Introduction
5-Fluorouracil (5-FU) is widely used as an
anticancer agent. It has been commonly used either alone or in
combination with other drugs and/or radiation for the treatment of
colorectal, breast, head and neck and other types of cancers
(1). 5-FU belongs to the class of
antimetabolite chemotherapeutics and is thought to be an inhibitor
of the enzyme thymidylate synthase (TS) which plays a role in
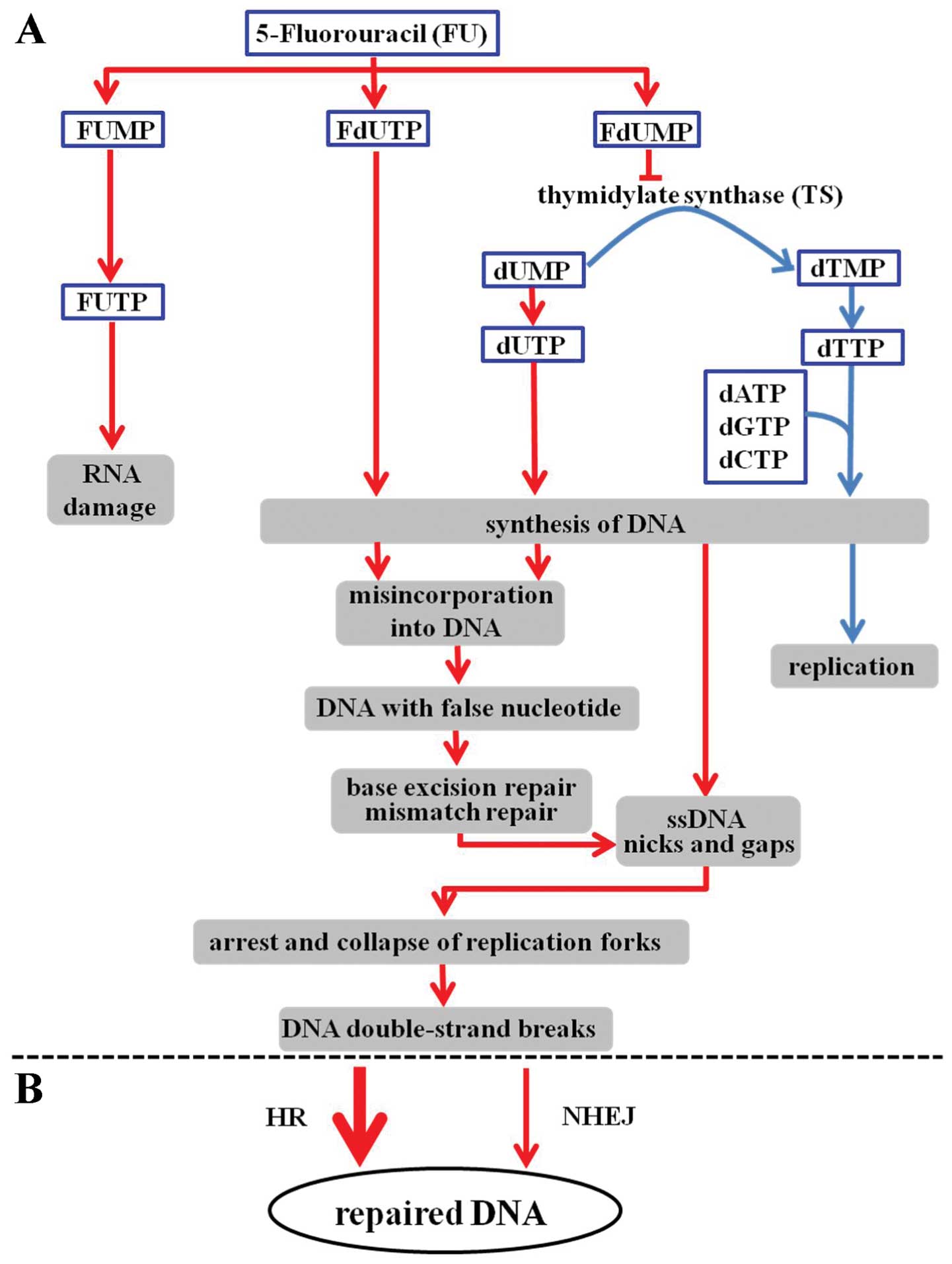
nucleotide synthesis (Fig. 1A)
(2,3). 5-FU is also converted to several
active metabolites (Fig. 1A),
including fluorouridine triphosphate (FUTP), fluorodeoxyuridine
triphosphate (FdUTP) and fluorodeoxyuridine monophosphate (FdUMP).
These active metabolites compromise global RNA metabolism through
the incorporation of FUMP during RNA and DNA metabolism. This
occurs through FdUMP-mediated inhibition of TS and incorporation of
FdUMP into DNA. Inhibition of TS occurs through the formation of a
ternary covalent complex consisting of TS-FdUMP-5,
10-methylenetetrahydrofolate. Once this complex is formed, cells
are unable to synthesize dTMP from dUMP and cellular dUTP levels
increase at the expense of dTTP. The resulting dUTP/dTTP imbalance
causes massive mis-incorporation of dUMP or FdUMP during DNA
replication (4). Although DNA
damage is considered to be one of the main triggers of the tumor
cell killing effect of 5-FU (5,6), it is
not fully understood how mis-incorporated dUMP or FdUMP is
processed and contributes to cytotoxicity. Mis-incorporated FdUMP
or dUMP is recognized and excised from DNA through base excision
repair (BER) or mismatch repair (MMR) (7). The repair of uracil-containing or
5-FU-containing DNA is mediated by the BER enzyme
uracil-DNA-glycosylase (8).
However, this repair mechanism is futile in the presence of high
FdUTP/dTTP ratios, and only results in additional false nucleotide
incorporation. MMR plays an important role in correcting
replication errors. The removal of FdUMP or dUMP by BER and MMR
produces nicks and gaps in single-strand DNA (ssDNA) (5). Recently, these nicks and gaps were
reported to act as triggers for the initial activation of an
ATR-Chk1 signaling pathway. Chk1 molecules are activated and then
stop DNA replication. During these processes, the fork of the
stalled replication complex was coated with replication protein A
(RPA). This event induces unstable conformations in the DNA
structure. Therefore, double-strand breaks (DSBs) are subsequently
induced when too many SSBs are present at stalled replication forks
in 5-FU treated cells (9).
DSBs are the most important DNA lesions which occur
in cells after treatment with chemotherapy and/or radiation. The
repair of these DSBs largely determines the outcome of cancer
therapy. If incorrectly repaired or unrepaired themselves through
the use of siRNA which targets a repair gene, DSBs may lead to cell
death.
The research described here was designed to
ascertain which components in DNA repair pathways significantly
contribute to the repair of DSBs induced by 5-FU (Fig. 1B).
Materials and methods
Cell lines
Chinese hamster lung fibroblast cell lines were used
in the present study: V79 (BRCA2 wild-type and XRCC2
wild-type); V-C8 (BRCA2-deficient), V-C8+BRCA2
(BRCA2 revertant, V-C8 containing a BAC with the murine
BRCA2 gene) (10–12), irs1 (XRCC2-deficient), V79B
(Ku80 wild-type), and XR-V15B (Ku80-deficient).
Chinese hamster ovary cell lines used in the present study were
CHO-K1 (DNA-PKcs wild-type) and XR-C1 (DNA-PKcs
deficient). These cells were kindly provided by Drs M.Z.
Zdzienicka, L.H. Thompson and A. Yasui. The human oral squamous
cell carcinoma cell lines used were SAS and HSC3. Cells were
obtained from the Japanese Collection of Research Bioresources
(Health Science Research Resources Bank, Osaka, Japan). The cells
were cultured at 37°C in a humidified 5% CO2 incubator,
and were grown in Dulbecco’s modified Eagle’s medium containing 10%
(v/v) fetal bovine serum, penicillin (50 U/ml), streptomycin (50
μg/ml) and kanamycin (50 μg/ml).
Drugs and drug treatments
5-FU (Kyowa Hakko, Tokyo, Japan) was dissolved at a
stock concentration of 100 mM in phosphate-buffered saline (PBS).
5-FU stock solutions were stored at −20°C until used. Cells were
treated with medium containing 5-FU at various concentrations for
24 h and then rinsed twice with PBS.
Colony forming assays
Cell survival was measured using a standard colony
forming assay as previously described (13). The sensitivity of each cell line was
assessed by its D50 value, i.e. from the 5-FU dose which
reduced cell survival to 50%. In order to accurately compare
sensitivities to 5-FU in the repair defective cell lines, the
relative D50 values were normalized using the
D50 value of the parental cell lines.
Immunohistochemistry
Cells were grown on glass slides in 100-mm dishes,
fixed in 100% methanol (Nacalai Tesque, Inc., Kyoto, Japan) for 20
min at 4°C. The cells were then permeabilized for 10 sec at 4°C in
100% acetone (Nacalai Tesque) and blocked in PBS with 3% skim milk
(Nacalai Tesque) for 1 h at 37°C. Cells were then incubated with
anti-phospho-H2AX (Ser 139) mouse monoclonal antibody (Millipore,
Billerica, MA, USA) for 1 h at 1:300 dilutions in PBS containing 1%
BSA, and washed three times in PBS containing 1% BSA for 10 min.
The cells were incubated with AlexaFluor 488-conjugated anti-mouse
IgG secondary antibody (Invitrogen, Carlsbad, CA, USA) for 1 h at
room temperature at 1:400 dilutions in PBS containing 1% BSA, and
washed three times for 10 min in PBS. Cover-glasses were mounted at
1:1,000 dilutions of 4,6-diamidino-2-phenylindole. Fluorescent
images were captured for analysis using a fluorescence microscope
(Keyence, Tokyo, Japan).
Flow cytometry
Cells were fixed in cold 70% methanol after a 100 μM
5-FU treatment for 24 h, and maintained at 4°C for up to 1 week
before analysis. The overall levels of phosphorylated H2AX (γH2AX)
were measured with flow cytometry as previously described (14).
Cell cycle analysis
After irradiation, cells were fixed with cold 70%
methanol and stored at 4°C for 3 days before analysis. For cell
cycle analysis, the cells were incubated for 30 min at room
temperature with 1 mg/ml RNase and 50 μg/ml propidium iodoide (PI),
and were analyzed using a flow cytometer. The cell cycle
distribution was assayed by determining the DNA content twice and
calculating the average values.
siRNA transfection
The siRNA sequences used for human BRCA2 and
its non-specific negative control were AACAAC AAUUACGAACCAAACUU and
UAUUCGCGCGUAUAG CGGUUU, respectively (12,15).
The siRNA duplexes were synthesized by Japan Bio Services Co., Ltd.
(Saitama, Japan) and provided as a purified and annealed duplex.
Transfections were carried out using Lipofectamine RNAiMAX in
accordance with the manufacturer’s instructions (Invitrogen).
Briefly, cells were seeded at 5×104 cells per 10-cm
plate for 24 h without antibiotics. The siRNA was diluted in
Opti-MEM I (Invitrogen) to produce a final siRNA concentration of
10 nM in a 1 ml final transfection volume. In a separate tube, 10
μl of Lipofectamine RNAiMAX was added to 490 μl of Opti-MEM I. The
Lipofectamine RNAiMAX dilution was mixed with the diluted siRNA and
incubated at room temperature for 15 min. The complex was then
added drop-wise onto the cells. The cells were incubated for 36 h
before further processing. These cells were then trypsinized for
colony forming assays.
Western blotting
Western blotting was carried out as previously
described in detail (16). The
membranes were then incubated with mouse monoclonal anti-BRCA2
antibody (Ab-4; Calbiochem, Darmstadt, Germany) or goat polyclonal
anti-actin antibody (I-19; Santa Cruz Biotechnology, Santa Cruz,
CA, USA) for the primary antibody for 1 h at room temperature. The
membranes were washed with TPBS buffer three times and incubated
with a secondary antibody conjugated to horseradish peroxidase for
1 h. After washing three times, the blots were visualized by using
an enhanced chemiluminescence method (GE Healthcare UK,
Buckinghamshire, UK) following the manufacturer’s protocol. The
protein in the samples was quantified by scanning profiles using
the ImageJ program (NIH, Bethesda, MD, USA). The relative ratio of
the intensity of the two bands was used to determine BRCA2 protein
expression levels, and these values were the average of three
experiments using densitometry measurements following β-actin
normalization with and without BRCA2-siRNA.
Statistical analysis
Data were compared statistically using the
two-tailed Student’s t-test.
Results
Repair genes which respond to
5-FU-induced DNA damage
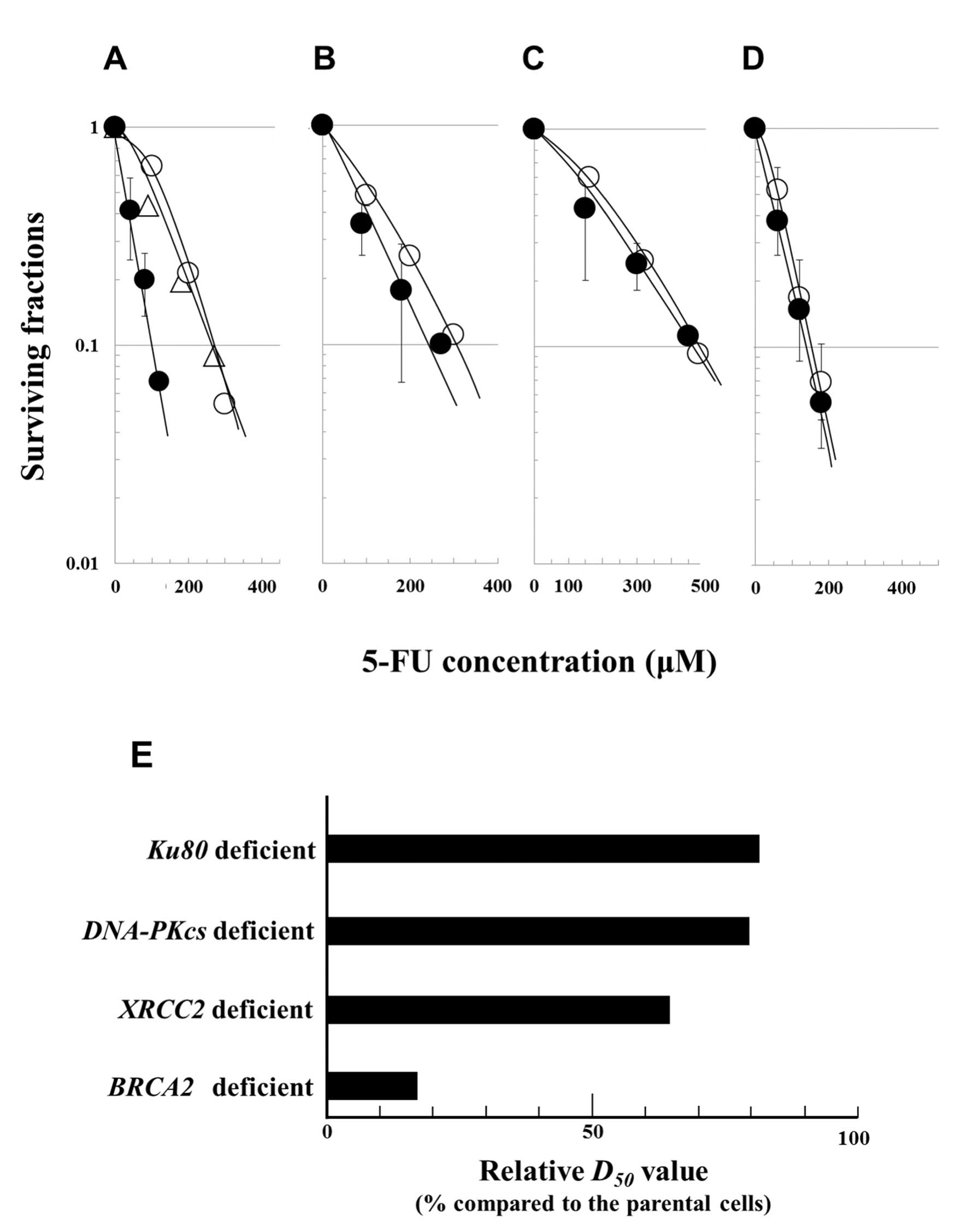
In order to determine the relative contributions of
homologous recombination (HR) and non-homologous end joining (NHEJ)
repair pathways, cellular responses to 5-FU were examined using
clonogenic survival assays after a 24-h exposure to 5-FU, using
different cell lines deficient in DSB repair pathways. The
sensitivity of each cell line was assessed according to its
D50 value, i.e. from the 5-FU dose which reduced cell
survival to 50%. Each D50 value was calculated from the
cell survival data shown in Fig.
2A–D. In order to accurately compare 5-FU sensitivity in the
repair defective cell lines, the relative D50 values
were normalized using the D50 value of the corresponding
proficient cell lines. The relative D50 values are
listed sequentially in order of their increasing values (reflecting
decreasing sensitivities to 5-FU) and are: BRCA2-deficient
cells (17%) < XRCC2-deficient cells (64%) <
DNA-PKcs-deficient cells (79%) < Ku80-deficient
cells (82%) (Fig. 2E). In summary,
the relative D50 value of the BRCA2-deficient
cells was the lowest after treatment with 5-FU, reflecting the fact
that these cells had the highest sensitivity to 5-FU.
Immunocytochemical staining of γH2AX
foci
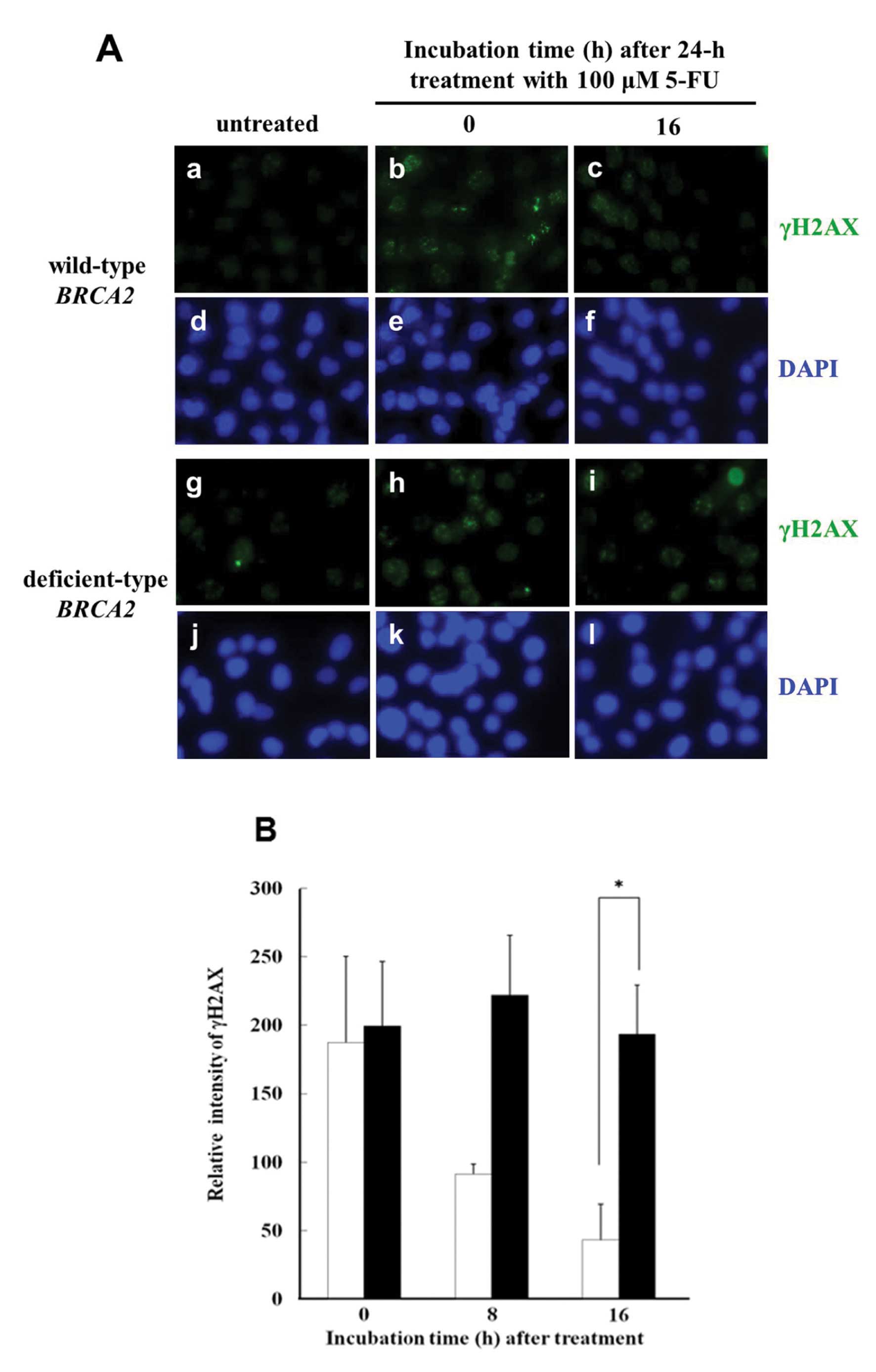
γH2AX immunocytochemical staining is an extremely
sensitive method by which to detect DSBs, and was used to examine
the presence of γH2AX foci induced by 5-FU. A typical image
(Fig. 3A) shows γH2AX foci in
BRCA2-deficient cells and in the parental cells after a 24-h
treatment with 100 μM 5-FU. In the parental cells, γH2AX foci in
the nucleus disappeared at 16 h after a 5-FU treatment. In
contrast, in BRCA2-deficient cells, the γH2AX foci in the
nucleus were still present at 16 h after 5-FU treatment.
Phosphorylation of histone H2AX
To quantify the γH2AX-positive foci, the optical
intensity of γH2AX was measured using flow cytometry. When cells
were fixed with methyl alcohol immediately after the 24-h treatment
with 100 μM 5-FU, the intensity of γH2AX in the
BRCA2-deficient cells was similar to that in the parental
wild-type cells (Fig. 3B). After a
16-h incubation, the intensity had decreased to ~25% in the
parental wild-type cells, while there was almost no change in the
intensity of γH2AX in the BRCA2-deficient cells.
Cell cycle histogram and
distribution
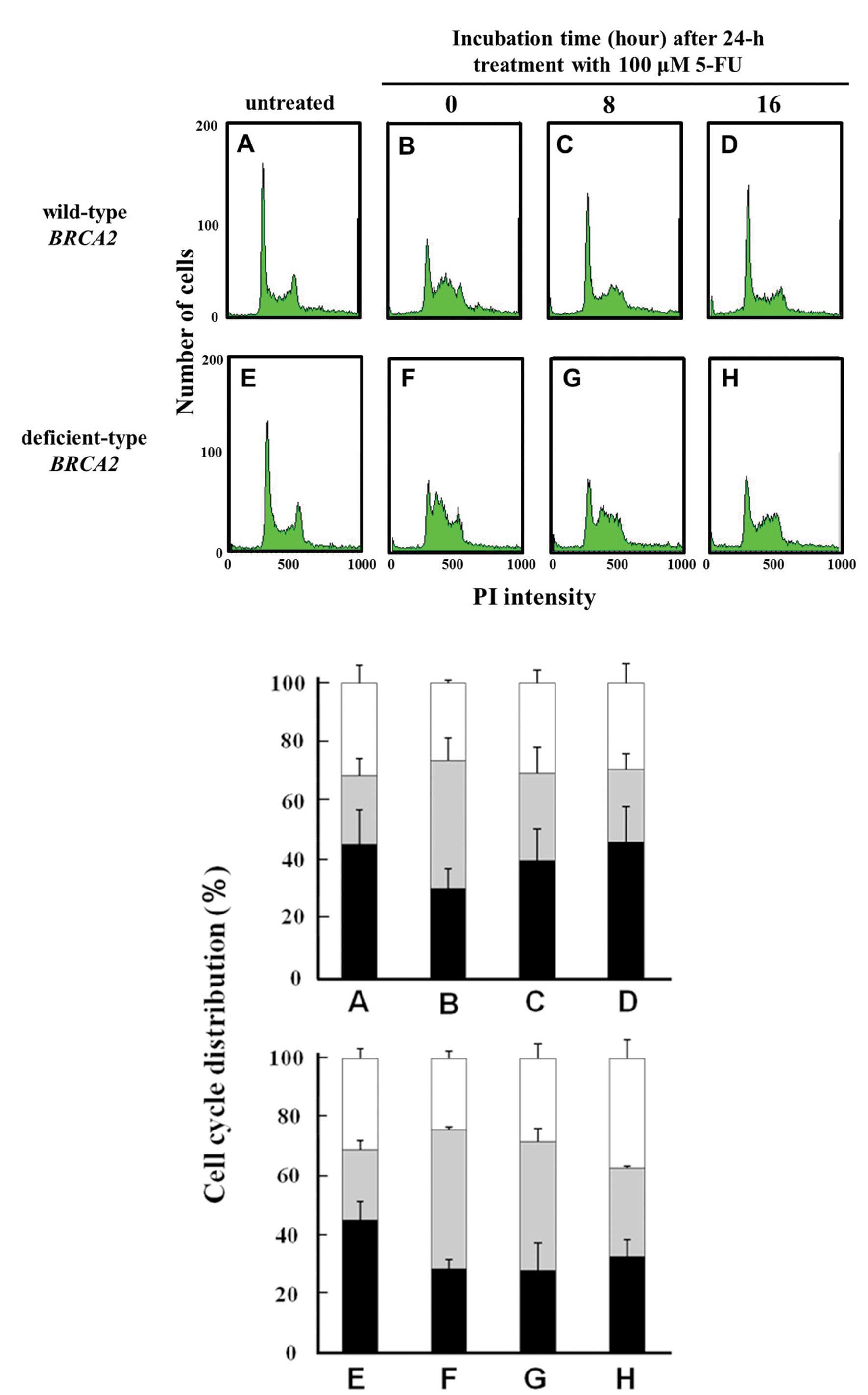
In the untreated control cells, the fraction of the
cell population in the G2/M phase in the
BRCA2-deficient cells and in the parental cells was 30 and
31%, respectively (Fig. 4E and A).
Immediately following 5-FU treatment, the fraction of the
population in the G2/M phase was almost the same or
~25%, in both cell lines. At 8 h following the 5-FU treatment, the
G2/M phase cell fractions were between 28 and 31%
(Fig. 4G and C) in both cell lines.
At 16 h following the 5-FU treatment, the G2/M phase
cell fractions were 37 or 29%, respectively. A G2/M
phase arrest was, thus, observed in the BRCA2-deficient
cells (Fig. 4H), but not in the
parental cells (Fig. 4D).
Effect of the silencing of BRCA2 on
cellular sensitivity to 5-FU in human oral cancer cells
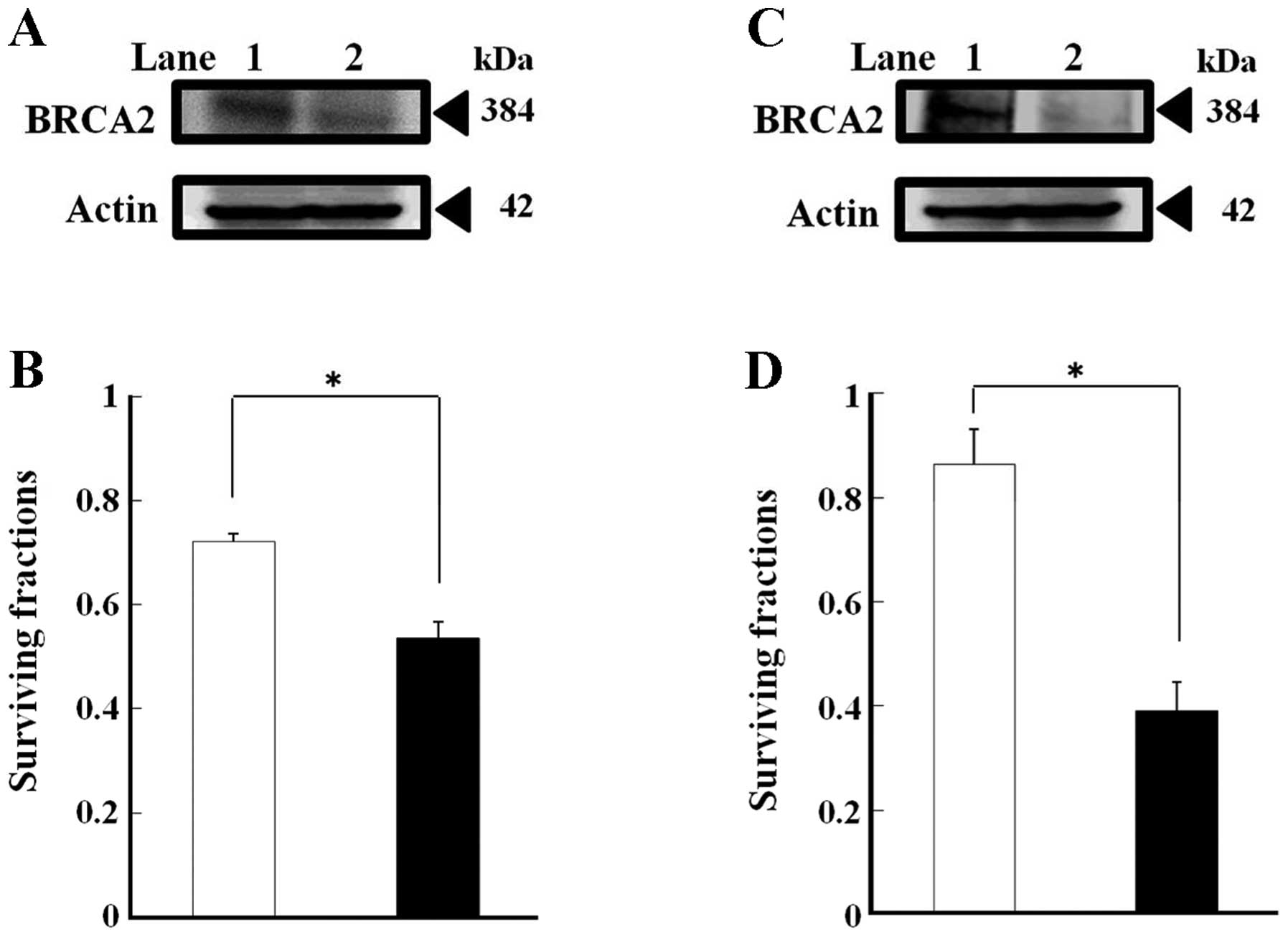
The quantity of BRCA2 protein in the
BRCA2-siRNA-transfected SAS and HSC3 cells was 55 and 37%,
respectively, of the levels observed in the control cells
transfected with the non-specific negative control siRNA (Fig. 5A and C).
To assess whether this result was pertinent to
chemotherapy used against human oral cancer cells, BRCA2
expression was silenced in human oral cancer SAS and HSC3 cells
using siRNA, and clonogenic survival assays were then conducted
with the silenced cells. In the colony formation assays, following
the 5-FU treatment, BRCA2 silencing caused an approximate
25% reduction in SAS cells and an approximate 55% reduction in HSC3
cells when compared to the cells transfected with the non-specific
negative control siRNA. These results indicate that in the SAS and
HSC3 cells, BRCA2 silencing increased cellular sensitivity
to 5-FU (Fig. 5B and D).
Discussion
5-FU has been widely used in cancer therapy for
colorectal, breast, head and neck, and other types of cancers. 5-FU
belongs to the class of antimetabolite chemotherapeutics, and is
thought to be an inhibitor of TS which is involved in thymidine
nucleotide synthesis. Recently, several reports have described
about 5-FU-induced DNA lesions and their repair in eukaryotic cells
such as yeast (17) and mammalian
cells (18). The repair mechanism
and lesions have been studied at the molecular level, and it is
possible that 5-FU induces DSBs (Fig.
1A) (9,18). When incorrect dUMP or FdUMP
nucleotides are incorporated into the newly synthesized DNA strands
during DNA replication, segments of ssDNA are formed from the nicks
and gaps generated during the BER and MMR repair processes. These
processes also result in the formation of unstable conformations in
the DNA structure through the activation of the ATR-Chk1 signaling
pathway, and these events can then lead to the formation of
DSBs.
Quantitative analysis of DNA damage is possible by
utilizing observation of γH2AX foci, and such measurements of γH2AX
focus formation are extremely sensitive. γH2AX foci are believed to
be specific indicators for the existence of DSBs induced by
ionizing radiation; specifically, one γH2AX focus correlates with
one DSB (19,20). In the research described here, flow
cytometry was used to quantitate relative repair activity with
fluorescent measurements of γH2AX-positive foci rather than by
counting the number of positive foci per nucleus (Fig. 3A and B).
The two major DSB repair pathways are HR and NHEJ
(21,22). HR operates mainly by using intact
sister chromatids during late S and G2 phases, but not
during G1 phase (23,24).
Proteins involved in HR in vertebrate cells include BRCA2, Rad52,
Rad54 and Rad51 paralogs such as Rad51C-XRCC3 and
Rad51B-Rad51C-Rad51D-XRCC2 (25).
Rad51 activity is regulated by BRCA2 which is an upstream protein
(26). Mutations in the
BRCA2 gene have been frequently observed in hereditary
breast (27) and ovarian cancers
(28). In contrast, NHEJ is
independent of cell cycle position, although its highest activity
is observed in the G1 phase (20,21).
The main components of the NHEJ repair pathway are the DNA-PK
complex (consisting of Ku70, Ku80 and DNA-PKcs) and the
XRCC4/ligase IV/XLF complex (20).
An aim of the research described in the present
study was to observe details in the repair pathways which repair
5-FU-induced DSBs. Cell survival was examined following 5-FU
treatment using colony forming assays with Chinese hamster lung
fibroblast cells, or with Chinese hamster ovary cells which are
deficient in components of the NHEJ machinery (DNA-PKcs and
Ku80) or in components of the HR machinery (BRCA2 and
XRCC2). The results indicated that HR enzymes, and
BRCA2 in particular, were responsible for a large
contribution to 5-FU resistance (Figs.
1B and 2A–E). In light of this
observation, attention was focused on the relationships between the
BRCA2 gene and DSBs induced by 5-FU. In BRCA2
wild-type cells, the intensity of γH2AX foci decreased to ~25% of
the original value by 16 h following the 5-FU treatment. In
contrast, γH2AX focus intensity showed almost no change in the
BRCA2-deficient cells. These results suggest that BRCA2
makes a major contribution to the repair of DSBs induced by 5-FU
(Fig. 3A and B).
DSBs induced by 5-FU may be generated during DNA
replication in the S phase of the cell cycle. In addition, it was
found that the cell cycle was arrested in the G2/M phase
in the BRCA2-deficient cells (Fig. 4H), but not in the parental cells
(Fig. 4D). These results support to
the idea that DSBs can induce a G2/M phase arrest when
HR repair does not progress.
In addition, observations showed that knockdown of
the BRCA2 gene by small interference RNA increased the
cellular sensitivity to 5-FU in human oral cancer SAS and HSC3
cells (Fig. 5A and C). These
results lead to the conclusion that disrupting BRCA2 protein
synthesis may be a potentially useful strategy for improving the
therapeutic efficacy of 5-FU for human oral cancer.
In summary, these observations suggest that the
BRCA2 gene product may serve as a molecular target for
improving the efficacy of 5-FU therapy. For future therapeutic
efforts, a combination of a loco-regional delivery system and the
simultaneous downregulation of the BRCA2 gene may be capable
of providing an effective tool to enhance the efficacy of 5-FU
chemotherapy for oral cancer patients.
Acknowledgements
The present study was supported by Grants-in-Aid for
Scientific Research from the Ministry of Education, Culture,
Sports, Science and Technology of Japan.
References
|
1
|
Loehrer P, Turner S, Kubilis P, et al:
Prospective randomized trial of fluorouracil versus fluorouracil
plus cisplatin in the treatment of metastatic colorectal cancer: a
Hoosier Oncology Group trial. J Clin Oncol. 6:642–648. 1988.
|
|
2
|
Welsh J, Hobbs S and Aherne G: Expression
of uracil DNA glcocylase does not affect cellular sensitivity to
thymidylate synthase (TS) inhibition. Eur J Cancer. 39:378–387.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kehler S and Ladner R: Small interfering
RNA-mediated suppression of dUTPase sensitizes cancer cells to
thymidylate synthase inhibition. Mol Pharmacol. 66:620–626.
2004.PubMed/NCBI
|
|
4
|
Longley D, Harkin D and Johnston P:
5-Fluorouracil: mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Waytt M and Wilson D III: Participation of
DNA repair in the response to 5-fluorouracil. Cell Mol Life Sci.
66:788–799. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li L, Morales J, Veigl M, et al: DNA
mismatch repair (MMR)-dependent 5-fluorouracil cytotoxicity and the
potential for new therapeutic targets. Br J Pharmacol. 158:679–692.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meyers M, Hwang A, Wagner M, et al: A role
for DNA mismatch repair in sensing and responding to
fluoropyrimidine damage. Oncogene. 22:7376–7388. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lindahl T: An N-glycosidase from
Escherichia coli that releases free uracil from DNA
containing deaminated cytosine residues. Proc Natl Acad Sci USA.
71:3649–3653. 1974.PubMed/NCBI
|
|
9
|
Fujinaka Y, Matsuoka K, Iimori M, et al:
ATR-Chk1 signaling pathway and homologous recombinational repair
protect cells from 5-fluorouracil cytotoxicity. DNA Repair.
11:247–258. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kraakman M, Overkamp W, Lange R, et al:
Brca2 (XRCC11) deficiency results in radioresistant DNA synthesis
and a higher frequency of spontaneous deletions. Mol Cell Biol.
22:669–679. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wiegant W, Overmeer R, Godthelp B, van
Buul P and Zdzienicka M: Chinese hamster cell mutant, V-C8, a model
for analysis of Brca2 function. Mutat Res. 600:79–88. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kondo N, Takahashi A, Mori E, et al:
FANCD1/BRCA2 plays predominant role in the repair of DNA
damage induced by ACNU or TMZ. PLoS One. 6:e196592011. View Article : Google Scholar
|
|
13
|
Kondo N, Takahashi A, Mori E, et al: DNA
ligase IV as a new molecular target for temozolomide. Biochem
Biophys Res Commun. 387:656–660. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Takahashi A, Matsumoto H, Nagayama K, et
al: Evidence for the involvement of double-strand breaks in
heat-induced cell killing. Cancer Res. 64:8839–8845. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bruun D, Folias A, Akkari Y, et al: siRNA
depletion of BRCA1 but not BRCA2, causes increased genome
instability in Fanconi anemia cells. DNA Repair. 2:1007–1013. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yamakawa N, Takahashi A, Mori E, et al:
High LET radiation enhances apoptosis in mutated p53 cancer
cells through caspase-9 activation. Cancer Sci. 99:1455–1460. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lauren S, Pawel J, Miral D and James T:
Linking uracil base excision repair and 5-fluorouracil toxicity in
yeast. Nucleic Acids Res. 34:140–151. 2004.
|
|
18
|
Raafat A, Ekram M and Jachen D: Targeting
DNA double-strand break repair: is it the right way for sensitizing
cells to 5-fluorouracil? Anticancer Drugs. 40:160–170.
2010.PubMed/NCBI
|
|
19
|
Rothkamm K and Lobrich M: Evidence for a
lack of DNA double-strand break repair in human cells exposed to
very low x-ray doses. Proc Natl Acad Sci USA. 100:5057–5062. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Takahashi A and Ohnishi T: Does γH2AX
focus formation depend on the presence of DNA double strand breaks?
Cancer Lett. 229:171–179. 2005.
|
|
21
|
Jackson SP: Sensing and repairing DNA
double-strand breaks. Carcinogenesis. 23:687–696. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kanar R, Hoeijimakers JH and Van Gent DC:
Molecular mechanisms of DNA double-strand break repair. Trends Cell
Biol. 8:483–489. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rothkamm K, Kruger I, Thompson LH and
Lobrich M: Pathways of DNA double-strand break repair during the
mammalian cell cycle. Mol Cell Biol. 23:5706–5715. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hinz JM, Yamada NA, Salazar EP, Tebbs RS
and Thompson LH: Influence of double-strand-break repair pathways
on radiosensitivity throughout the cell cycle in CHO cells. DNA
Repair. 4:782–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thompson L and Schild D: Homologous
recombinational repair of DNA ensures mammalian chromosome
stability. Mut Res. 477:131–153. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Davies A, Masson J, McIlwraith M and
Stasiak A and Stasiak A: Role of BRCA2 in control of the RAD 51
recombination and DNA repair protein. Mol Cell. 7:273–282. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ottini L, Masala G, D’Amico C, et al:
BRCA1 and BRCA2 mutation status and tumor
characteristics in male breast cancer: a population-based study in
Italy. Cancer Res. 63:342–347. 2003.
|
|
28
|
Schrader K, Hurlburt J, Kalloger S, et al:
Germline BRCA1 and BRCA2 mutations in ovarian cancer:
utility of a histology-based referral strategy. Obstet Gynecol.
120:235–240. 2012.
|