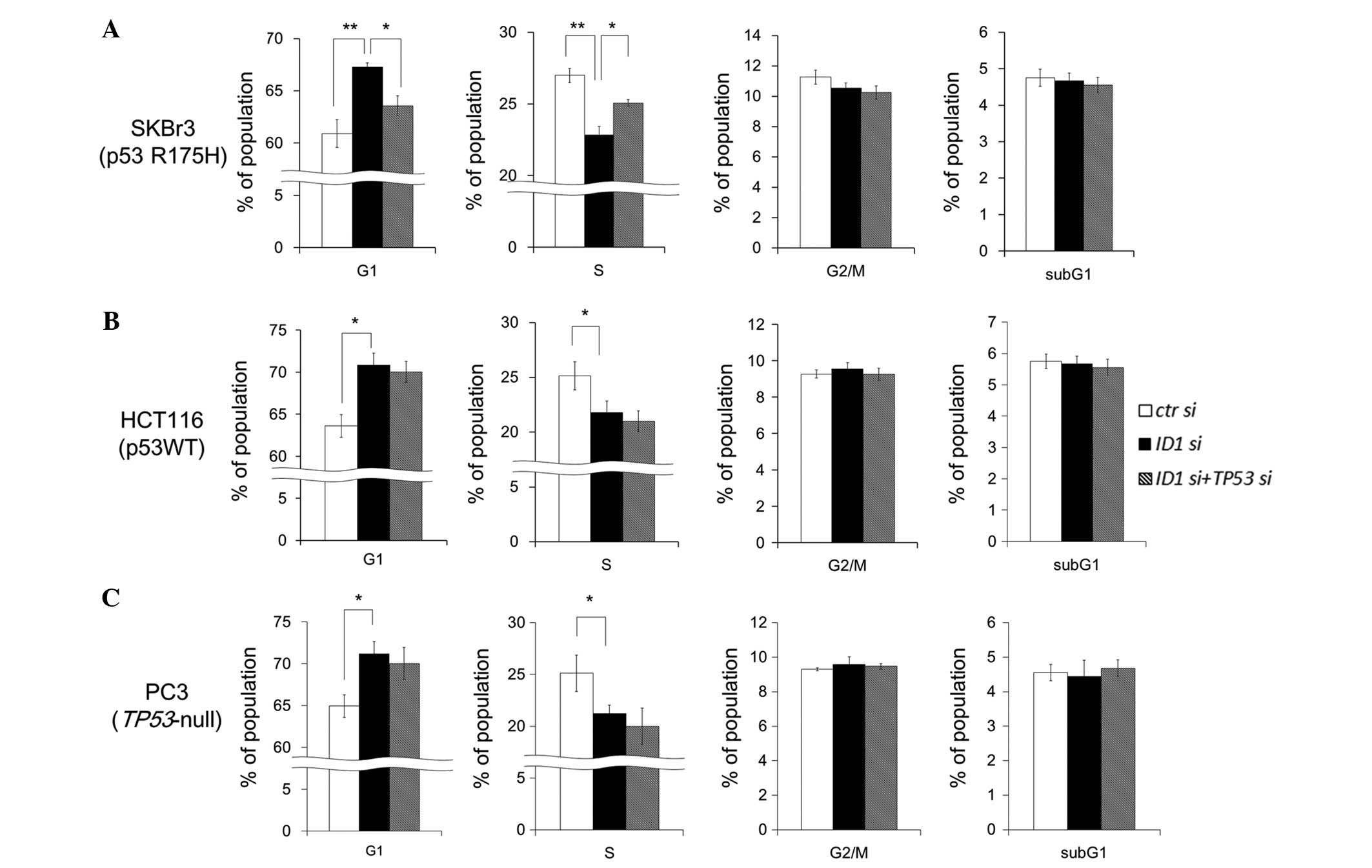
Cell cycle analysis under ID1 suppression
and ID1/TP53 double suppression
To examine whether ID1 and/or TP53
suppression change the proportion of cell cycle phases, FACS
analysis was performed in SKBr3 cells. ID1 suppression did
not change the sub-G1 fraction, but significantly decreased the S
phase fraction and increased the G1 phase fraction (Fig. 5A). ID1/TP53 double
suppression significantly restored the proportion of S phase and G1
phase fractions. These results suggest that p53 R175H potentiates
G1 arrest by ID1 suppression. In HCT116 (wild-type p53) and
PC3 (TP53-null) cells, ID1 suppression increased the
G1 phase fraction and decreased the S phase fraction. However,
unlike in SKBr3 cells, ID1/TP53 double suppression
did not restore the proportion of S phase and G1 phase fractions
(Fig. 5B and C). These results
suggest that ID1 suppression induces G1 arrest and the
arrest is specifically accelerated by p53 R175H expression.
Discussion
We identified ID1 as a synthetic sick/lethal
gene that caused cell growth inhibition in the presence of p53
R175H. Id1 is a member of the helix-loop-helix protein family
expressed in actively proliferating cells and regulates gene
transcription by hetero-dimerization with the basic
helix-loop-helix (bHLH) transcription factor (25). The homodimer of the bHLH
transcription factor activates the differentiation, whereas the
heterodimer, composed of Id1 and the bHLH transcription factor,
attenuates their ability to bind DNA and consequently inhibits cell
differentiation (26). Supporting
this finding, stable Id1 expression was found to block B cell
maturation (27). Moreover, Id1 can
inhibit differentiation of muscle and myeloid cells by associating
in vivo with E2A proteins (28,29).
It has also been reported that Id1 was immunohistochemically
expressed in majority of non-small cell lung cancer (NSCLC) samples
(30). Furthermore, Id1 protein
expression in prostate cancer cells mediated resistance to
apoptosis induced by TNFα (31).
These lines of evidence also indicate that ID1 may play an
essential role in carcinogenesis.
In the present study, we demonstrated that
ID1 suppression resulted in cell growth inhibition that was
independent of TP53 status. However, cell growth inhibition
caused by ID1 suppression was accelerated specifically by
the p53 R175H mutant protein. If the accelerated cell growth
inhibition is attributable only to loss-of-function in p53 R175H,
this phenomenon should also be observed in TP53-null cells
and other cells expressing loss-of-function mutations other than
p53 R175H. Some p53 mutant proteins acquire additional functions
called gain-of-function (32). For
example, ectopic expression of p53 R175H resulted in
transactivation of genes that are not usually activated by
wild-type p53 (33–35). On the basis of these observations,
we concluded that the acceleration of cell growth inhibition was
likely attributable to gain-of-function of p53 R175H.
To date, synthetic sickness/lethality has been
classified into 2 types based on the initial genetic event. The
first type is attributable to a loss-of-function mutation in a
target gene and the second type is attributable to a
gain-of-function or an activated mutation in a target gene. For
example, the synthetic lethal interaction between loss-of-function
mutations in BRCA1 and BRCA2 genes and PARP
inhibition (8) are a former type,
and gain-of-function mutations in the KRAS gene and STK33
inhibition (6) are a latter type.
Based on our results, it is clear that the synthetic sick/lethal
interaction between p53 R175H expression and ID1 suppression
is of the latter type. However, there is a clear difference between
the activated KRAS-STK33 interaction and the p53 R175H-Id1
interaction. Gain-of-function in activated K-ras depends on STK33
and is therefore blocked by STK33 suppression. By contrast, the
accelerated cell growth inhibition observed here cannot be
explained only by blockade of gain-of-function in p53 R175H by
ID1 suppression. By contrast, it is necessary for
accelerated growth inhibition by ID1 suppression. Taken
together, these results suggest that the synthetic
sickness/lethality of p53 R175H with ID1 suppression may be
through a gain-of-function mechanism that is distinct from the
previously identified gain-of-function mechanisms. Since both
expression and suppression of p53 R175H had no effect on the amount
of Id1 protein, p53 R175H may cooperate with downstream factor(s)
that are altered by ID1 suppression and may promote
synthetic sickness/lethality in cooperation with protein(s)
downstream of ID1.
The precise molecular mechanisms of the synthetic
sickness/lethality of ID1 suppression and p53 R175H
expression remain to be elucidated. In conclusion, Id1 and its
associated signaling pathway is one of the molecular targets of
cancer cells expressing the common p53 mutant R175H.
Acknowledgements
The authors thank Atsuko Sato, Eri Yokota and Satoko
Aoki for their technical assistance. This study was supported in
part by grants-in-aid from the Ministry of Education, Culture,
Sports, Science and Technology (12217010 to S.K., 17015002 to C.I.,
and 20390163 to C.I.) and the Gonryo Medical Foundation to C.I. and
Sapporo Bioscience Foundation to S.K.
References
|
1
|
Molenaar JJ, Ebus ME, Geerts D, et al:
Inactivation of CDK2 is synthetically lethal to MYCN
over-expressing cancer cells. Proc Natl Acad Sci USA.
106:12968–12973. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Canaani D: Methodological approaches in
application of synthetic lethality screening towards anticancer
therapy. Br J Cancer. 100:1213–1218. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kaelin WG Jr: The concept of synthetic
lethality in the context of anticancer therapy. Nat Rev Cancer.
5:689–698. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bandyopadhyay N, Ranka S and Kahveci T:
Sslpred: predicting synthetic sickness lethality. Pac Symp
Biocomput. 7–18. 2012.
|
|
5
|
Luo J, Emanuele MJ, Li D, et al: A
genome-wide RNAi screen identifies multiple synthetic lethal
interactions with the Ras oncogene. Cell. 137:835–848. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Scholl C, Fröhling S, Dunn IF, et al:
Synthetic lethal interaction between oncogenic KRAS dependency and
STK33 suppression in human cancer cells. Cell. 137:821–834. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Farmer H, McCabe N, Lord CJ, et al:
Targeting the DNA repair defect in BRCA mutant cells as a
therapeutic strategy. Nature. 434:917–921. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kaye SB, Lubinski J, Matulonis U, et al:
Phase II, open-label, randomized, multicenter study comparing the
efficacy and safety of olaparib, a poly (ADP-ribose) polymerase
inhibitor, and pegylated liposomal doxorubicin in patients with
BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin
Oncol. 30:372–379. 2012. View Article : Google Scholar
|
|
9
|
Hollstein M, Sidransky D, Vogelstein B and
Harris CC: p53 mutations in human cancers. Science. 253:49–53.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Levine AJ: p53, the cellular gatekeeper
for growth and division. Cell. 88:323–331. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lill NL, Grossman SR, Ginsberg D, DeCaprio
J and Livingston DM: Binding and modulation of p53 by p300/CBP
coactivators. Nature. 387:823–827. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sakaguchi K, Herrera JE, Saito S, et al:
DNA damage activates p53 through a phosphorylation-acetylation
cascade. Genes Dev. 12:2831–2841. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Soussi T, Dehouche K and Béroud C: p53
website and analysis of p53 gene mutations in human cancer: forging
a link between epidemiology and carcinogenesis. Hum Mutat.
15:105–113. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Olivier M, Eeles R, Hollstein M, Khan MA,
Harris CC and Hainaut P: The IARC TP53 database: new online
mutation analysis and recommendations to users. Hum Mutat.
19:607–614. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Soussi T, Ishioka C, Claustres M and
Béroud C: Locus-specific mutation databases: pitfalls and good
practice based on the p53 experience. Nat Rev Cancer. 6:83–90.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kato S, Han SY, Liu W, et al:
Understanding the function-structure and function-mutation
relationships of p53 tumor suppressor protein by high-resolution
missense mutation analysis. Proc Natl Acad Sci USA. 100:8424–8429.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Olive KP, Tuveson DA, Ruhe ZC, et al:
Mutant p53 gain of function in two mouse models of Li-Fraumeni
syndrome. Cell. 119:847–860. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lang GA, Iwakuma T, Suh YA, et al: Gain of
function of a p53 hot spot mutation in a mouse model of Li-Fraumeni
syndrome. Cell. 119:861–872. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Song H, Hollstein M and Xu Y: p53
gain-of-function cancer mutants induce genetic instability by
inactivating ATM. Nat Cell Biol. 9:573–580. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu G, McDonnell TJ, Montes de Oca Luna R,
et al: High metastatic potential in mice inheriting a targeted p53
missense mutation. Proc Natl Acad Sci USA. 97:4174–4179. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Watanabe G, Kato S, Nakata H, Ishida T,
Ohuchi N and Ishioka C: αB-crystallin: a novel p53-target gene
required for p53-dependent apoptosis. Cancer Sci. 100:2368–2375.
2009.
|
|
22
|
Shiraishi K, Kato S, Han SY, et al:
Isolation of temperature-sensitive p53 mutations from a
comprehensive missense mutation library. J Biol Chem. 279:348–355.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dong Z, Wei F, Zhou C, et al: Silencing
Id-1 inhibits lymphangiogenesis through down-regulation of VEGF-C
in oral squamous cell carcinoma. Oral Oncol. 47:27–32. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kakudo Y, Shibata H, Otsuka K, Kato S and
Ishioka C: Lack of correlation between p53-dependent
transcriptional activity and the ability to induce apoptosis among
179 mutant p53s. Cancer Res. 65:2108–2114. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ling MT, Wang X, Zhang X and Wong YC: The
multiple roles of Id-1 in cancer progression. Differentiation.
74:481–487. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Benezra R, Davis RL, Lockshon D, Turner DL
and Weintraub H: The protein Id: a negative regulator of
helix-loop-helix DNA binding proteins. Cell. 61:49–59. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun XH: Constitutive expression of the Id1
gene impairs mouse B cell development. Cell. 79:893–900. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jen Y, Weintraub H and Benezra R:
Overexpression of Id protein inhibits the muscle differentiation
program: in vivo association of Id with E2A proteins. Genes Dev.
6:1466–1479. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kreider BL, Benezra R, Rovera G and
Kadesch T: Inhibition of myeloid differentiation by the
helix-loop-helix protein Id. Science. 255:1700–1702. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rothschild SI, Kappeler A, Rastchiller D,
et al: The stem cell gene ‘inhibitor of differentiation 1’ (ID1) is
frequently expressed in non-small cell lung cancer. Lung Cancer.
71:306–311. 2011.
|
|
31
|
Ling MT, Wang X, Ouyang XS, Xu K, Tsao SW
and Wong YC: Id-1 expression promotes cell survival through
activation of NF-κB signalling pathway in prostate cancer cells.
Oncogene. 22:4498–4508. 2003.PubMed/NCBI
|
|
32
|
Brosh R and Rotter V: When mutants gain
new powers: news from the mutant p53 field. Nat Rev Cancer.
9:701–713. 2009.PubMed/NCBI
|
|
33
|
Roger L, Jullien L, Gire V and Roux P:
Gain of oncogenic function of p53 mutants regulates E-cadherin
expression uncoupled from cell invasion in colon cancer cells. J
Cell Sci. 123:1295–1305. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yan W and Chen X: Identification of GRO1
as a critical determinant for mutant p53 gain of function. J Biol
Chem. 284:12178–12187. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu DP, Song H and Xu Y: A common gain of
function of p53 cancer mutants in inducing genetic instability.
Oncogene. 29:949–956. 2010. View Article : Google Scholar : PubMed/NCBI
|