Introduction
The use of imatinib in the treatment of patients
with chronic myeloid leukemia (CML) must be ranked as one of the
great medical success stories of the past 30 years (1). However, ~20% of patients with CML do
not respond to treatment with imatinib either initially or as a
result of acquired resistance (2).
Dasatinib and nilotinib are treatment options for CML patients for
whom treatment with imatinib has failed owing to resistance and/or
intolerance. However, 22–33% of those patients had to discontinue
by two years due to adverse events, treatment failure or other
causes (3). Resistance to tyrosine
kinase inhibitors (TKIs) in patients with CML and Philadelphia
chromosome-positive acute lymphoblastic leukemia (Ph-positive ALL)
is frequently caused by mutations in the BCR-ABL kinase domain
(4). Ponatinib (the chemical
structure is shown in Fig. 1) was
designed by ARIAD using a computational and structure-based drug
design platform to inhibit the activity of BCR-ABL with very high
potency and broad specificity. Ponatinib targets not only native
BCR-ABL but also its isoforms that carry mutations which confer
resistance to treatment with existing TKIs, including the T315I
which causes uniform resistance to TKIs (5). The Food and Drug Administration (FDA)
approved ponatinib to treat two forms of drug-resistant leukemia,
CML and Ph-positive ALL, in December 2012.
The ATP-Binding Cassette (ABC) transporter
superfamily is one of the largest and most highly conserved protein
families, with structural features and mechanisms of action
conserved from prokaryotes to humans (6). ABC transporters are known to account
for the multidrug resistant (MDR) phenotype of various cancer
cells. They are capable of recognizing and extruding a broad range
of compounds, without relation to chemical structure or cellular
target (7). When overexpressed in
cancer cells, ABC transporters reduce intracellular drug
concentrations below the effective cytotoxic threshold and induce
drug resistance (8). The major ABC
proteins that are widely accepted to be responsible for the MDR
phenotype of cancer cells are P-glycoprotien (P-gp, also called
MDR1 or ABCB1), multidrug resistance proteins (MRPs, also called
ABCCs) and breast cancer resistance protein (BCRP, also called
ABCG2, BCRP, MXR or ABCP) with each having promiscuous and
overlapping substrate recognition spectra (9).
ABC transporters have recently been recognized as
important determinants of the general ADME-Tox (absorption,
distribution, metabolism, excretion and toxicity) properties of
small molecule TKIs, as well as key factors in resistance against
targeted anticancer therapeutics (10). The interaction of numerous
clinically applied TKIs with various ABC transporters is
complicated. TKIs may be extruded out of the cell by ABC
transporters. However, TKIs may also directly inhibit ABC
transporters, thereby inducing sensitization to chemotherapy agents
(11,12). In some cases, TKI compounds may
behave both as substrates and inhibitors of a given transporter,
depending on the concentration range applied (13). Imatinib was reported to be able to
inhibit the transport of substrates of P-gp, BCRP, MRP1 and MRP7
(14–16). Nilotinib was shown to be an
inhibitor of P-gp, BCRP and MRP7 both in vitro and in
vivo (16–18). Similarly, lapatinib, which is used
for the treatment of Her-2 positive advanced or metastatic breast
cancer, is an inhibitor of P-gp, BCRP and MRP7 (19,20).
The third-generation TKI ponatinib also enhances uptake of
substrates of BCRP and P-gp, but not MRP1, with a greater effect on
BCRP than on P-gp (21).
MRP7 (also called ABCC10) belongs to the C subfamily
of ABC transporters. It confers in vitro resistance to a
wide range of clinically used anticancer drugs, including taxanes,
vinca alkaloids, nucleoside analogs and epothilone B (22). In vivo, absence of this
transporter also sensitized animals to taxanes in an
Abcc10−/− mouse model, indicating that MRP7 may function
as a major determinant of taxane sensitivity (23). These findings suggest that
modulation of MRP7 may have clinical value in management of human
cancers which are treated with taxane-contained regimens. Whether
ponatinib has potential efficacy in reversing MRP7-mediated MDR was
explored in the present study. We found that ponatinib was able to
reverse MRP7-mediated MDR at a low concentration by both blocking
MRP7 function and downregulating its expression.
Materials and methods
Reagents
Paclitaxel, docetaxel, vincristine, vinblastine,
dimethyl sulfoxide (DMSO) and
1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan (MTT) were
purchased from Sigma-Aldrich. Cepharanthine was kindly provided by
the Kakenshoyaku Co. [3H]-paclitaxel (38.9 Ci/mmol) was
obtained from Moravek Biochemicals. Ponatinib was acquired from
Selleck Chemicals.
Cell lines and cell culture
MRP7 expression vector (HEK/MRP7) and parental
plasmid (HEK/pcDNA) were transfected into human embryonic kidney
293 (HEK293) cells by electroporation as we previously reported
(24). Transfected cells were
selected in DMEM containing 2 mg/ml G418. MRP7 protein was detected
by immunoblot analysis. All cell lines were grown as adherent
monolayers in DMEM supplemented with 10% fetal bovine serum (FBS),
200 U/ml penicillin and 200 U/ml streptomycin (HyClone). All cell
lines were grown at 37°C in 5% CO2 under humidifying
conditions.
MTT assay
The sensitivity of cells to anticancer drugs was
measured by an MTT colorimetric assay with minor modifications from
that previously described (25).
Cells were harvested with trypsin and resuspended at a final
concentration of 6×103 cells/well. After incubation in
DMEM supplemented with 10% FBS at 37°C for 24 h, ponatinib (0.1,
0.25 or 0.5 μM, 20 μl/well) or the MRP7 inhibitor cepharanthine
(26) (2.5 μM, 20 μl/well) were
added 1 h prior to the addition of anticancer drugs at different
concentrations (20 μl/well). After 68 h of incubation, 20 μl of MTT
solution (4 mg/ml) was added to each well, and the plate was
incubated for another 4 h, allowing viable cells to convert the
yellow-colored MTT into dark blue formazan crystals. Then the
medium was aspirated, and 100 μl DMSO was added to each well to
dissolve the formazan crystals. The absorbance was determined at
570 nm and 630 nm by an Opsys microplate reader (Dynex
Technologies). The degree of resistance was calculated by dividing
the IC50 (concentrations required to inhibit growth by
50%) for the MDR cells by that of the parental sensitive cells. The
IC50 values were calculated from the survival curves
using the Bliss method (27).
[3H]-paclitaxel accumulation
and efflux
Intracellular paclitaxel accumulation and efflux
were measured in HEK/pcDNA cells and HEK/MRP7 cells. For the
accumulation assay, the cells were trypsinized and three aliquots
(5×106 cells) from each cell line were resuspended in
medium. To measure drug accumulation, cells were preincubated in
DMEM in the presence or absence of ponatinib (at 0.5 μM) or
cepharanthine (at 2.5 μM) for 2 h, washed and then incubated with
0.01 μM [3H]-paclitaxel with or without reversing agents
for another 2 h at 37°C. The cells were pelleted at 4°C, washed
twice with 10 ml ice-cold PBS, and lysed in 10 mM lysis buffer (pH
7.4, containing 1% Triton X-100 and 0.2% SDS). Radioactivity was
measured in a liquid scintillation counter. For the efflux study,
cells were incubated with 0.01 μM [3H]-paclitaxel
according to the method for the accumulation study. After being
washed twice with cold PBS, the cells were cultured in fresh DMEM
with or without 0.5 μM ponatinib at 37°C. After 0, 30, 60 or 120
min, aliquots of cells were removed and immediately washed with
ice-cold PBS. The cell pellets were collected for radioactivity
measurement in a Packard TRI-CARB® 1900CA liquid
scintillation analyzer (Packard Instrument Co.).
Western immunoblot analysis
To determine the effect of ponatinib on the
expression of MRP7, HEK/MRP7 cells were incubated with 0.5 μM
ponatinib for 0, 2, 4, 6, 8, 12 and 24 h or 0, 0.1, 0.25 and 0.5 μM
for 24 h. After treatment, the cells were harvested and rinsed
three times with ice-old PBS. Cell extracts were prepared by
incubating cells for 30 min on ice with radioimmunoprecipitation
assay (RIPA) buffer (PBS with 0.1% SDS, 1% Nonidet P-40, 0.5%
sodium deoxycholate and 100 mg/ml p-aminophenylmethylsulfonyl
fluoride) with occasional rocking, followed by centrifugation
(12,000 g, 4°C for 20 min). The supernatant containing total cell
lysates was stored at −80°C prior to experiments. Cell lysates
containing identical amounts of total protein (25 μg for different
time treatment group and 60 μg for different concentration
treatment group) were resolved by sodium dodecyl sulfate
polycrylamide gel electrophoresis (SDS-PAGE) and transferred onto
polyvinylidene fluoride (PVDF) membranes. After incubation in a
blocking solution containing 5% non-fat milk in TBST buffer [10
mmol/l Tris-HCl (pH 8.0), 150 mmol/l NaCl, and 0.1% Tween-20] for 2
h at room temperature, the membranes were immunoblotted overnight
with primary antibodies against MRP7 (1:200 dilution; Santa Cruz
Biotechnology) or GAPDH (1:1,000 dilution; Cell Signaling
Technology) at 4°C, and then the membranes were washed five times
for 5 min/each time with TBST buffer and incubated at room
temperature with horseradish peroxidase (HRP)-conjugated secondary
antibody (1:1,000 dilution) for 2 h. The protein-antibody complex
was detected by chemiluminescence. Densitometry analysis was
performed using the Quantity One software (Bio-Rad) and MRP7
immunoblots were normalized to GAPDH immunoblots for each sample
analyzed.
Reverse transcription polymerase chain
reaction (RT-PCR)
Total RNA was extracted from HEK/pcDNA and HEK/MRP7
cells using TRIzol according to the manufacturer’s instructions.
Reverse transcription was performed with 2 μg total RNA in a volume
of 20 μl by Transcription System (Promega) according to the
manufacturer’s instructions. The sequences of the MRP7 and GAPDH
primers were as follows: MRP7 (303 bp) sense:
5′-GGCTCCGGCAAGTCTTCCCTGTT-3′ and antisense:
5′-AGATAAGCTCCGGCCCCCCTCACC-3′. GAPDH (322 bp) sense:
5′-CGGGAAGCTTGTCATCAA TGG-3′ and antisense
5′-GGCAGTGATGGCATGGACTG-3′. PCR was carried out with
GoTaq® Master Mixes and the reaction conditions were
94°C for 30 sec, 60°C for 40 sec (GAPDH) or 65°C for 40 sec (MRP7),
72°C for 45 sec with 35 cycles. The PCR products were separated by
agarose gel electrophoresis. The gel was stained with 0.5 μg/ml
ethidium bromide and the bands were visualized under UV light.
Statistical analysis
All experiments were repeated at least three times.
Statistical differences were determined by the two-tailed Student’s
t-test, and were deemed significant if P-value was <0.05.
Results
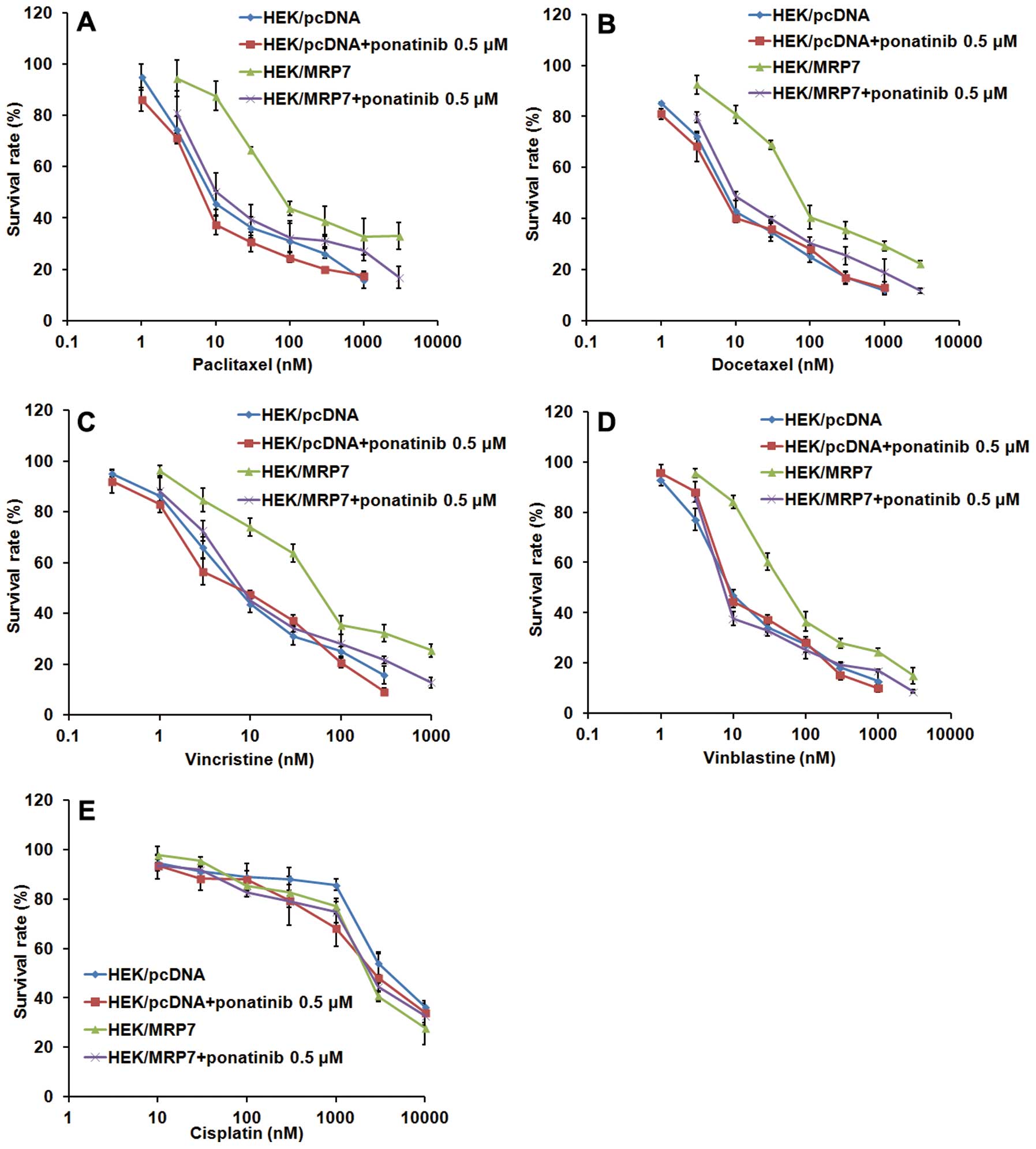
Ponatinib significantly enhanced the drug
sensitivity of MRP7-overexpressing cells
In order to determine if ponatinib could reverse ABC
transporter-mediated MDR, we treated both the parental cell line
HEK/pcDNA and the resistant cell lines HEK/MRP7 with ponatinib 1 h
prior to anticancer drugs, then measured the IC50 of
anticancer drugs in parental cells and resistant cells using the
MTT assay. Compared to parental HEK/pcDNA cells, HEK/MRP7 cells
exhibited significant resistance to various MRP7 substrates,
including paclitaxel (9.14-fold), docetaxel (8.75-fold),
vincristine (5.65-fold) and vinblastine (5.99-fold). As shown in
Table I, ponatinib at 0.1, 0.25 and
0.5 μM produced a concentration-dependent increase in sensitivity
to paclitaxel, docetaxel, vincristine and vinblastine in
MRP7-overexpressing cells. However, ponatinib did not significantly
alter the cytotoxity of the tested drugs in the parental sensitive
HEK/pcDNA cells. In addition, ponatinib did not significantly alter
the IC50 value of cisplatin, which is not a substrate of
MRP7. Curves clearly shifted significantly to the left side after
coincubation of HEK/MRP7 cells with ponatinib at 0.5 μM (Fig. 2).
 | Table IEffect of ponatinib and cepharanthine
on the cytotoxicity of paclitaxel, docetaxel, vincristine,
vinblastine, and cisplatin in MRP7-transfected cells. |
Table I
Effect of ponatinib and cepharanthine
on the cytotoxicity of paclitaxel, docetaxel, vincristine,
vinblastine, and cisplatin in MRP7-transfected cells.
| IC50
±SDa (resistance fold) |
|---|
|
|
|---|
| Compounds | HEK/pcDNA | HEK/MRP7 |
|---|
| Paclitaxel
(μM) | 9.16±0.53
(1.00) | 83.75±3.77
(9.14) |
| Ponatinib
(0.10) | 9.11±0.53
(0.99) | 19.37±1.01
(2.11)c |
| Ponatinib
(0.25) | 8.87±0.60
(0.97) | 10.02±1.57
(1.09)c |
| Ponatinib
(0.50) | 7.65±0.68
(0.84) | 9.25±0.22
(1.01)c |
| Cepharanthine
(2.5) | 7.79±0.97
(0.85) | 8.46±1.07
(0.92)c |
| Docetaxel (μM) | 8.74±0.82
(1.00)b | 76.44±5.53
(8.75) |
| Ponatinib
(0.10) | 7.78±1.05
(0.89) | 16.30±1.02
(1.86)c |
| Ponatinib
(0.25) | 7.66±0.47
(0.88) | 10.30±0.85
(1.18)c |
| Ponatinib
(0.50) | 8.26±1.04
(0.95) | 7.16±0.67
(0.82)c |
| Cepharanthine
(2.5) | 8.12±0.88
(0.93) | 7.50±1.35
(0.86)c |
| Vincristine
(μM) | 7.82±0.85
(1.00)b | 44.19±3.73
(5.56) |
| Ponatinib
(0.10) | 8.84±0.79
(1.13) | 21.46±2.36
(2.74)c |
| Ponatinib
(0.25) | 7.38±0.93
(0.94) | 9.02±0.75
(1.15)c |
| Ponatinib
(0.50) | 8.12±0.44
(1.04) | 7.87±0.82
(1.01)c |
| Cepharanthine
(2.5) | 7.21±0.42
(0.92) | 8.28±0.79
(1.06)c |
| Vinblastine
(μM) | 9.49±0.69
(1.00)b | 56.79±7.41
(5.99) |
| Ponatinib
(0.10) | 8.17±0.72
(0.86) | 11.00±0.99
(1.16)c |
| Ponatinib
(0.25) | 8.33±0.69
(0.88) | 6.66±0.94
(0.70)c |
| Ponatinib
(0.50) | 8.96±1.16
(0.94) | 4.03±0.34
(0.43)c |
| Cepharanthine
(2.5) | 8.55±0.94
(0.90) | 4.93±0.71
(0.52)c |
| Cisplatin (μM) | 4354.43±358.66
(1.00)b | 4627.31±341.80
(1.06) |
| Ponatinib
(0.10) | 4417.11±145.05
(1.01) | 5395.15±159.79
(1.24) |
| Ponatinib
(0.25) | 4625.90±444.78
(1.06) | 5115.46±229.46
(1.17) |
| Ponatinib
(0.50) | 4394.95±252.00
(1.01) | 5286.64±319.97
(1.21) |
| Cepharanthine
(2.5) | 4489.97±379.32
(1.03) | 5304.49±427.60
(1.22) |
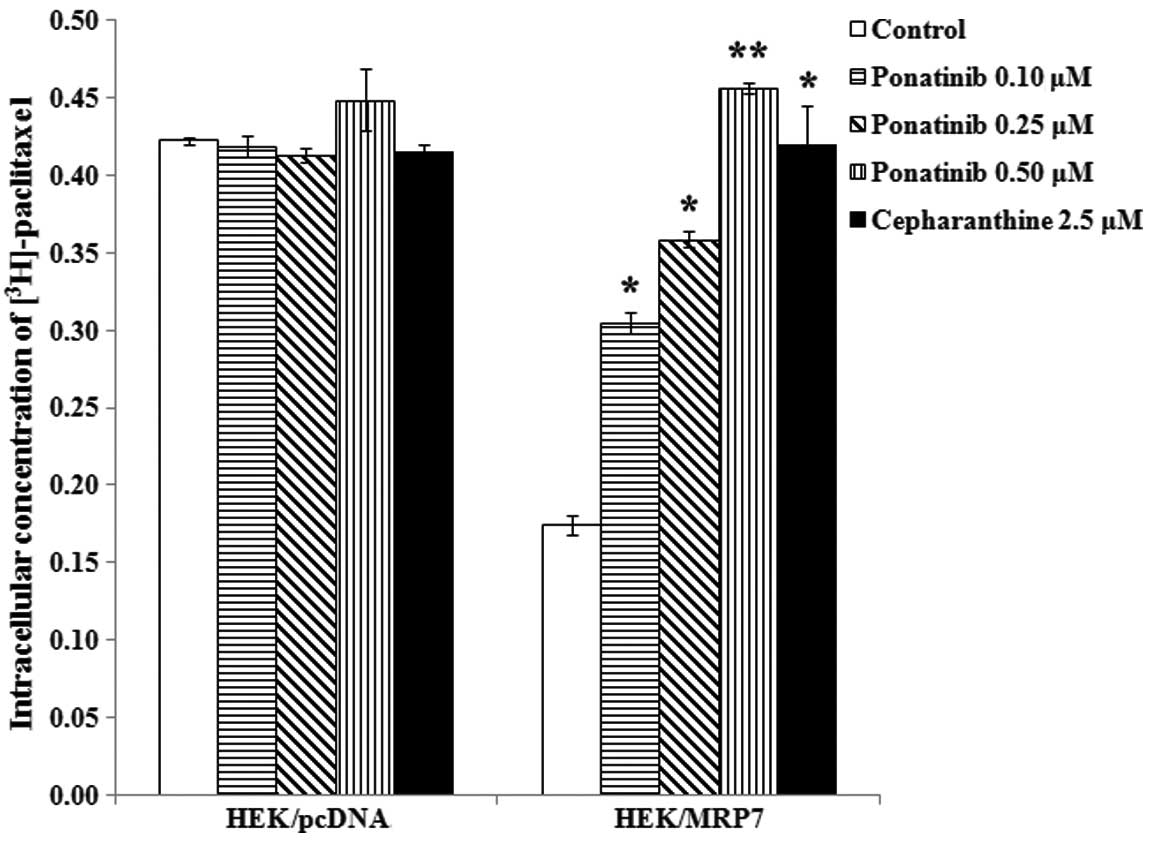
Ponatinib significantly increases the
accumulation of intracellular [3H]-paclitaxel in
MRP7-overexpressing cells
In order to determine the effect of ponatinib on the
function of MRP7, we measured the accumulation of
[3H]-paclitaxel with or without ponatinib in HEK/pcDNA
and HEK/MRP7 cells. The intracellular concentration of
[3H]-paclitaxel in HEK/MRP7 was ~41.2% of that in
parental HEK/pcDNA cells. After the cells were incubated with
ponatinib at 0.1, 0.25 and 0.5 μM and cepharanthine at 2.5 μM for 4
h, intracellular [3H]-paclitaxel accumulation was
significantly increased in HEK/MRP7 by 1.75-, 2.1-, 2.6- and
2.4-fold, respectively. However, the intracellular level of
[3H]-paclitaxel in HEK/pcDNA was not altered by either
ponatinib or cepharanthine (Fig.
3).
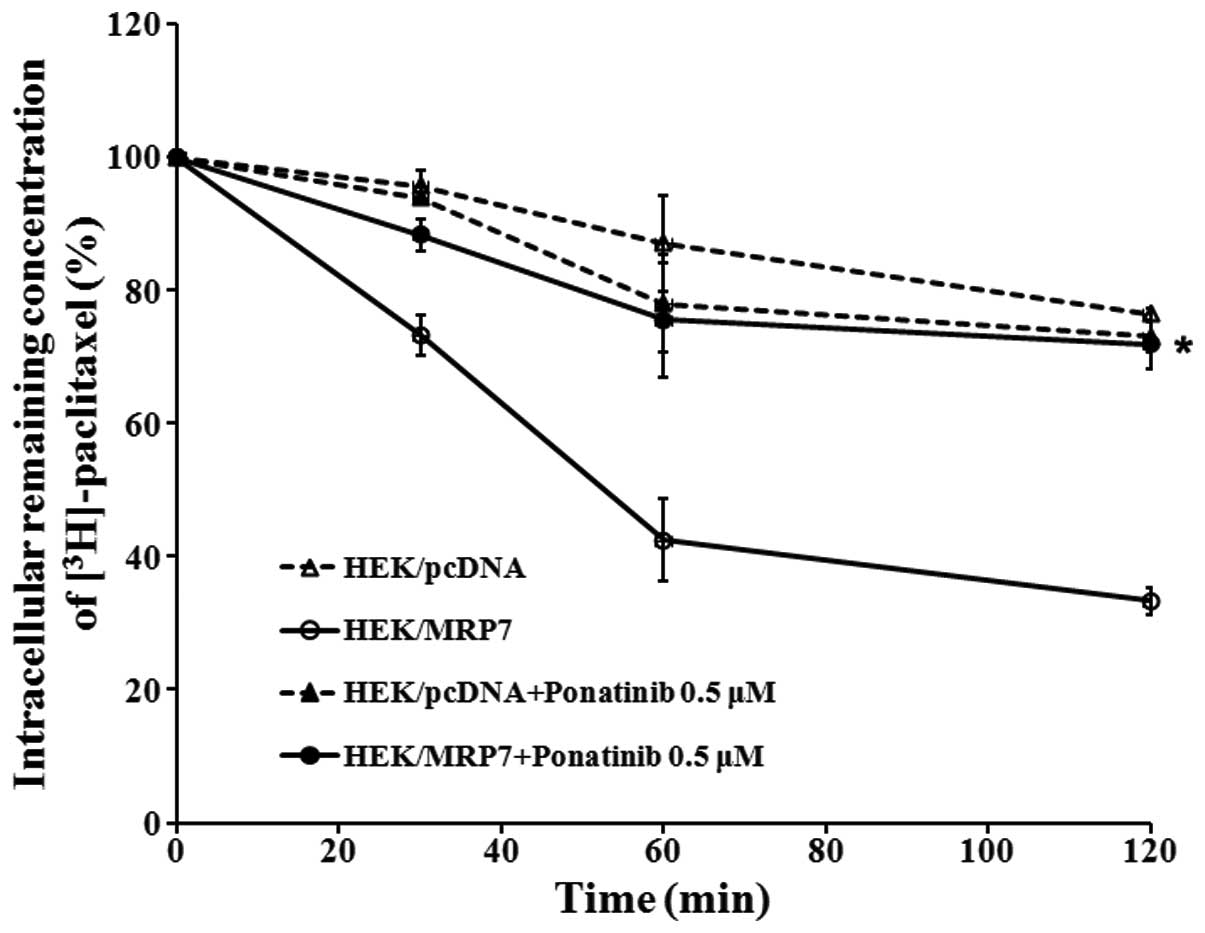
Ponatinib blocks the efflux of
[3H]-paclitaxel in MRP7-overexpressing cells
To establish whether the increased intracellular
[3H]-paclitaxel accumulation in MRP7-overexpressing
cells caused by ponatinib was due to inhibition of
[3H]-paclitaxel efflux, we performed a time course study
to determine the remaining intracellular concentration of
[3H]-paclitaxel in the presence of ponatinib. As
expected, HEK/MRP7 cells released a significantly higher percentage
of accumulated [3H]-paclitaxel compared to HEK/pcDNA
cells. When ponatinib at 0.5 μM was added to HEK/MRP7 cells, it
significantly blocked the intracellular [3H]-paclitaxel
efflux at different time periods (0, 30, 60 and 120 min), but had
no effct in the parental HEK/pcDNA cells. The accumulation of
[3H]-paclitaxel at 0 min was set at 100% and at 30, 60
and 120 min, the percentages of the intracellular
[3H]-paclitaxel that remained in HEK/MRP7 cells were
73.32, 42.51 and 33.28%, respectively, in the absence of ponatinib.
When HEK/MRP7 cells were incubated with ponatinib, the percentages
at 30, 60 and 120 min increased to 88.35, 75.54 and 71.89%,
respectively (Fig. 4).
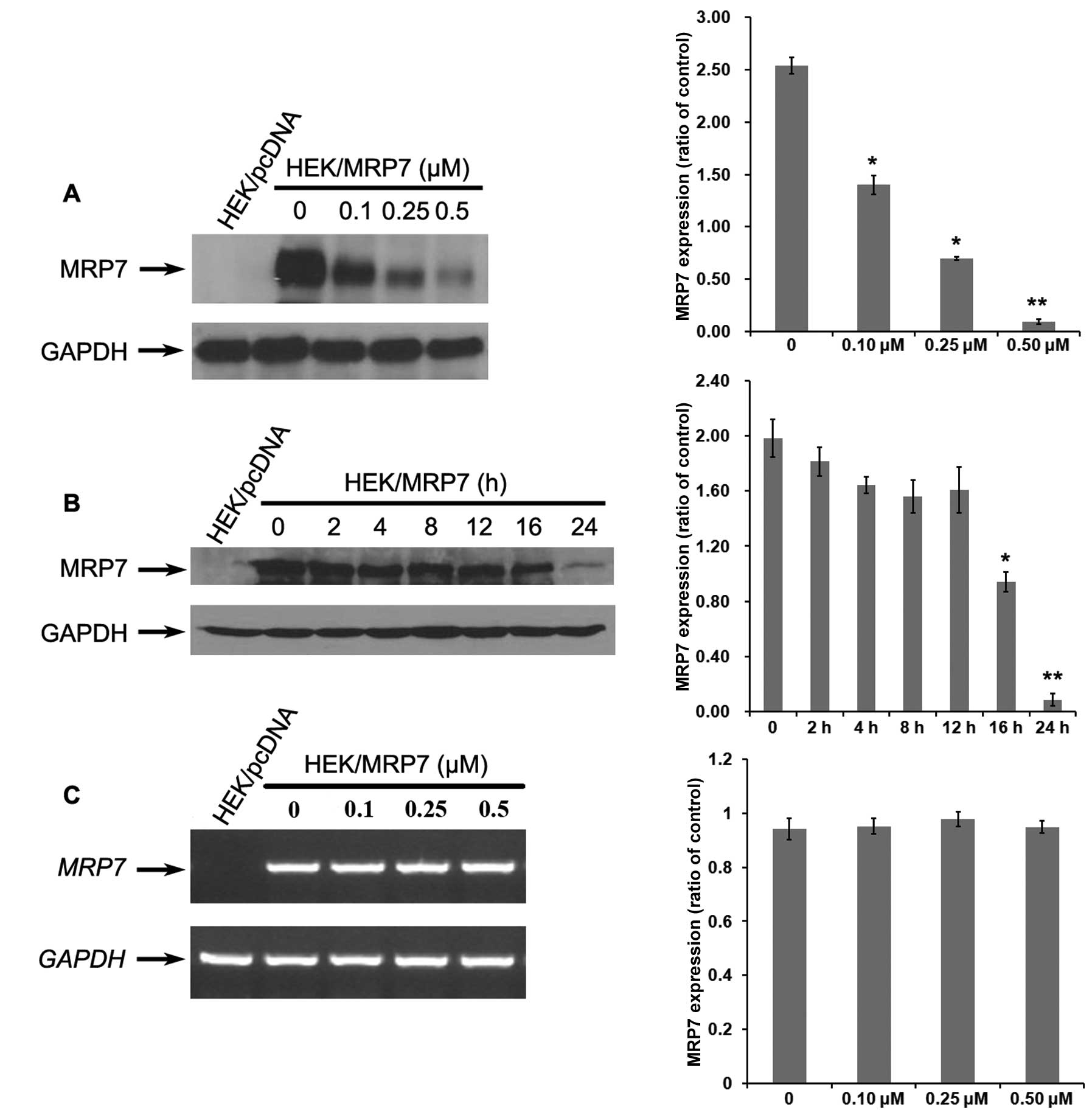
Ponatinib downregulates MRP7 protein
expression in a concentration- and time-dependent manner
MRP7-mediated MDR can be reversed by either
inhibiting its transport function or decreasing the protein
expression level of MRP7. To further ascertain the mechanism of
reversal by ponatinib, we also treated MRP7-overexpressing cells
with ponatinib at 0.5 μM for 0, 2, 4, 6, 8, 12 and 24 h or at 0,
0.1, 0.25 and 0.5 μM for 24 h to test the expression of MRP7.
Treatment of HEK/MRP7 cells with ponatinib at 0, 0.1, 0.25 and 0.5
μM for 24 h led to downregulation of MRP7 expression in a
concentration-dependent manner (Fig.
5A). The results shown in Fig.
5B indicated that ponatinib treatment for more than 16 h
significantly downregulated MRP7 protein expression in a
time-dependent manner.
Protein downregulation could occur either at the
transcriptional or the post-transcriptional level. RT-PCR was
conducted to ascertain whether the mRNA level of MRP7 was also
downregulated in HEK/MRP7 cells treated with ponatinib at 0.5 μM.
As shown in Fig. 5C, mRNA levels of
MRP7 did not decrease significantly in the presence of ponatinib
even after 24 h. These results indicated that ponatinib
downregulated MRP7 expression at the post-transcriptional
level.
Discussion
Increased expression and functionality of ABC
transporters are common features of cancer cells and often underlie
chemoresistance (28–30). Thus, the persistence of cancer cells
expressing abnormally high levels of ABC transporters is associated
with dismal prognosis (31,32). ABC pumps constitute an attractive
therapeutic target, as revealed by multiple preclinical and
clinical studies showing that inhibitors of drug efflux sensitize
cancer cells to chemotherapy (31,33).
Nevertheless, the clinical use of ABC transporter inhibitors has
been limited by unacceptable side-effects, drug interactions or
concerns about long-term safety (34–36).
The attractive feature of the novel BCR-ABL inhibitor ponatinib is
that its toxicities were generally mild. The common side-effects
were rashes, dry skin, abdominal pain, headache and constipation,
all seen in ~40% of patients, and most cases were reported to be
mild (37). Ponatinib has been
found to increase the cytotoxicity of substrates of BCRP and P-gp
in BCRP- and P-gp overexpressing cells (21).
In the present study, we found that ponatinib was a
potent reversal agent for MRP7-mediated MDR. It could increase the
cytotoxic response of MRP7-expressing cells to chemotherapeutic
agents, including paclitaxel, docetaxel, vincristine and
vinblastine, at a clinically achievable concentration. Consistent
with the cytotoxicity result, drug accumulation studies also showed
that ponatinib significantly increased the intracellular
accumulation of [3H]-paclitaxel in MRP7-overexpressing
cells in a concentration-dependent manner. Efflux data indicated
that this increased intracellular accumulation of
[3H]-paclitaxel in a short time period (4 h) was caused
by direct inhibition of MRP7 transport function, as MRP7 protein
downregulation caused by ponatinib only occurred after 16-h
incubation. These findings indicate that the potent BCR-ABL
inhibitor ponatinib could reverse MRP7-mediated MDR by inhibiting
the function of MRP7 in a short time period and also by
downregulating protein expression after longer incubation.
The molecular mechanisms underlying the protein
regulation of MRP7 still need further study. Our RT-PCR data showed
that MRP7 mRNA level did not decrease in conjunction with
protein downregulation, which indicates that protein regulation
occurred at a post-transtriptional level. Protein degradation and
translocation may be considered and confirmed by further studies.
Niu et al (38) reported
that low molecular weight heparin (LMWH) prevented chemoresistance
of lung SP cells by reducing BCRP protein expression through the
ubiquitin-proteasome pathway, as the proteasomal inhibitor MG132
restored ABCG2 protein expression, while the lysosomal inhibitors
leupeptin and pepstatin A did not. In addition, it was also shown
that significant enhancement of the intracellular accumulation of
calcein was able to induce downregulation of P-gp (39). PI3k/Akt and NF-κB are the common
pathways involved in the regulation of ABC transporter expression.
PAPP-A decreased the expression of ABCA1 and ABCG1 in THP-1
macrophage-derived foam cells through the PI3K/Akt signaling
pathway (40). Bortezomib reversed
leukemia cell MDR in a concentration-dependent manner as the result
of reduction of P-gp expression through the NF-κB pathway (41).
MRP7 expression is increased in many cancers in
association with stage and prognosis. MRP7 expression level was
upregulated in non-small cell lung cancer (NSCLC) compared to
normal lung tissues, and the higher expression was correlated with
advanced pathological grades and TNM stage in adenocarcinoma
(42). Oguri et al (43) found that MRP7 can affect in
vivo tissue sensitivity to taxanes, and could be used as a
predictive marker of resistance to paclitaxel in NSCLC. A similar
phenomenon was also observed in hepatocellular carcinoma; the MRP7
expression level was also elevated compared to normal adjacent
healthy liver samples (44). These
findings indicate that MRP7 expression might be a biomarker or
regulator of treatment response in certain cancers and modulation
of MRP7 expression and function may have clinical value in cancer
treatment.
In conclusion, we showed here for the first time
that ponatinib inhibits MRP7 function and downregulates MRP7
protein expression, hence facilitating the intracellular
accumulation of selected chemotherapeutic agents and increasing
their cytotoxicity. Clearly, additional studies of mechanism and
animal study are needed, and it will be worthwhile to explore
whether ponatinib can increase the cytotoxicity of anticancer
therapeutic agents in vivo. The clinical application of
ponatinib in MDR cancer patients may have great potential.
Acknowledgements
We thank Dr Gary D. Kruh (University of Illinois)
for kindly providing the HEK293 cell line and the MRP7 cDNA,
Kakenshoyaku Co. for providing cepharanthine and Drs Blasé Billack
and Woon-Kai Low (St. John’s University, Queens, New York, NY, USA)
for advice on RT-PCR. The present study was supported by funds from
NIH (No. 1R15CA143701) and St. John’s University Research Seed
Grant (No. 579-1110-7002) to Z.S.C.
References
|
1
|
Goldman JM: Ponatinib for chronic myeloid
leukemia. N Engl J Med. 367:2148–2149. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gromicho M, Dinis J, Magalhaes M, et al:
Development of imatinib and dasatinib resistance: dynamics of
expression of drug transporters ABCB1, ABCC1, ABCG2, MVP, and
SLC22A1. Leuk Lymphoma. 52:1980–1990. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Breccia M and Alimena G: Refining targeted
therapies in chronic myeloid leukemia: development and application
of nilotinib, a step beyond imatinib. Onco Targets Ther. 1:49–58.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shah NP, Nicoll JM, Nagar B, et al:
Multiple BCR-ABL kinase domain mutations confer polyclonal
resistance to the tyrosine kinase inhibitor imatinib (STI571) in
chronic phase and blast crisis chronic myeloid leukemia. Cancer
Cell. 2:117–125. 2002. View Article : Google Scholar
|
|
5
|
Cortes JE, Kantarjian H, Shah NP, et al:
Ponatinib in refractory Philadelphia chromosome-positive leukemias.
N Engl J Med. 367:2075–2088. 2012. View Article : Google Scholar
|
|
6
|
Jones PM and George AM: The ABC
transporter structure and mechanism: perspectives on recent
research. Cell Mol Life Sci. 61:682–699. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Davidson AL, Dassa E, Orelle C and Chen J:
Structure, function, and evolution of bacterial ATP-binding
cassette systems. Microbiol Mol Biol Rev. 72:317–364. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun YL, Patel A, Kumar P and Chen ZS: Role
of ABC transporters in cancer chemotherapy. Chin J Cancer.
31:51–57. 2012. View Article : Google Scholar
|
|
9
|
Szakacs G, Varadi A, Ozvegy-Laczka C and
Sarkadi B: The role of ABC transporters in drug absorption,
distribution, metabolism, excretion and toxicity (ADME-Tox). Drug
Discov Today. 13:379–393. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brozik A, Hegedus C, Erdei Z, et al:
Tyrosine kinase inhibitors as modulators of ATP binding cassette
multidrug transporters: substrates, chemosensitizers or inducers of
acquired multidrug resistance? Expert Opin Drug Metab Toxicol.
7:623–642. 2011. View Article : Google Scholar
|
|
11
|
Hegedus T, Orfi L, Seprodi A, Varadi A,
Sarkadi B and Keri G: Interaction of tyrosine kinase inhibitors
with the human multidrug transporter proteins, MDR1 and MRP1.
Biochim Biophys Acta. 1587:318–325. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ozvegy-Laczka C, Hegedus T, Varady G, et
al: High-affinity interaction of tyrosine kinase inhibitors with
the ABCG2 multidrug transporter. Mol Pharmacol. 65:1485–1495. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hegedus C, Ozvegy-Laczka C, Apati A, et
al: Interaction of nilotinib, dasatinib and bosutinib with ABCB1
and ABCG2: implications for altered anti-cancer effects and
pharmacological properties. Br J Pharmacol. 158:1153–1164. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Burger H, van Tol H, Boersma AW, et al:
Imatinib mesylate (STI571) is a substrate for the breast cancer
resistance protein (BCRP)/ABCG2 drug pump. Blood. 104:2940–2942.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brendel C, Scharenberg C, Dohse M, et al:
Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity
interaction with ABCG2 on primitive hematopoietic stem cells.
Leukemia. 21:1267–1275. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shen T, Kuang YH, Ashby CR, et al:
Imatinib and nilotinib reverse multidrug resistance in cancer cells
by inhibiting the efflux activity of the MRP7 (ABCC10). PLoS One.
4:e75202009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tiwari AK, Sodani K, Wang SR, et al:
Nilotinib (AMN107, Tasigna) reverses multidrug resistance by
inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR
transporters. Biochem Pharmacol. 78:153–161. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tiwari AK, Sodani K, Dai CL, et al:
Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-,
ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer
Lett. 328:307–317. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dai CL, Tiwari AK, Wu CP, et al: Lapatinib
(Tykerb, GW572016) reverses multidrug resistance in cancer cells by
inhibiting the activity of ATP-binding cassette subfamily B member
1 and G member 2. Cancer Res. 68:7905–7914. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kuang YH, Shen T, Chen X, et al: Lapatinib
and erlotinib are potent reversal agents for MRP7 (ABCC10)-mediated
multidrug resistance. Biochem Pharmacol. 79:154–161. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sen R, Natarajan K, Bhullar J, et al: The
novel BCR-ABL and FLT3 inhibitor ponatinib is a potent inhibitor of
the MDR-associated ATP-binding cassette transporter ABCG2. Mol
Cancer Ther. 11:2033–2044. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Malofeeva EV, Domanitskaya N, Gudima M and
Hopper-Borge EA: Modulation of the ATPase and transport activities
of broad-acting multidrug resistance factor ABCC10 (MRP7). Cancer
Res. 72:6457–6467. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hopper-Borge EA, Churchill T, Paulose C,
et al: Contribution of Abcc10 (Mrp7) to in vivo paclitaxel
resistance as assessed in Abcc10−/− mice. Cancer Res.
71:3649–3657. 2011.PubMed/NCBI
|
|
24
|
Chen ZS, Hopper-Borge E, Belinsky MG,
Shchaveleva I, Kotova E and Kruh GD: Characterization of the
transport properties of human multidrug resistance protein 7 (MRP7,
ABCC10). Mol Pharmacol. 63:351–358. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Carmichael J, DeGraff WG, Gazdar AF, Minna
JD and Mitchell JB: Evaluation of a tetrazolium-based semiautomated
colorimetric assay: assessment of chemosensitivity testing. Cancer
Res. 47:936–942. 1987.PubMed/NCBI
|
|
26
|
Zhou Y, Hopper-Borge E, Shen T, et al:
Cepharanthine is a potent reversal agent for MRP7(ABCC10)-mediated
multidrug resistance. Biochem Pharmacol. 77:993–1001. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shi Z, Liang YJ, Chen ZS, et al: Reversal
of MDR1/P-glycoprotein-mediated multidrug resistance by
vector-based RNA interference in vitro and in vivo. Cancer Biol
Ther. 5:39–47. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
van den Heuvel-Eibrink MM, van der Holt B,
Burnett AK, et al: CD34-related coexpression of MDR1 and
BCRP indicates a clinically resistant phenotype in patients
with acute myeloid leukemia (AML) of older age. Ann Hematol.
86:329–337. 2007.PubMed/NCBI
|
|
29
|
Wulf GG, Wang RY, Kuehnle I, et al: A
leukemic stem cell with intrinsic drug efflux capacity in acute
myeloid leukemia. Blood. 98:1166–1173. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lainey E, Sebert M, Thepot S, et al:
Erlotinib antagonizes ABC transporters in acute myeloid leukemia.
Cell Cycle. 11:4079–4092. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Steinbach D and Legrand O: ABC
transporters and drug resistance in leukemia: was P-gp nothing but
the first head of the Hydra? Leukemia. 21:1172–1176. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schaich M, Soucek S, Thiede C, Ehninger G
and Illmer T: MDR1 and MRP1 gene expression are
independent predictors for treatment outcome in adult acute myeloid
leukaemia. Br J Haematol. 128:324–332. 2005. View Article : Google Scholar
|
|
33
|
List AF, Kopecky KJ, Willman CL, et al:
Benefit of cyclosporine modulation of drug resistance in patients
with poor-risk acute myeloid leukemia: a Southwest Oncology Group
study. Blood. 98:3212–3220. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Coley HM: Overcoming multidrug resistance
in cancer: clinical studies of P-glycoprotein inhibitors. Methods
Mol Biol. 596:341–358. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
van der Holt B, Lowenberg B, Burnett AK,
et al: The value of the MDR1 reversal agent PSC-833 in addition to
daunorubicin and cytarabine in the treatment of elderly patients
with previously untreated acute myeloid leukemia (AML), in relation
to MDR1 status at diagnosis. Blood. 106:2646–2654. 2005.PubMed/NCBI
|
|
36
|
Tang R, Faussat AM, Perrot JY, et al:
Zosuquidar restores drug sensitivity in P-glycoprotein expressing
acute myeloid leukemia (AML). BMC Cancer. 8:512008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cortes JE, Pinilla-Ibarz J, le Coutre P,
Paquette R, Chuah C, Nicolini FE, Apperley J and Khoury HJ: A
pivotal phase 2 trial of ponatinib in patients with chronic myeloid
leukemia (CML) and Philadelphia chromosome-positive acute
lymphoblastic leukemia (Ph+ALL) resistant or intolerant to
dasatinib or nilotinib, or with the T315I BCR-ABL mutation:
12-Month Follow-up of the PACE Trial J, 2012. https://ash.confex.com/ash/2012/webprogram/Paper48561.html.
|
|
38
|
Niu Q, Wang W, Li Y, et al: Low molecular
weight heparin ablates lung cancer cisplatin-resistance by inducing
proteasome-mediated ABCG2 protein degradation. PLoS One.
7:e410352012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Han HK and Van Anh LT: Modulation of
P-glycoprotein expression by honokiol, magnolol and
4-O-methylhonokiol, the bioactive components of Magnolia
officinalis. Anticancer Res. 32:4445–4452. 2012.PubMed/NCBI
|
|
40
|
Tang SL, Chen WJ, Yin K, et al: PAPP-A
negatively regulates ABCA1, ABCG1 and SR-B1 expression by
inhibiting LXRα through the IGF-I-mediated signaling pathway.
Atherosclerosis. 222:344–354. 2012.PubMed/NCBI
|
|
41
|
Wang H, Wang X, Li Y, et al: The
proteasome inhibitor bortezomib reverses P-glycoprotein-mediated
leukemia multi-drug resistance through the NF-κB pathway.
Pharmazie. 67:187–192. 2012.PubMed/NCBI
|
|
42
|
Wang P, Zhang Z, Gao K, et al: Expression
and clinical significance of ABCC10 in the patients with non-small
cell lung cancer. Zhongguo Fei Ai Za Zhi. 12:875–878. 2009.(In
Chinese).
|
|
43
|
Oguri T, Ozasa H, Uemura T, et al:
MRP7/ABCC10 expression is a predictive biomarker for the resistance
to paclitaxel in non-small cell lung cancer. Mol Cancer Ther.
7:1150–1155. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Borel F, Han R, Visser A, et al: Adenosine
triphosphate-binding cassette transporter genes up-regulation in
untreated hepatocellular carcinoma is mediated by cellular
microRNAs. Hepatology. 55:821–832. 2012. View Article : Google Scholar
|