Introduction
Warthin tumors (WTs) are the second most common
benign salivary gland tumors and account for ~15% of all epithelial
parotid gland tumors. They are well encapsulated lesions with
cystic and solid areas consisting of an oncocytic epithelial cell
and a variable stroma component with lymphoid tissue. Malignant
alteration is very rare and surgical resection is the most common
treatment modality (1–3).
To date, tumorigenesis of WTs is unknown. Two
different theories for WT development are currently being
discussed. One is the hypothesis of heterotopia, which implies that
the tumor results from proliferating ductal cells of the salivary
gland that were entrapped in parotid lymph nodes during embryonal
life. The second theory suggests that a WT initially develops in
the parotid lymph nodes as an adenomatous epithelial proliferation
in response to a yet unidentified stimulus, such as tobacco, with
concomitant lymphocytic infiltration (1). The putative link between tobacco
consumption as one of the aetiologic factors associated with the
development of WTs is in line with the observation that the
incidence of WTs in smokers is eight times higher than that in
non-smokers (4).
To date, only limited cytogenetic information is
available for WTs. The majority of tumors analyzed to date by
conventional cytogenetics have an apparently normal karyotype. Only
10% of WTs exhibit genetic alterations (5–13).
Based on the detected genetic alterations, three main stemline
groups are proposed for WTs: i) normal karyotype, ii) numerical
changes only, i.e. loss of Y chromosome or trisomy/monosomy 5, and
iii) reciprocal translocations such as the t(11;19) translocation
resulting in the MAML2/CRTC1 fusion gene (11). This translocation is characteristic
for mucoepidermoid carcinoma (MEC) while a possible derivation of
certain MECs from WTs is under discussion (11–15).
Identification of 6p translocations suggests that the short arm of
chromosome 6 contains a region involved in the origin of WTs
(6,8). Identification of clonal alterations in
almost half of the cases further supports that this lesion is a
‘true’ neoplasm rather than an autoimmune or hypersensitivity
related tumor-like proliferation, as previously suggested (16).
To the best of our knowledge, only one molecular
cytogenetic study using comparative genomic hybridization (CGH)
analysis on WTs has been published to date. The authors of this
previous study observed several chromosomal gains and losses in a
cohort of 15 tumors. Frequent chromosomal losses were reported at
12q (47%), 17p (53%) and 22q (73%), whereas frequent chromosomal
gains were located at 4q (60%), 6q (33%) and 13q (67%) (17). This high frequency of detected
alterations by molecular cytogenetic analysis compared to
conventional cytogenetics underlines the need and importance for
investigating native tumor material other than cell culture
preparations to detect chromosomal alterations.
Herein, we present a second molecular cytogenetic
study of WTs with the largest cohort of 30 tumors to date in order
to confirm previous reported alterations and to detect new
significant chromosomal aberrations because of the higher number of
tumor samples.
Materials and methods
Patient characteristics
A total of 30 patients (14 female and 16 male), with
a median age of 57 years (19–87 years), who received a
parotidectomy with the purpose to remove a parotid gland mass
between the period 2000 and 2006 at the Department of
Otolaryngology, University Hospital Homburg/Saar, Germany was
investigated. All surgeries were primary surgeries treating no
recurrent disease. Prior to surgery, all patients were informed in
regards to the protocol and provided their written consent for
donating tissue for research purposes. Tissue was snap frozen
immediately after removal at −80°C for CGH analysis. The diagnosis
of benign WT was confirmed by detailed histopathological evaluation
in all tumors. To date, none of the patients has developed a tumor
recurrence.
CGH
We obtained DNA using phenol/chloroform extraction.
Tumor DNA and reference DNA were labeled with biotin and
digoxigenin by nick translation according to the manufacturer’s
protocol (Roche Diagnostics, Mannheim, Germany). Tumor DNA and
reference DNA, 600 ng each, were hybridized with Cot1-DNA (Roche
Diagnostics) to normal chromosome metaphase spreads from peripheral
blood lymphocytes prepared following standard procedures. After
three days of hybridization at 37°C, post-hybridization washes were
performed at a stringency of 50% formamide/2× standard saline
citrate (SSC), 2× SSC and 0.1× SSC at 45°C. Tumor DNA was
visualized with fluorescein-isothiocyanate (Vector Laboratories,
Inc., Burlingame, CA, USA), reference DNA with rhodamine (Roche
Diagnostics) and counterstained with an anti-fade solution
containing DAPI (4,6-diamidino-2-phenylindole) (Vector
Laboratories). Fluorescence images were captured using an Olympus
BX 61 fluorescence microscope with a cooled charged-coupled device
camera. Image processing was performed by using the computerized
ISIS digital image analysis software system (MetaSystems,
Altlussheim, Germany). Average ratio profiles were determined from
analysis of 12–15 metaphases. The thresholds used for ratio
profiles were 1.2 for gains, and 0.8 for losses.
Due to the suppression with Cot1-DNA, the
fluorescence intensities were not representative at chromosome
regions with tandem repetitive DNA clusters, that is at the
heterochromatic blocks on chromosomes 1, 9, 16, and Y at the
centromeric regions and along the short arms of acrocentric
chromosomes. These areas and the whole sex chromosomes were
excluded from CGH evaluation. Additionally, the chromosomal
regions, such as 1p34pter, 19 and 22 were interpreted cautiously,
particularly as they are prone to problems associated with the
different hybridizability of their GC-rich regions (18).
Results
CGH
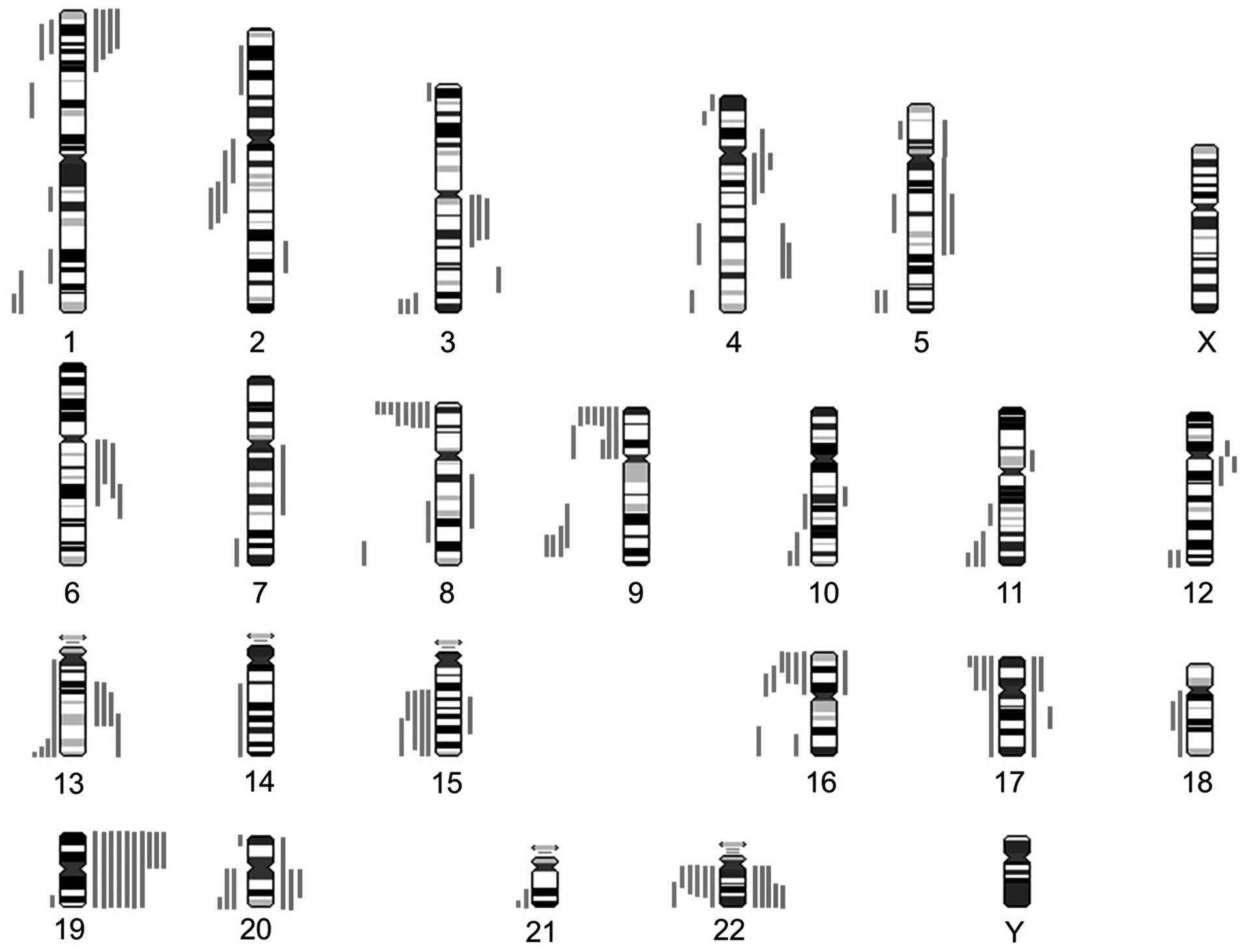
Chromosomal alterations were detected in a
non-random distribution in 27 out of the 30 hybridized WTs by CGH
analysis. The mean number of imbalances was 5.3 (range from 0 to
21). Detection of losses was more frequent than gains (93 vs. 67,
mean 3.1 vs. 2.3). The graphical representation of the genetic
alterations is shown in Fig. 1. The
clinical data and chromosomal imbalances are summarized in Table I.
 | Table IResults obtained from CGH analysis of
30 Warthin tumors. |
Table I
Results obtained from CGH analysis of
30 Warthin tumors.
| Tumor | Age/Gender | Chromosomal
gains | Chromosomal
losses | No. of gains | No. of losses | Total no. of
imbalances |
|---|
| 1 | 75/F | 3q11q21, 4q28q31,
6q11q22.2, 12q11q13 | 22q11q12 | 4 | 1 | 5 |
| 2 | 56/F | 3q11q13.3, 4q26q31.3,
8q13q22, 13q14q22 | | 4 | 0 | 4 |
| 3 | 52F | - | - | 0 | 0 | 0 |
| 4 | 58/M | 5q14q23, 6q21q22 | - | 2 | 0 | 2 |
| 5 | 50/M | 13q22qter | | 1 | 0 | 1 |
| 6 | 53/M | - | - | 0 | 0 | 0 |
| 7 | 50/M | - | 8q24.1qter | 0 | 1 | 1 |
| 8 | 57/M | - | 5p14 | 0 | 1 | 1 |
| 9 | 87/F |
4q11q13.1,12q11q12 | 1p33p36.1,
16p11.1p12 | 2 | 2 | 4 |
| 19 | 56/M | 19 | - | 2 | 0 | 2 |
| 20 | 39/M | 19p | 2q12q23,
8p23.1pter | 1 | 2 | 3 |
| 25 | 56/M | - | 9p11p21 | 0 | 1 | 1 |
| 26 | 51/M | - | 1p22.1p31.1,
10q24.3qter | 0 | 2 | 2 |
| 27 | 42/M | - | - | 0 | 0 | 0 |
| 28 | 64/F | - | 17p | 0 | 1 | 1 |
| 22 | 71/F | 1p33pter, 17p11pter, 19,
20q11.1q13.1,
22q | 9p22pter, 13q33qter,
20p13pter | 6 | 3 | 9 |
| 23 | 56/F | 1p32.3pter, 19p, 22q12.2qter | 3q28qter, 4q33qter,
5q14q21, 9p11p13,
9p23pter | 3 | 5 | 8 |
| 24 | 66/F | 1p34.1pter, 19,
20 | 1q43qter, 9p | 5 | 2 | 7 |
| 29 | 19/M | 1p35pter, 17p, 19, 22 | 5p11p12 | 5 | 1 | 6 |
| 30 | 57/F | 11p11.1p12, 19,
22 | 2q21.3q24.3,
4q26q31.1 | 4 | 2 | 6 |
| 21 | 61/F | 15q21.2q24, 17, 19,
20q, 22q12.3qter | 2q21.1q24.2,
8p23.1pter, 9p | 6 | 3 | 9 |
| 10 | 61/F | 2q32.1q33,
3q25.3q26.3, 4p11p14, 4q11q21.3 |
15q21.1q22.3, 16p13.1pter,
22q11.1q13.1 | 4 | 3 | 7 |
| 11 | 62/M | 5p11p14, 5q11q23.3,
6q11q22.1 | 3p24.3pter,
5q34qter, 7q33qter, 8p23.1pter, 9q32q34.1,
11q23.1qter, 12q24.2qter, 13q32qter, 15q21.1qter,
16p12pter, 16q23qter, 17, 18q, 19q13.3qter, 20q,
21q22.1qter, 22q12.2qter | 3 | 18 | 21 |
| 12 | 45/F | 3q11.2q13.3,
4q11q22, 6q12q21, 13q14.3q22 | 8p23.1pter,
9q32q34.1, 10q26.1qter, 11q23.3qter, 12q24.2qter, 14q21qter,
15q23qter, 16q22qter, 17p, 20q13.2qter,
22q11.1q13.2 | 4 | 11 | 15 |
| 13 | 64/M | 19p | 1p34.1p36.2,
1q21.2q23, 1q32.1q41, 2p16p23, 8p21.3pter, 8q21.3q23,
9q22.1q33, 10q22.1q24.1, 11q14.1q22.1, 15q21.1q25,
16p11.2p13.1, 18q12.1q21.3, 20q, 22 | 1 | 13 | 14 |
| 18 | 63/M | 12p11p12.1 | 2q11.1q14.3,
3q28qter, 8p21.3pter, 15q21.3qter, 22q11.1q12.3 | | 5 | 6 |
| 14 | 61/F | 7q11.1q31.2,
10q21.2q22.2, 13q14.1q22 | 1q41qter,
8p22pter, 16p | 3 | 3 | 6 |
| 15 | 80/F | 17q21.2q22, 19 | 5q34qter, 9p23pter,
13q34qter | 3 | 3 | 6 |
| 16 | 57/M | - | 3q27qter, 4p16pter,
8p22pter, 9q31q34.1, 16p12pter, 17p13pter,
21q22.3qter | 0 | 7 | 7 |
| 17 | 46/M | 19 | 8p23.1pter,
9p23pter, 11q24qter | 2 | 3 | 5 |
The most commonly observed alterations were
deletions of the short arm of chromosome 8 (30%), followed by
deletions of 9p (23%). Further representative changes were
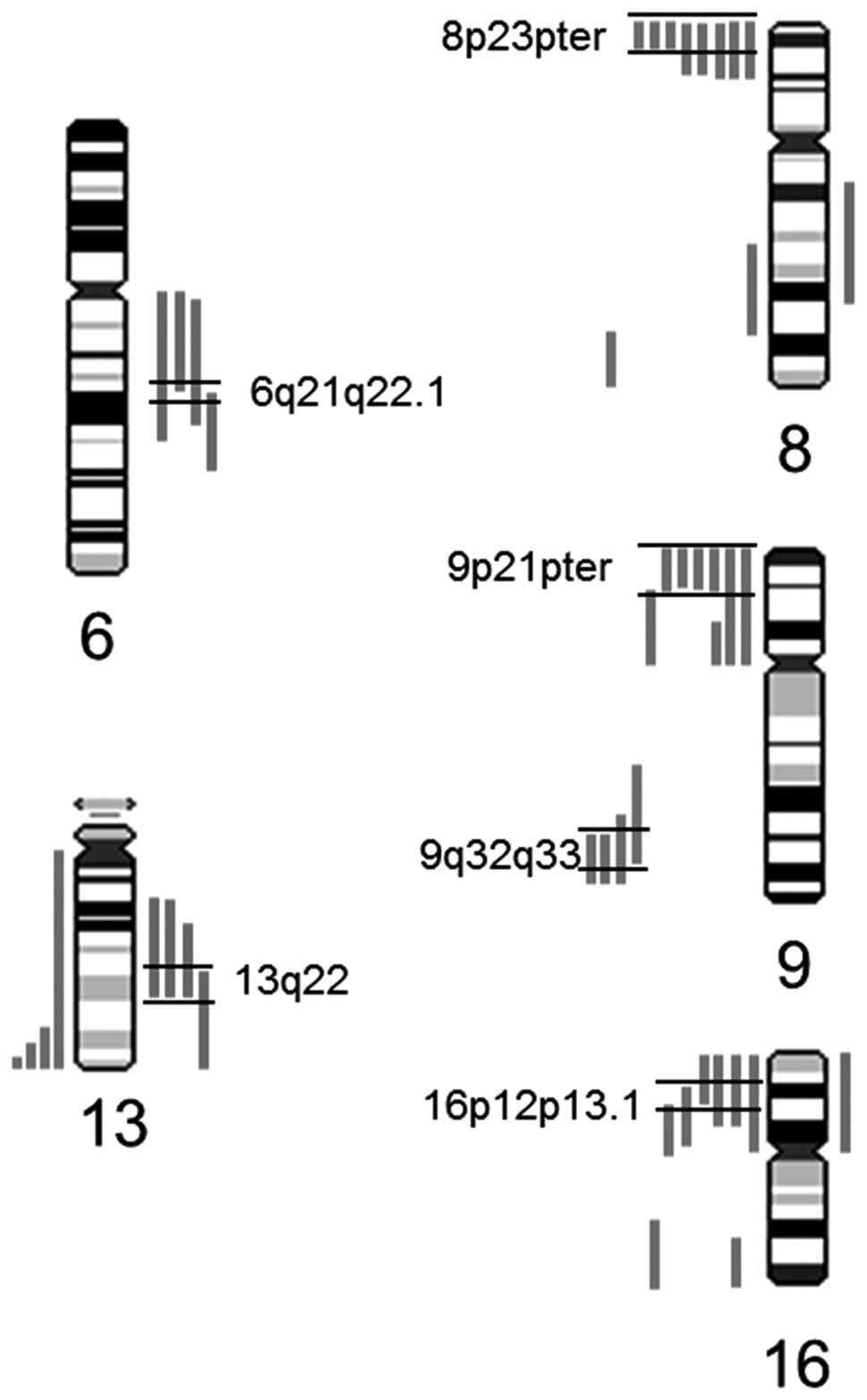
deletions on chromosome arm 16p and 22q with the minimal
overlapping region at 16p12p13.1 and 22q12.1q12.3 in 20% of the
investigated tumors (Fig. 2).
Moreover, in 17% of the cases, gains at 4q, 15q and 22q were
detected. At a lower frequency, deletions on 2q, 9q and 17p and
gains on 6q and 13q were observed (13% each), with the minimal
overlapping regions at 13q22 and 6q21q22.1. An overview of the most
commonly observed alterations and putative candidate genes is shown
in Table II.
| Table IIChromosomal alterations and putative
target genes observed by CGH. |
Table II
Chromosomal alterations and putative
target genes observed by CGH.
| Chromosome | No. of cases/30
(%) | Candidate
genes |
|---|
| Deletions |
| 2q | 4 (13) | ? |
| 8p23.1pter | 9 (30) | ANGPT2,
MCPH1, PINXI |
| 9p | 7 (23) | CDKN2A,
CDKN2B |
| 9q | 4 (13) | TSC1,
GAS1 |
| 11q24qter | 3 (10) | TBRG1,
CHEK1 |
| 15q | 5 (17) | ? |
| 16p | 6 (20) | MAPK3,
ERCC4, LITAF |
| 17p | 4 (13) | TP53 |
| 22q12.1q12.3 | 6 (20) | CHEK2,
TIMP3 |
| Gains |
| 4q | 5 (17) | EGF,
C-Kit |
| 6q21 | 4 (13) | FYN,
HDAC2 |
| 13q22 | 4 (13) | KLF12 |
| 22q | 5 (17) | PDGFB |
Our data indicate that almost all tumors with a
higher number of copy number alterations (6 or more) are able to be
distinguished by two different patterns of alterations. The first
one is characterized by deletions affecting 8p, 9q, 11q 15q, 16p
and 22, whereas in the second one gains of 22 were partly apparent
with gains on 1p and 20q and losses on 9p (Table I).
Discussion
Herein, we present our CGH study on 30 WTs that
displayed numerous chromosomal alterations. In regard to only one
available CGH study of 15 WTs, previous reported chromosomal
aberrations but also novel regions of interest in this tumor have
been observed.
Giefing et al (17) revealed in his subset of 15 WTs, that
losses of chromosome 22, 17p and 12q (73, 53 and 47%, respectively)
were the most consistent alterations. Chromosomal gains were
observed most frequently on 13q, 4q, 6q and 2q (67, 60, 33 and 27%,
respectively). The present study confirms these commonly observed
losses and gains; however, at an approximately one-third lower
incidence of the affected tumors when compared to Giefing et
al (17).
Deletions of the terminal region of 9q were observed
in 13% of our tumors and were also noted by Giefing et al
(17) in their smaller cohort at a
percentage of 13% in their tumors. In this region, GAS1 on
9q21.33 and TSC1 on 9q34.13 are located. The
tumor-suppressor gene GAS1 has previously been shown to play
a role in other entities such as myeloid malignancies (19). Loss of TSC genes has been
reported to result in tumor development by its constitutive
activation of MTOR and downstream signaling elements (20).
In accordance to Giefing et al (17), our study revealed additional
deletions affecting chromosomes 16, 17 and 22 with the minimal
overlapping region on 16p12p13.1, 17p13 and 22q12. Close to the
minimal overlapping region on 16p12p13.1 (Fig. 2), several significant candidate
genes are located which are known to be involved in cell cycle
regulation and apoptosis, for example MAPK3, LITAF
and ERCC4. Furthermore, for other tumors such as breast
carcinomas, colorectal tumors and anaplastic thyroid cancers, the
important role of deletions of the short arm of chromosome 16 has
been previously demonstrated (21–23).
Notably, Giefing et al (17) as well as our study revealed gains on
6q as a frequent event in WTs. In the delineated consensus region
on 6q21, which was affected in 33% of our tumors, the FYN
oncogene is located. FYN belongs to the Src kinases, which
are key upstream mediators of both the PI3-K and MAPK signaling
pathways, and have been shown to play important roles in cell
proliferation, migration and survival (24). In contrast, cytogenetic examination
of the cultured tumor cells showed normal karyotypes in the
majority of WTs. Nevertheless, one study also revealed non-clonal
deletions of chromosome 6 as well as a deletion of 6q21 in 3 of 13
cultured tumors (10). Notable, in
malignant salivary gland tumors, i.e. adenoid cystic carcinomas and
mucoepidermoid carcinomas, deletions or rare translocations
involving the terminal region of the long arm of chromosome 6, were
found to be the most consistent alterations (25–28).
In addition, certain types of solid tumors, and several types of
leukemias and lymphomas are characterized by various deletions of
6q (29).
As novel findings, the present study detected
frequent alterations on 8p (30%) and 9p (23%) in this tumor entity.
Alterations on 8p may indicate genes involved in DNA damage
response and tumorigenesis such as tumor-suppressor genes
MCPH1, ANGPT2 and PINX1 at 8p23.1pter.
CDKN2A and CDKN2B, located at 9p21, are also
important candidate genes known to be involved in cell cycle
regulation (Fig. 2). As these
observations were not detected in previous conventional karyotyping
analyses (6,8,10) or
comparative genomic hybridization analysis (17), they warrant particular interest in
further studies.
In addition to the deletions on 22q12, gains of
chromosome 22 were also a frequent event in our WT cohort. The
potential candidate tumor-suppressor genes on 22q12 CHEK2
and TIMP3 were previously shown to be associated with
increased risk of prostate cancer (30) and pancreatic endocrine tumors
(31). Moreover, the growth factor
PDGFB was found to play an essential role in the regulation of cell
proliferation, cell migration and survival (32). Due to the identification of specific
gains at 22q12.3qter, further analysis of PDGFB in WTs is
warranted.
Furthermore, conventional karyotyping of WTs
revealed a translocation t(11;19)(q21;p13.1) (5,8,9,11).
This translocation results in the fusion gene MAML2/CRTC1,
which is common in most cases of mucoepidermoid carcinoma (MEC) and
possibly indicates the derivation of certain MECs from WTs
(11,15). Since balanced translocations are not
detectable with CGH, no conclusions are possible concerning
presentation of the t(11;19) translocation in our cohort.
Notably, a study analyzing a cohort of 15 primary
MECs by CGH analysis revealed losses on 15q in 4 of their tumors,
partly together with deletions on 8p and 22 (33). Furthermore, a microarray analysis of
a salivary duct carcinoma arising in WT also revealed losses of 8,
15 and 22 in addition to other alterations (34). We also observed this pattern of
alterations in our WTs, particularly in those with a high number of
chromosomal alterations possibly indicating chromosomal
instability. Notably, in a variety of other tumor entities such as
MEC, head and neck squamous cell carcinoma and prostate cancer,
defined copy number alterations, e.g. loss of 8p, as well as the
total number of imbalances are of general importance in the
progression and also the metastatic potential of these tumors
(28,35). Collectively, all these findings
support a potential role of this pattern of alterations for a more
pronounced tumor type for WT. As none of the patients in this
series presented with a tumor recurrence to date, we cannot address
this issue in the present study. Nevertheless, our findings need to
be interpreted with caution reflecting on the morphological
heterogeneity in WTs. In the selected study design, it was not
considered to which extent lymphoid or epithelial tumor components
were analyzed. Therefore, the identification of different
chromosomal aberrations may reflect the different tumor components.
However, in defining significant candidate genes based on the CGH
findings, one should be aware that chromosomal alterations can
point to important genes in tumorigenesis, but may also be the
result of chromosomal instability.
To the best of our knowledge, the present study
presents the largest cohort of 30 WTs analyzed by CGH to date. Our
molecular cytogenetic analysis confirmed the findings of previous
cytogenetic studies and identified new recurrent alterations as
well as different patterns of chromosomal aberrations with the
potential for a diagnostic impact. The presented data identified
significant consensus regions that may harbor candidate genes of
importance in the tumor biology of WTs. These warrant further study
to assess their possible involvement in WT tumorigenesis.
References
|
1
|
Ellis GL and Auclair PL: Tumors of the
salivary glands. 3rd edition. Armed Forces Institute of Pathology;
Washington: 1996
|
|
2
|
Teymoortash A: Head and neck: Salivary
gland: Warthin’s Tumors. Atlas Genet Cytogenet Oncol Haematol.
April;2008.http://AtlasGeneticsOncology.org
|
|
3
|
Thangarajah T, Reddy VM,
Castellanos-Arango F and Panarese A: Current controversies in the
management of Warthin tumour. Postgrad Med J. 85:3–8. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yu GY, Liu XB, Li ZL and Peng X: Smoking
and the development of Warthin’s tumour of the parotid gland. Br J
Oral Maxillofac Surg. 36:183–185. 1998.
|
|
5
|
Mark J, Dahlenfors T, Stenman G and
Nordquist A: A human adenolymphoma showing the chromosomal
aberrations del(7)(p12;p14–15) and t(11;19)(q21;p12–13). Anticancer
Res. 9:1565–1566. 1989.PubMed/NCBI
|
|
6
|
Mark J, Dahlenfors R, Stenman G and
Nordquist A: Chromosomal patterns in Warthin’s tumor. A second type
of human benign salivary gland neoplasm. Cancer Genet Cytogenet.
46:35–39. 1990.
|
|
7
|
Mark HF, Hanna I and Gnepp DR: Cytogenetic
analysis of salivary gland type tumors. Oral Surg Oral Med Oral
Pathol Oral Radiol Endod. 82:187–192. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Martins C, Fonseca I, Roque L and Soares
J: Cytogenetic characterisation of Warthin’s tumour. Oral Oncol.
33:344–347. 1997.
|
|
9
|
Bullerdiek J, Haubrich J, Meyer K and
Bartnitzke S: Translocation t(11;19)(q21;p13.1) as the sole
chromosome abnormality in a cystadenolymphoma (Warthin’s tumor) of
the parotid gland. Cancer Genet Cytogenet. 35:129–132. 1988.
|
|
10
|
Nordkvist A, Mark J, Dahlenfors R, Bende M
and Stenman G: Cytogenetic observations in 13 cystadenolymphomas
(Warthin’s tumours). Cancer Genet Cytogenet. 76:129–135. 1994.
|
|
11
|
Enlund F, Behboudi A, Andrén Y, Oberg C,
Lendahl U, Mark J and Stenman G: Altered Notch signaling resulting
from expression of a WAMTP1-MAML2 gene fusion in
mucoepidermoid carcinomas and benign Warthin’s tumors. Exp Cell
Res. 292:21–28. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tirado Y, Williams MD, Hanna EY, Kaye FJ,
Batsakis JG and El-Naggar AK: CRTC1/MAML2 fusion transcript
in high grade mucoepidermoid carcinomas of salivary and thyroid
glands and Warthin’s tumors: implications for histogenesis and
biologic behavior. Genes Chromosomes Cancer. 46:708–715. 2007.
View Article : Google Scholar
|
|
13
|
Fehr A, Roser K, Belge G, Loening T and
Bullerdiek J: A closer look at Warthin tumors with respect to the
t(11;19). Cancer Genet Cytogenet. 180:135–139. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tonon G, Modi S, Wu L, Kubo A, Coxon AB,
Komiya T, O’Neil K, Stover K, El-Naggar A, Griffin JD, Kirsch IR
and Kaye FJ: t(11;19)(q21;p13) translocation in mucoepidermoid
carcinoma creates a novel fusion product that disrupts a Notch
signaling pathway. Nat Genet. 33:208–213. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bell D, Luna MA, Weber RS, Kaye FJ and
El-Naggar AK: CRTC1/MAML2 fusion transcript in Warthin’s tumor and
mucoepidermoid carcinoma: evidence for a common genetic
association. Genes Chromosomes Cancer. 47:309–314. 2008.
|
|
16
|
Ogawa Y, Hong S, Toyosawa S, Chang CK and
Yagi T: Expression of major histocompatibility complex class II
antigens and interleukin-I by epithelial cells of Warthin’s tumor.
Cancer. 66:2111–2117. 1990.PubMed/NCBI
|
|
17
|
Giefing M, Wierzbicka M, Rydzanicz M,
Cegla R, Kujawski M and Szyfter K: Chromosomal gains and losses
indicate oncogene and tumor suppressor gene candidates in salivary
gland tumors. Neoplasma. 55:55–60. 2008.PubMed/NCBI
|
|
18
|
Karhu R, Kähkönen M, Kuukasjärvi T,
Pennanen S, Tirkkonen M and Kallioniemi O: Quality control of CGH:
impact of metaphase chromosomes and the dynamic range of
hybridization. Cytometry. 28:198–205. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Evdokiou A, Webb GC, Peters GB, Dobrovic
A, O’Keefe DS, Forbes IJ and Cowled PA: Localization of the human
growth arrest-specific gene (GAS1) to chromosome bands
9q21.3–q22, a region frequently deleted in myeloid malignancies.
Genomics. 18:731–733. 1993.PubMed/NCBI
|
|
20
|
Orlova KA and Crino PB: The tuberous
sclerosis complex. Ann NY Acad Sci. 1184:87–105. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lininger RA, Park WS, Man YG, Pham T,
MacGrogan G, Zhuang Z and Tavassoli FA: LOH at 16p13 is a novel
chromosomal alteration detected in benign and malignant
microdissected papillary neoplasms of the breast. Hum Pathol.
29:1113–1118. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kadota M, Tamaki Y, Sakita I, Komoike Y,
Miyazaki M, Ooka M, Masuda N, Fujiwara Y, Ohnishi T, Tomita N,
Sekimoto M, Ohue M, Ikeda T, Kobayashi T, Horii A and Monden M:
Identification of a 7-cM region of frequent allelic loss on
chromosome band 16p13.3 that is specifically associated with
anaplastic thyroid carcinoma. Oncol Rep. 7:529–533. 2000.PubMed/NCBI
|
|
23
|
Alcock HE, Stephenson TJ, Royds JA and
Hammond DW: Analysis of colorectal tumor progression by
microdissection and comparative genomic hybridization. Genes
Chromosomes Cancer. 37:369–380. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Summy JM and Gallick GE: Src family
kinases in tumor progression and metastasis. Cancer Metastasis Rev.
22:337–358. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sandros J, Stenman G and Mark J:
Cytogenetic and molecular observations in human and experimental
salivary gland tumors. Cancer Genet Cytogenet. 44:153–167. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jin Y, Mertens F, Limon J, Mandahl N,
Wennerberg J, Dictor M, Heim S and Mitelman F: Characteristic
karyotypic features in lacrimal and salivary gland carcinomas. Br J
Cancer. 7:42–47. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Persson M, Andrén Y, Moskaluk CA, Frierson
HF Jr, Cooke SL, Futreal PA, Kling T, Nelander S, Nordkvist A,
Persson F and Stenman G: Clinically significant copy number
alterations and complex rearrangements of MYB and NFIB in head and
neck adenoid cystic carcinoma. Genes Chromosomes Cancer.
51:805–817. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jee KJ, Persson M, Heikinheimo K,
Passador-Santos F, Aro K, Knuutila S, Odell EW, Mäkitie A, Sundelin
K, Stenman G and Leivo I: Genomic profiles and CRTC1-MAML2 fusion
distinguish different subtypes of mucoepidermoid carcinoma. Mod
Pathol. 26:213–222. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mitelman F, Johansson B and Mertens F:
Mitelman Database of Chromosome Aberrations and Gene Fusions in
Cancer. http://cgap.nci.nih.gov/Chromosomes/Mitelman.
2012
|
|
30
|
Cybulski C, Wokołorczyk D, Huzarski T,
Byrski T, Gronwald J, Górski B, Debniak T, Masojć B, Jakubowska A,
Gliniewicz B, Sikorski A, Stawicka M, Godlewski D, Kwias Z, Antczak
A, Krajka K, Lauer W, Sosnowski M, Sikorska-Radek P, Bar K, Klijer
R, Zdrojowy R, Małkiewicz B, Borkowski A, Borkowski T, Szwiec M,
Narod SA and Lubiński J: A large germline deletion in the Chek2
kinase gene is associated with an increased risk of prostate
cancer. J Med Genet. 43:863–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wild A, Ramaswamy A, Langer P, Celik I,
Fendrich V, Chaloupka B, Simon B and Bartsch DK: Frequent
methylation-associated silencing of the tissue inhibitor of
metalloproteinase-3 gene in pancreatic endocrine tumors. J Clin
Endocrinol Metab. 88:1367–1373. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Romashkova JA and Makarov SS: NF-κB is a
target of AKT in anti-apoptotic PDGF signalling. Nature. 401:86–90.
1999.
|
|
33
|
Verdorfer I, Fehr A, Bullerdiek J, Scholz
N, Brunner A, Krugmann J, Hager M, Haufe H, Mikuz G and Scholtz A:
Chromosomal imbalances, 11q21 rearrangement and MECT1-MAML2 fusion
transcript in mucoepidermoid carcinomas of the salivary gland.
Oncol Rep. 22:305–311. 2009.PubMed/NCBI
|
|
34
|
Kim HJ, Yoo YS, Park K, Kwon JE, Kim JY
and Monzon FA: Genomic aberrations in salivary duct carcinoma
arising in Warthin tumor of parotid gland: DNA microarray and HER2
fluorescence in situ hybridization. Arch Pathol Lab Med.
135:1088–1091. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gebhart E: Comparative genomic
hybridization (CGH): ten years of substantial progress in human
solid tumor molecular cytogenetics. Cytogenet Genome Res.
104:352–358. 2004.PubMed/NCBI
|