Introduction
B7-H1 (CD274, PD-L1) is a cell-surface glycoprotein
belonging to the B7 family of costimulatory molecules, which was
first identified in 1999 (1). B7-H1
expression is found not only in antigen-presenting cells but also
in activated T cells and in a number of tumour cells. Many studies
have confirmed that B7-H1 is widely distributed in various types of
human cancers, including melanoma, ovarian, colon and lung cancers
(2), pancreatic (3), oesophageal (4), gastric carcinoma (5) and gliomas (6). Cancer cell-associated B7-H1 is closely
correlated with poor prognosis and a higher grade of malignancy.
Moreover, B7-H1 has been shown to be involved in the protection of
cancer cells from activated tumour antigen-specific T lymphocytes
through the induction of T lymphocyte anergy or apoptosis after
ligation to the T lymphocyte receptor PD-1 or other receptors
(7,8). In addition, B7-H1 expression is
induced on the tumour cell surface by cytokines, such as interferon
(IFN)-γ. The blockade of the B7-H1/PD-1 pathway by neutralising
antibodies effectively enhances T lymphocyte responses against
human cancer and reduces tumour growth (9–11).
Thus, B7-H1/PD-1 interaction apparently regulates antitumour
immunity.
However, other interpretations for this molecular
function have been revealed by several recent studies. The
expression of B7-H1 in breast cancer patients is directly
associated with the proliferative Ki-67 marker, which is
independent of the PD-1 expression in the host (12). A similar phenomenon was also found
in myeloma cells (13). In
addition, in skin carcinoma, B7-H1 was confirmed to regulate
epithelial-mesenchymal transition (EMT) and to enhance
carcinogenesis (14), and in
mastocytoma cells, B7-H1 can act as a receptor to transmit a signal
from T cells to tumour cells that leads to resistance to cell lysis
(15). Moreover, our laboratory
previously found that the PD-1/PD-L1 complex transfers a reverse
signal to B7-H1+ pancreatic cancer cells upon
drug-induced apoptosis in vitro (16). All of these results may indicate
that B7-H1 not only mediates the inhibition of the activated T cell
response by binding to PD-1; but also accelerates carcinogenesis in
solid tumour cells.
Nevertheless, the roles of B7-H1 and the exact
mechanism of action of this protein during cancer onset and
progression are largely undefined. In particular, it is not known
whether and how B7-H1 molecules play a role in the pathophysiology
of pancreatic neoplasms. The current data suggest the use of tumour
B7-H1 expression as a predictive biomarker. However, the
designation of positive B7-H1 expression remains confounded by
tumour heterogeneity, antibody diversity and assay variability.
Therefore, in the present study, B7-H1 was overexpressed and
depleted in several pancreatic cancer cell lines to investigate its
effects on biological function and to elucidate the possible
underlying mechanisms. These observations support the conclusion
that B7-H1 may have effects on tumourigenesis in human pancreatic
cancer and may be an effective therapeutic target for this deadly
disease in the clinical setting.
Materials and methods
Patients and tissue samples
Pancreatic carcinoma specimens (histopathologically
confirmed primary pancreatic duct adenocarcinomas) were surgically
removed from 30 pancreatic carcinoma patients at Zhejiang
Provincial People’s Hospital. Surgically resected pancreatic cancer
specimens were stored at −80°C for protein extraction.
Formalin-fixed and paraffin-embedded sections were used for
immunohistochemical staining. Two healthy pancreas tissue samples
obtained through an organ donor program were used as the controls
in the present study which was approved by the local ethics
committee. All patients in the study provided informed consent.
Immunohistochemical staining for B7-H1
expression in pancreatic carcinoma tissues
Paraffin-embedded 4-μm-thick tissue sections were
dried for 40 min at 58°C, deparaffinised in xylene, and rehydrated
through sequential incubation in EtOH/water solutions. The sections
were treated with 3% hydrogen peroxide in methanol for 15 min at
room temperature in the dark to quench the endogenous peroxidase
activity. After rinsing in water and phosphate-buffered saline
(PBS), the sections were blocked with serum (diluted 1:20 in PBS)
from a non-immunised animal for 30 min to reduce non-specific
binding. Subsequently, the sections were incubated with a primary
monoclonal antibody against B7-H1 (R&D Systems; diluted 1:50 in
PBS) in a moist chamber at 4°C overnight. After washing in PBS,
biotinylated anti-mouse immunoglobulin (Dako) at a dilution of
1:200 was added for 45 min, and the sections were then washed and
incubated with the ABC reagent (Strept ABC complex/HRP; Dako) for
45 min. The peroxidase reaction was developed with 0.02%
3,3′-diaminobenzidine tetrahydrochloride (Sigma, St. Louis, MO,
USA) in PBS containing 0.06% hydrogen peroxide for 1 min. The
sections were then rinsed with water and counterstained with
Harris’ hematoxylin. Immunoreactivity in >10% of the cells in
one field was defined as positive.
Cell lines and cultures
Four human pancreatic cancer cell lines (BxPC-3, MIA
PaCa-2, SW1990 and PANC-1) and the human embryonic kidney cell line
293T were obtained from the American Type Culture Collection (ATCC;
Manassas, VA, USA). The human pancreatic cancer cell lines were
cultured in RPMI-1640 medium (Gibco, Carslbad, CA, USA)
supplemented with 10% fetal bovine serum (FBS; Gibco) and
antibiotics (100 μg/ml streptomycin and 100 IU/ml of penicillin) at
37°C in a humidified incubator containing 5% CO2. The
293T cells were maintained in high-glucose DMEM (Gibco) containing
10% FBS. The cells were grown on sterile tissue culture dishes and
were passaged every two days using 0.25% trypsin (Invitrogen).
Generation of stable B7-H1-overexpressing
and -silenced pancreatic cell lines
The complete cDNA sequence of the human B7-H1 was
generated by PCR using 5′-GGAATTCAT GAGGATATTTGCTGTC-3′ as the
forward primer and 5′-TT TGCGGCCGCTTACGTCTCCTCCA-3′ as the reverse
primer. The sequence was then cloned into a pLenO-GTP plasmid
(Invabio Biotechnology Ltd., Shanghai, China). The plasmids were
sequenced with the primers to confirm the correctness of the
constructed plasmid. An empty vector was used as a negative
control. Replication-defective lentiviruses were produced by the
transient transfection of 293T cells using Lipofectamine Plus
(Invitrogen), pLenO-GTP-B7-H1, pRsv-REV, pMDlg-pRRE and pMD2G
(Invabio Biotechnology Ltd). After transfection, samples of the
viruses were harvested at 72 h and filtered through a Millex-HV
0.45-μm PVDF filter (Millipore, Billerica, MA, USA). The
transduction of PANC-1 cells was performed by 6 h of exposure to
dilutions of the viral supernatant in the presence of Polybrene (5
μg/ml). The stably transfected cells were selected by culture in
puromycin (2 μg/ml).
To suppress B7-H1 expression, the shRNA-expressing
pGPU6 lentivirus system was used (Shanghai GenePharma, Shanghai,
China) to transfect the BxPC-3 cells. Briefly, the B7-H1 shRNA
targeting sequence (B7-H1 shRNA) was 5′-GGATCCAGTCACCTCTGAACA-3′,
and the negative control shRNA targeting sequence (control shRNA)
was 5′-TTCTCCGAACGTGTCACGT-3′. Cells seeded in a 6-well culture
plate were transfected with either B7-H1-shRNA or control-shRNA
lentivirus according to the manufacturer’s protocol. The stable
suppression of the target genes was achieved by the selection of
cells in 1 mg/ml of G418 (Life Technologies).
Cell proliferation and colony formation
assay
For the quantification of the cell viability,
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxy-methoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium,
inner salt (MTS) assays were performed according to the
manufacturer’s instructions (CellTiter 96 AQueous Non-Radioactive
Cell Proliferation assay; Promega, Madison, WI, USA). Briefly, the
cells were plated in 96-well plates in medium containing 10% FBS at
a density of ~3,000 cells/well. The absorbance was measured at 490
nm using a microtitration plate spectrophotometer.
For the colony formation assays, dissociated cells
were planted into 6-cm cell culture dishes (100 cells/dish) and
incubated for 12 days. The plates were washed with PBS and stained
with Giemsa. The number of colonies consisting of >50 cells was
counted.
Cell cycle analysis
The cells (5×105) were seeded into a 6-cm
tissue culture dish. At the indicated time-points, the cells were
harvested, fixed in 1% paraformaldehyde, washed with PBS, and
stained with 5 mg/ml propidium iodide in PBS supplemented with
RNase A (MultiSciences Biotech Co., Ltd., Hangzhou, China) for 30
min at room temperature. The stained cells were collected and
analysed using BD flow cytometry systems.
Western blotting
The total protein from cultured cells was extracted
in cell lysis buffer (Beyotime Institute of Biotechnology, Haimen,
China) and quantified using the Bradford method. Proteins (40 μg)
were loaded and separated on 10% SDS-PAGE. After the proteins were
transferred to a polyvinylidene fluoride membrane (Millipore), the
membrane was incubated overnight at 4°C with antibodies against
B7-H1 (R&D Systems, Minneapolis, MN, USA), cyclin D1, p-Rb,
CDK4, CDK6, phosphorylated extracellular signal-regulated kinase
(p-ERK), ERK, phosphorylated c-Jun N-terminal protein kinase
(p-JNK), JNK, phosphorylated p38 (p-p38), p38, β-actin and GAPDH
(1:1,000; Cell Signaling Technology, Beverly, MA, USA). After
incubation with peroxidase-conjugated anti-mouse or rabbit IgG
(Santa Cruz Biotechnology) at 37°C for 2 h, the bound proteins were
visualised using ECL and detected using the ImageQuant LAS 4000
(Fujifilm, Tokyo, Japan). The relative protein levels were
calculated in reference to GAPDH as the loading control.
RNA extraction and quantitative real-time
PCR
The total RNA was isolated from cultured cells using
the TRIzol reagent (Invitrogen) according to the manufacturer’s
recommended procedure. The reverse transcription of 1 μg of RNA was
performed using the SuperRT Reverse Transcriptase reagent kit
(Beijing CoWin Biotech Co., Ltd., Beijing, China) following the
manufacturer’s instructions. The quantitative real-time PCR was
performed in a total volume of 25 μl using the SYBR-Green PCR
Master Mix and a 7500 Fast Real-Time PCR System (Applied
Biosystems). The reaction conditions were as follows: 95°C for 10
min and 40 cycles of 95°C for 15 sec and 60°C for 1 min. A
dissociation step was applied to generate a melting curve in order
to confirm the specificity of the amplification. GAPDH transcripts
were amplified and served as the reference. The relative levels of
gene expression were represented as ΔCt = Ct(gene) - Ct(reference),
and the fold-change of gene expression was calculated using the
2−ΔΔCt method. The experiments were performed in
triplicate. The primer sequences are shown in Table I.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Name | Primer sequences |
|---|
| B7-H1 forward |
5′-GGTGCCGACTACAAGCGAAT-3′ |
| B7-H1 reverse |
5′-GGTGACTGGATCCACAACCAA-3′ |
| GAPDH forward |
5′-ACGGATTTGGTCGTATTGGG-3′ |
| GAPDH reverse |
5′-CCTGGAAGATGGTGATGGGATT-3′ |
| PTEN forward |
5′-GGCACCGCATATTAAAACGTA-3′ |
| PTEN reverse |
5′-ATGCCATTTTTCCATTTCCA-3′ |
| E-cadherin
forward |
5′-TCAGCGTGTGTGACTGTGAA-3′ |
| E-cadherin
reverse |
5′-CCTCCAAGAATCCCCAGAAT-3′ |
| Slug forward |
5′-GCACTGTGATGCCCAGTCTA-3′ |
| Slug reverse |
5′-CAGTGAGGGCAAGAGAAAGG-3′ |
| Twist forward |
5′-CTTCTCCGTCTGGAGGATGG-3′ |
| Twist reverse |
5′-CACGCCCTGATTCTTGTGAA-3′ |
| Snail forward |
5′-CTTGTGTCTGCACGACCTGT-3′ |
| Snail reverse |
5′-CTTCACATCCGAGTGGGTTT-3′ |
| Vimentin forward |
5′-TCTCTGGCACGTCTTGACCTT-3′ |
| Vimentin reverse |
5′-GCTCCTGGATTTCCTCTTCGT-3′ |
Statistical analysis
Each experiment was performed at least three times
independently, and the data are presented as the means ± SD. The
significance of the differences between the groups was judged using
a two-tailed Student’s t-test. All of the tests were considered
significant if the P-value was <0.05.
Results
B7-H1 expression is upregulated in
pancreatic carcinoma specimens
Previous research has revealed that the expression
of B7-H1 is increased in a variety of human tumours. In the present
study, a similar increase in B7-H1 expression was detected in
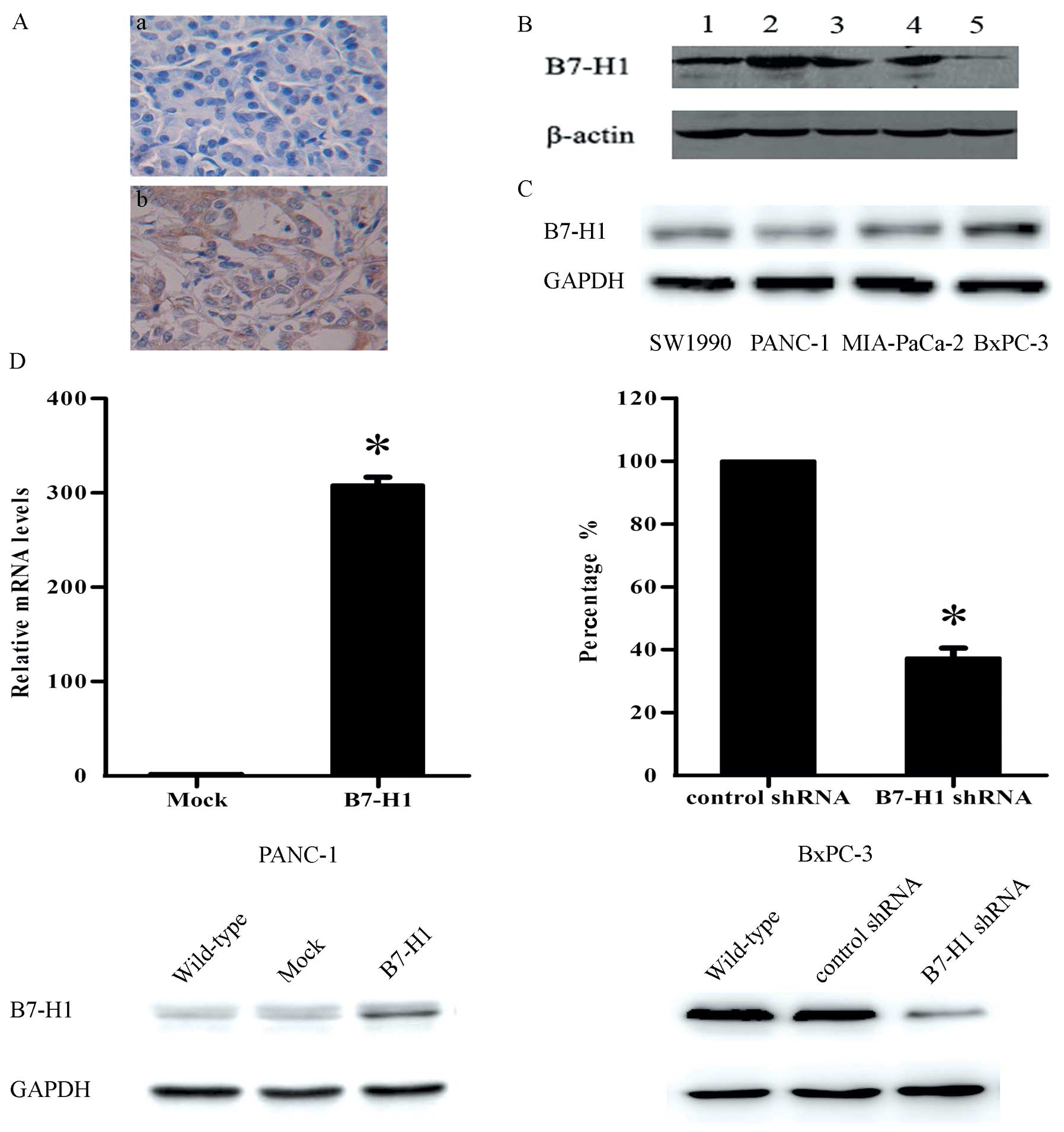
pancreatic carcinoma. The immunohistochemical analysis showed that
62.4±9.1% of the tumour samples expressed B7-H1 and that this
protein was primarily located in the cytoplasm of the tumour cells
(Fig. 1A). The western blot
analysis further confirmed these results (Fig. 1B).
Expression of B7-H1 in human pancreatic
cancer cell lines
We detected the B7-H1 expression levels in four
human pancreatic cancer cell lines by western blot analysis
(Fig. 1C). To explore whether B7-H1
plays a functional role in pancreatic tumour cell behaviour in
vitro, we chose to use BxPC-3 and PANC-1 cells in the
experiments. The two cell lines were genetically altered to express
different B7-H1 levels, as shown in Fig. 1D, to determine the effect of B7-H1
on tumour cell growth.
B7-H1 regulates cell proliferation, cell
cycle distribution and colony formation in pancreatic cancer cell
lines in vitro
To determine whether the overexpression of the B7-H1
gene affects the behaviour of pancreatic tumour cells in
vitro, the growth curve, cell cycle distribution and colony
formation were examined in cell lines that were genetically altered
to express different B7-H1 levels (Fig.
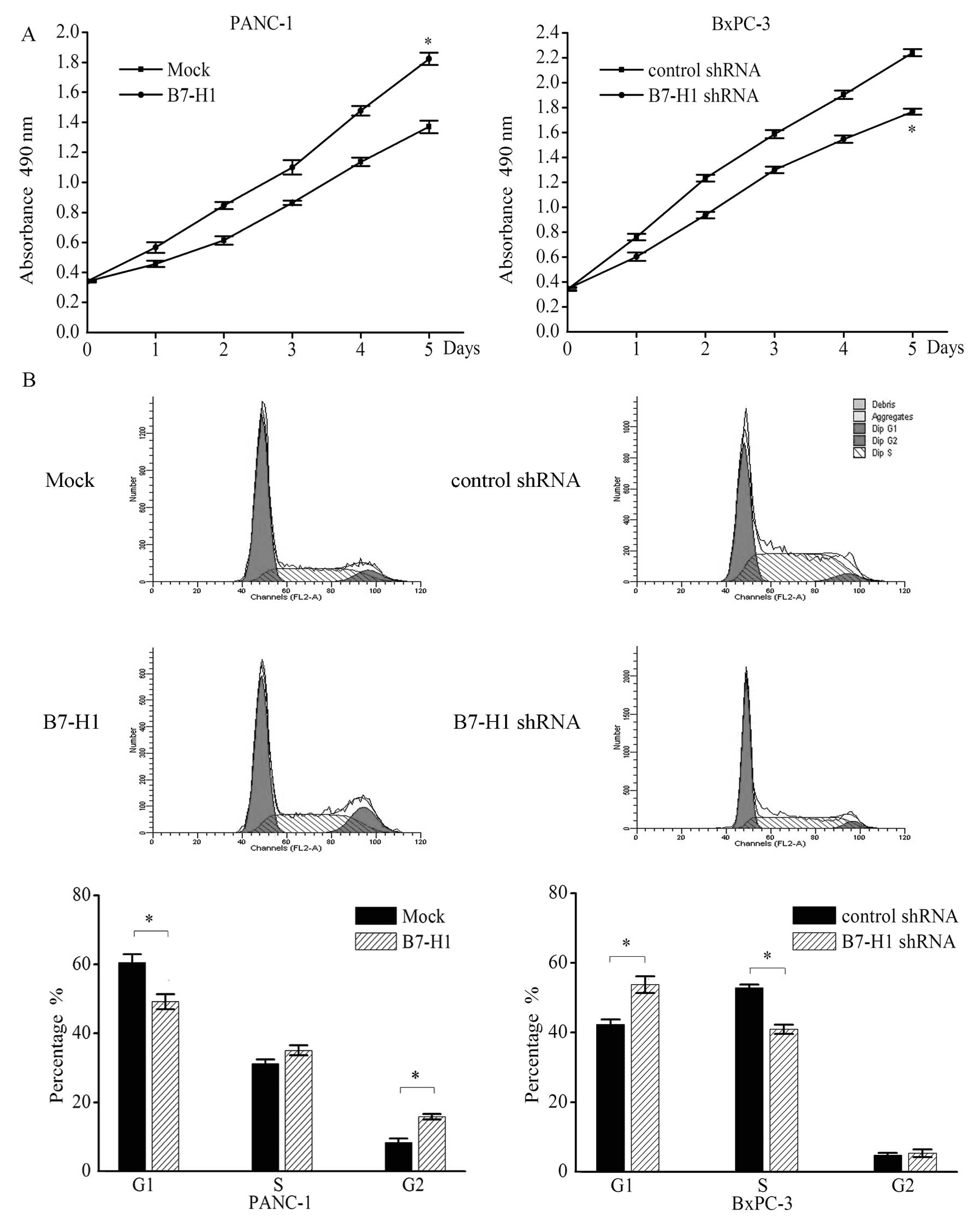
2). Compared with the mock-transfected cells (Mock), the PANC-1
cells transfected with the B7-H1 vector (B7-H1) exhibited an
increased cell growth rate on day 7. On day 5 the absorbance of the
B7-H1-overexpressing cells was >32.8% of that of the
mock-transfected cells. In contrast, knockdown of B7-H1 in BxPC-3
cells (B7-H1 shRNA) reduced the proliferation relative to the cells
transfected with a negative control shRNA (control shRNA; Fig. 2A).
In addition, results of the flow cytometry for the
cell cycle analysis confirmed the effect of B7-H1 on pancreatic
tumour cells. We found that the percentage of cells in the G1 phase
was significantly decreased in the B7-H1-overexpressing PANC-1
cells (Mock vs. B7-H1, 60.5±2.4 vs. 49.2±2.2%) and was
significantly increased in the B7-H1-depleted BxPC-3 cells (control
shRNA vs. B7-H1 shRNA, 42.3±1.4 vs. 53.8±2.4%), whereas the
percentage of cells in the S phase was increased in the
B7-H1-overexpressing PANC-1 cells (Mock vs. B7-H1, 31.2±1.2 vs.
35.1±1.4%) and was significantly decreased in the B7-H1-depleted
BXPC-3 cells (control shRNA vs. B7-H1 shRNA, 52.9±0.9 vs.
40.9±1.3%; Fig. 2B). In addition,
the percentage of cells in the G2 phase was significantly increased
in the B7-H1-overexpressing cells (Mock vs. B7-H1, 8.3±1.2 vs.
15.8±0.8%).
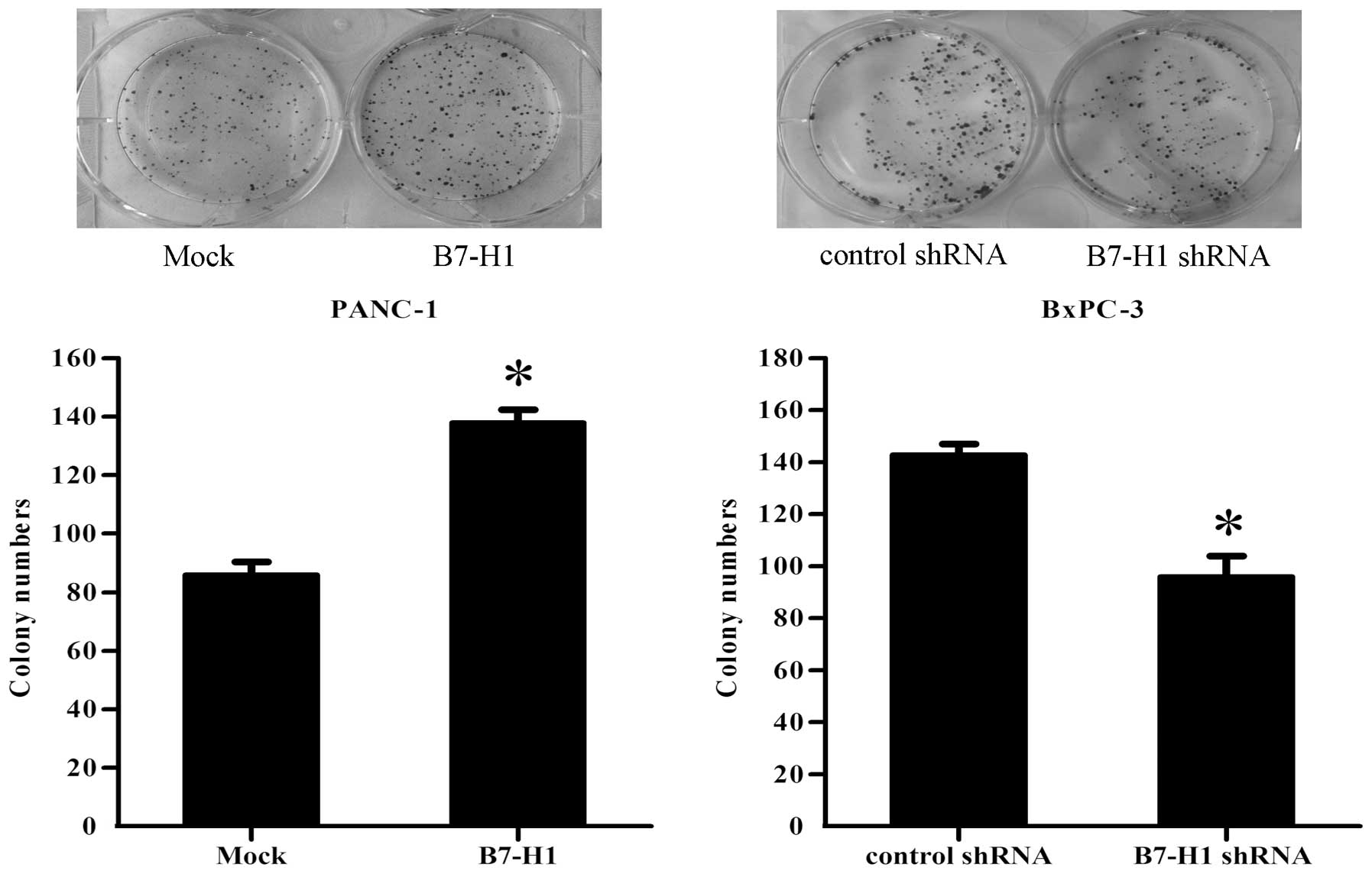
Finally, to document the ability of the different
groups of pancreatic cancer cells to form colonies, single-cell
suspensions were plated in 6-cm culture dishes at a density of 100
cells/dish. After 12 days, as shown in Fig. 3, the PANC-1 cells transfected with
the B7-H1 vector (B7-H1) exhibited a significant increase in the
colony formation numbers by 60%, when compared with the control
(Mock). However, the knockdown of B7-H1 in BxPC-3 cells (B7-H1
shRNA) resulted in a reduction of 32% in the ability of the cells
to form colonies compared with the cells transfected with the
control shRNA.
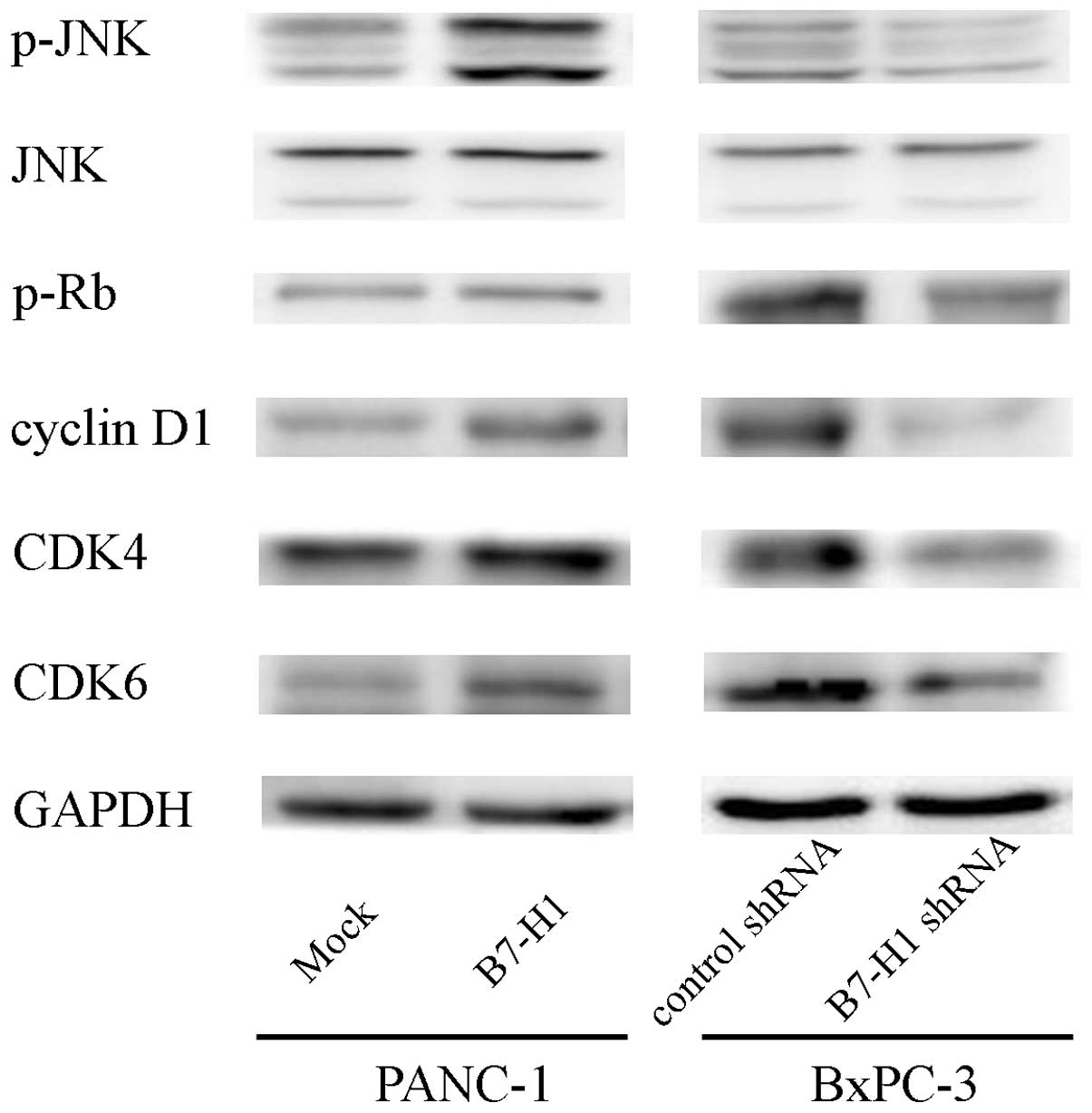
B7-H1 overexpression upregulates cyclin
D1 and p-JNK in pancreatic cancer cells
To investigate the possible mechanisms through which
B7-H1 affects pancreatic cancer cell proliferation, we tested the
effects of B7-H1 overexpression and knockdown on various cell
cycle-related proteins related to cell proliferation. As shown in
Fig. 4, B7-H1 overexpression caused
the upregulation of cyclin D1, CDK4/6 and Rb phosphorylation.
Furthermore, we explored the phosphorylation of the
mitogen-activated protein kinase (MAPK) signalling molecules (ERK,
JNK and p38) and found that the phosphorylation level of JNK was
increased, which may indicate that this protein is involved in the
process through which B7-H1 affects cell growth. In contrast, B7-H1
depletion reduced cyclin D1, CDK4/6, p-Rb and p-JNK expression.
Together, these results suggest that B7-H1 affects cell cycle
progression likely by regulating cyclin D1 and p-JNK expression in
pancreatic cancer cells.
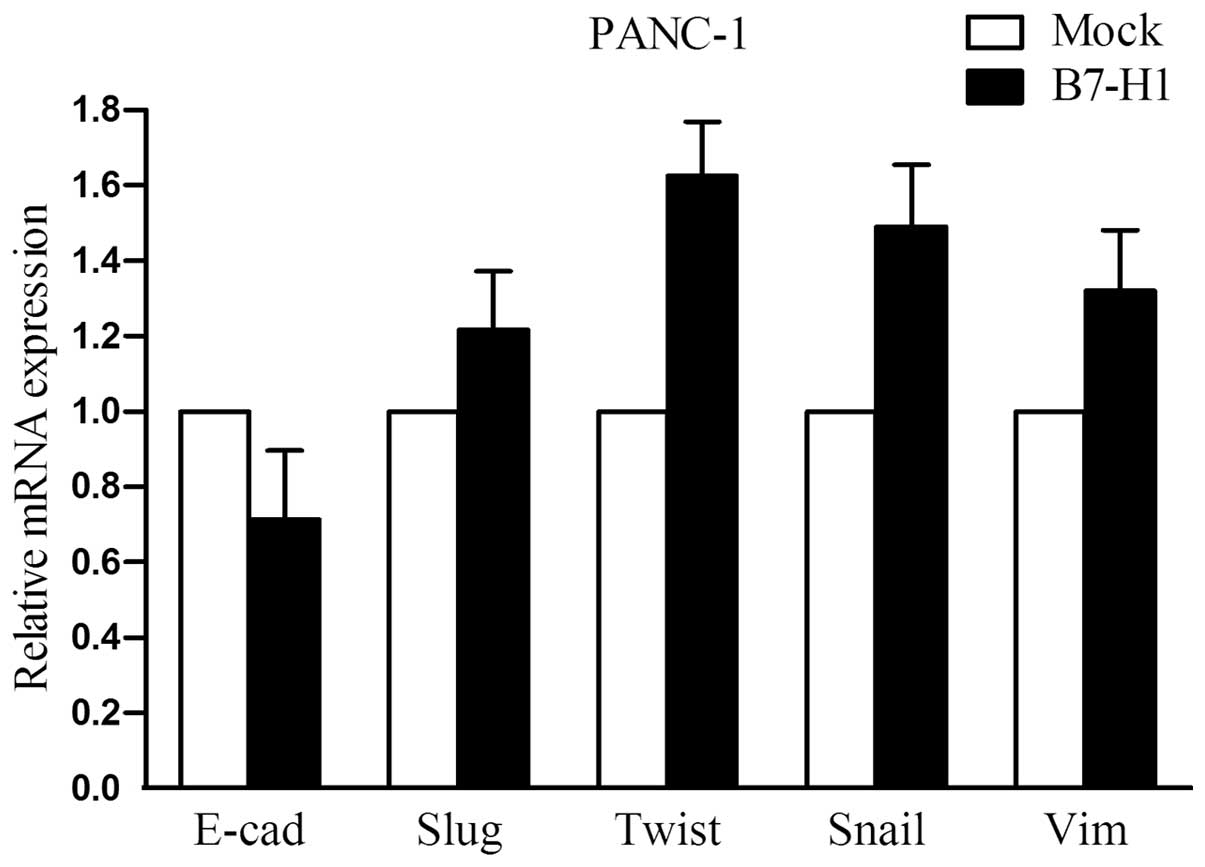
EMT-related molecules are not markedly
altered in B7-H1- overexpressing cells
To address the impact of B7-H1 on cellular
epithelial-mesenchymal transition (EMT), we explored the expression
of several important EMT-related molecules in B7-H1-overexpressing
PANC-1 cells. As shown in Fig. 5,
the mRNA levels of E-cadherin, Slug, Twist, Snail and Vimentin were
examined by quantitative real-time RT-PCR. However, we did not
observe any marked changes in their expression.
Discussion
In the present study, we demonstrated that the B7-H1
expression level is upregulated in pancreatic carcinoma tissues and
is a regulatory element in pancreatic tumour cell growth. The
proliferation of pancreatic tumour cells was promoted in the
B7-H1-overexpressing cell line, and the downregulation of B7-H1 by
shRNA in a pancreatic cancer cell line with high constitutive B7-H1
expression decreased cell growth. In addition, the upregulation of
cyclin D1 by B7-H1 overexpression was, at least in part, due to an
increase in the phosphorylation level of the JNK pathway. We
extended the results of previous studies; and further demonstrated
that the high expression of B7-H1 in pancreatic cancer cells
exhibits an intrinsic proliferative advantage in vitro.
Due to a lack of effective therapies, pancreatic
cancer is one of the most aggressive and intractable human
malignant tumours. Therefore, novel approaches against this lethal
disease need to be established to improve patient prognosis. Among
the proposed novel therapies, immunotherapy may be a potent
strategy. B7-H1, a member of the B7 family of proteins, is
upregulated in many tumours, including pancreatic cancer and
confers resistance to cytolysis by CD8+ cytolytic T
cells (CTLs) to allow cancer cells to escape the immune
surveillance of the host (2). In
addition, its expression is strongly associated with patient
clinicopathological parameters (3,11). In
pancreatic carcinoma, immunohistochemical staining revealed that
B7-H1 and IL-10 are almost completely expressed in tumour cells.
B7-H1 expression is significantly associated with poor tumour
differentiation and advanced tumour stage (17). Ample evidence demonstrates that
B7-H1 acts as a ligand for programmed death receptor-1 (PD-1), and
that their binding delivers an inhibitory signal to T cells that
induces apoptosis, anergy and exhaustion (18–20).
The ablation of the B7-H1 and PD-1 interaction by neutralising
antibodies could restore the CTL-mediated lysis of tumour cells
in vitro, which suggests that the B7-H1/PD-1 interaction
forms a barrier between tumour cells and CTLs (15). Recent studies, however, have
revealed that B7-H1 exhibits other molecular shield phenomena. For
instance, B7-H1 is a ubiquitous antiapoptotic receptor in
mastocytoma cells and exhibits greater proliferative capacity in
B7-H1+ MDS blasts than in B7-H1− MDS blasts
(15,21,22).
Our previous study also showed that B7-H1 transduces an
antiapoptotic signal and that the formation of the PD-1/PD-L1
complex inhibits drug-induced apoptosis in pancreatic cancer cells
in vitro (16). Accordingly,
in the present study, we used PANC-1 cells that were stably
transfected with B7-H1 and B7-H1-knockdown BxPC-3 cells to explore
the effect of B7-H1 on cell growth. Our results indicated that
B7-H1 indeed affected pancreatic cancer tumourigenesis and cell
cycle progression. Furthermore, the knockdown of B7-H1 decreased
the protein levels of cyclin D1 and p-Rb, and B7-H1 overexpression
increased cyclin D1 and p-Rb expression. Cyclin D1 was found to
interact with CDK4/6 to elicit its function. The cyclin D-CDK4/6
complex partially phosphorylates Rb, which is important for cell
cycle progression (23). All of
these data suggest that B7-H1 may be involved in early-stage
carcinogenesis through intrinsic cell changes before its binding to
PD-1 or other receptors to downregulate active tumour immunity.
This finding is consistent with our previous reports, which suggest
that the upregulation of B7-H1 expression in human pancreatic
carcinoma tissues may play a role in tumour progression and
invasiveness. Thus, B7-H1 may be an effective therapeutic target
for pancreatic cancer in the clinical setting.
As the B7-H1/PD-1 pathway plays an important role in
the evasion of the host antitumour immune response, immunotherapies
that regulate this pathway are expected to be beneficial. Initial
clinical studies of antibodies directed against PD-1 and B7-H1 have
shown marked antitumour activity in subsets of patients with
malignancies (24,25). In pancreatic ductal adenocarcinoma,
the blocking of B7-H1 increased the proliferation and function of
tumour-specific CTLs at the tumour sites. However, the function of
B7-H1 may not only be associated with the downregulation of T-cell
function. One study indicated that B7-H1 molecules in myeloma cells
exhibit dual activity; the induction of T-cell downregulation and
the enhancement of aggressive myeloma cell characteristics,
including increased proliferative ability and drug resistance.
Thus, the blocking of the signal to T cells alone may be
insufficient (13). In our
experiment, B7-H1 molecules in pancreatic tumour cells were also
associated with aggressive cell behaviour, including increased
proliferative potential and resistance to drug-induced apoptosis.
Thus, this finding must be taken into account when designing
clinical trials targeting B7-H1.
Previous research has identified a mechanism that
links the loss of the tumour suppressor ‘phosphatase and tensin
homologue on chromosome ten’ (PTEN) with immunoresistance, mediated
in part by B7-H1 (26). Thus, we
tested the expression of PTEN in B7-H1-overexpressing pancreatic
tumour cells and found no obvious difference (data not shown).
Another study reported that squamous cell carcinomas induced in
B7-H1 transgenic mice exhibited a loss of E-cadherin and the
elevated expression of the transcription factors Slug and Twist,
which drive the epithelial-mesenchymal transition (EMT) process
(14). In the present study, the
mRNA expression levels of E-cadherin, Slug, Twist, Snail and
Vimentin were found to be comparable between the control and the
B7-H1-overexpressing pancreatic cancer cells (Fig. 5). It is possible that B7-H1
overexpression in different tissues can produce different effects
on tumour cell characteristics or that posttranscriptional
regulation of relevant gene expression occurs. To summarise, these
results suggest that the overexpression of B7-H1 in pancreatic
tumour cells does not lead to cell EMT and PTEN expression
changes.
Although several studies using different types of
carcinomas have demonstrated that an elevated level of B7-H1 is
highly correlated with tumour cell proliferation, antiapoptosis and
EMT, the precise signalling involved in the B7-H1-mediated
intrinsic cell changes remain largely undefined (2,12,13).
Since B7-H1 most likely has another unidentified receptor, it may
receive a proliferative signal by interacting with this receptor.
Another possible mechanism is that B7-H1 molecules may not transmit
any signal to tumour cells; but may coincidentally link with the
activation of proliferative signals. In the present study, we
showed that B7-H1 induced both cyclin D1 and phospho-JNK
expression, which suggests that the upregulation of cyclin D1 by
B7-H1 overexpression was likely through the JNK pathway. JNK
signalling is reported to play an important role in pancreatic
cancer cell proliferation (27).
Further studies, including in vivo experiments and the in
situ detection of cell cycle-related molecules and B7-H1
expression in human pancreatic tumour samples, are required.
In conclusion, the B7-H1 expression level was
upregulated in pancreatic carcinoma tissues and the overexpression
of B7-H1 accelerated pancreatic tumour cell proliferation. Our data
provide a deeper understanding of B7-H1 function, namely, the
promotion of pancreatic cell growth by facilitating cell cycle
progression through the JNK-mediated upregulation of cyclin D1.
These results suggest that B7-H1 functions as a positive regulator
of pancreatic cancer proliferation.
Acknowledgements
The present study was supported by the Key Social
Development Project of Major Science and Technology (2011C13036-1).
All research was performed in the Biomedical Research Center,
Zhejiang Provincial People’s Hospital, Hangzhou, China.
References
|
1
|
Dong H, Zhu G, Tamada K and Chen L: B7-H1,
a third member of the B7 family, co-stimulates T-cell proliferation
and interleukin-10 secretion. Nat Med. 5:1365–1369. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dong H, Strome SE, Salomao DR, et al:
Tumor-associated B7-H1 promotes T-cell apoptosis: a potential
mechanism of immune evasion. Nat Med. 8:793–800. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nomi T, Sho M, Akahori T, et al: Clinical
significance and therapeutic potential of the programmed death-1
ligand/programmed death-1 pathway in human pancreatic cancer. Clin
Cancer Res. 13:2151–2157. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ohigashi Y, Sho M, Yamada Y, et al:
Clinical significance of programmed death-1 ligand-1 and programmed
death-1 ligand-2 expression in human esophageal cancer. Clin Cancer
Res. 11:2947–2953. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen XL, Cao XD, Kang AJ, Wang KM, Su BS
and Wang YL: In situ expression and significance of B7
costimulatory molecules within tissues of human gastric carcinoma.
World J Gastroenterol. 9:1370–1373. 2003.
|
|
6
|
Wintterle S, Schreiner B, Mitsdoerffer M,
et al: Expression of the B7-related molecule B7-H1 by glioma cells:
a potential mechanism of immune paralysis. Cancer Res.
63:7462–7467. 2003.PubMed/NCBI
|
|
7
|
Dong H and Chen L: B7-H1 pathway and its
role in the evasion of tumor immunity. J Mol Med. 81:281–287.
2003.PubMed/NCBI
|
|
8
|
Iwai Y, Ishida M, Tanaka Y, Okazaki T,
Honjo T and Minato N: Involvement of PD-L1 on tumor cells in the
escape from host immune system and tumor immunotherapy by PD-L1
blockade. Proc Natl Acad Sci USA. 99:12293–12297. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Flies DB, Sandler BJ, Sznol M and Chen L:
Blockade of the B7-H1/PD-1 pathway for cancer immunotherapy. Yale J
Biol Med. 84:409–421. 2011.PubMed/NCBI
|
|
10
|
Okudaira K, Hokari R, Tsuzuki Y, et al:
Blockade of B7-H1 or B7-DC induces an anti-tumor effect in a mouse
pancreatic cancer model. Int J Oncol. 35:741–749. 2009.PubMed/NCBI
|
|
11
|
Sznol M and Chen L: Antagonist antibodies
to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human
cancer. Clin Cancer Res. 19:1021–1034. 2013. View Article : Google Scholar
|
|
12
|
Ghebeh H, Tulbah A, Mohammed S, et al:
Expression of B7-H1 in breast cancer patients is strongly
associated with high proliferative Ki-67-expressing tumor cells.
Int J Cancer. 121:751–758. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tamura H, Ishibashi M, Yamashita T, et al:
Marrow stromal cells induce B7-H1 expression on myeloma cells,
generating aggressive characteristics in multiple myeloma.
Leukemia. 27:464–472. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cao Y, Zhang L, Kamimura Y, et al: B7-H1
overexpression regulates epithelial-mesenchymal transition and
accelerates carcinogenesis in skin. Cancer Res. 71:1235–1243. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Azuma T, Yao S, Zhu G, Flies AS, Flies SJ
and Chen L: B7-H1 is a ubiquitous antiapoptotic receptor on cancer
cells. Blood. 111:3635–3643. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang F, Ma J, Liu J, Jin H and Huang D:
Synthetic small peptides acting on B7H1 enhance apoptosis in
pancreatic cancer cells. Mol Med Rep. 6:553–557. 2012.PubMed/NCBI
|
|
17
|
Geng L, Huang D, Liu J, et al: B7-H1
up-regulated expression in human pancreatic carcinoma tissue
associates with tumor progression. J Cancer Res Clin Oncol.
134:1021–1027. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tsushima F, Yao S, Shin T, et al:
Interaction between B7-H1 and PD-1 determines initiation and
reversal of T-cell anergy. Blood. 110:180–185. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Goldberg MV, Maris CH, Hipkiss EL, et al:
Role of PD-1 and its ligand, B7-H1, in early fate decisions of CD8
T cells. Blood. 110:186–192. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Okazaki T and Honjo T: The PD-1-PD-L
pathway in immunological tolerance. Trends Immunol. 27:195–201.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kondo A, Yamashita T, Tamura H, et al:
Interferon-γ and tumor necrosis factor-α induce an immunoinhibitory
molecule, B7-H1, via nuclear factor-κB activation in blasts in
myelodysplastic syndromes. Blood. 116:1124–1131. 2010.
|
|
22
|
Ghebeh H, Lehe C, Barhoush E, et al:
Doxorubicin downregulates cell surface B7-H1 expression and
upregulates its nuclear expression in breast cancer cells: role of
B7-H1 as an anti-apoptotic molecule. Breast Cancer Res. 12:R482010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Knudsen KE, Diehl JA, Haiman CA and
Knudsen ES: Cyclin D1: polymorphism, aberrant splicing and cancer
risk. Oncogene. 25:1620–1628. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brahmer JR, Drake CG, Wollner I, et al:
Phase I study of single-agent anti-programmed death-1 (MDX-1106) in
refractory solid tumors: safety, clinical activity,
pharmacodynamics, and immunologic correlates. J Clin Oncol.
28:3167–3175. 2010. View Article : Google Scholar
|
|
25
|
Brahmer JR, Tykodi SS, Chow LQ, et al:
Safety and activity of anti-PD-L1 antibody in patients with
advanced cancer. N Engl J Med. 366:2455–2465. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Parsa AT, Waldron JS, Panner A, et al:
Loss of tumor suppressor PTEN function increases B7-H1 expression
and immunoresistance in glioma. Nat Med. 13:84–88. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Takahashi R, Hirata Y, Sakitani K, et al:
Therapeutic effect of c-Jun N-terminal kinase inhibition on
pancreatic cancer. Cancer Sci. 104:337–344. 2013. View Article : Google Scholar : PubMed/NCBI
|