Introduction
Metformin, a first-line oral anti-type II diabetes
agent used worldwide, displays an antitumorigenesis effect,
according to recent epidemic studies (1–3). As
compared to insulin or sulfonylureas administration, metformin use
may markedly reduce the cancer risk in patients with type II
diabetes. Recent studies have confirmed the anti-proliferation
effect on various human cancer cell types, such as pancreas
(4), prostate (5), breast (6), stomach (7) and liver (8). Metformin inhibits the
pro-proliferation effect of insulin receptor- and IGF
receptor-dependent signaling by reducing insulin resistance.
Furthermore, metformin activates AMP-activated protein kinase
(AMPK) and subsequently inhibits activation of mammalian target of
rapamycin (mTOR) to prevent proliferation of tumor cells, and
activates p53 protein-inducing cell cycle arrest of tumor cells.
Several studies have indicated that metformin can potentiate the
effect of chemotherapeutic agents or reverse drug resistance in
cancer cells (8–10). However, the mechanism of the
anti-cancer effects of metformin remains unclear.
A recent epidemiological study that included 1,828
potential intrahepatic cholangiocarcinoma (ICC) patients described
that metformin use was significantly associated with a 60%
reduction in ICC risk in diabetic patients (11). Cholangiocarcinoma (CC)categorized as
intrahepatic and extrahepatic cholangiocarcinoma (ECC) is highly
lethal. ICC is the second most common type of primary liver cancer
and its incidence and mortality rates have been rising in recent
decades (12–15). Less than 30% of patients with ICC
have the opportunity to have radical operation at diagnostic
presentation. Apart from radical operation, some treatment
approaches such as systemic chemotherapy, transarterial
chemoembolization and radiofrequency ablation may be applied at
advanced stages of ICC; however, none of the approaches can
significantly improve the prognosis of ICC. Thus, new treatment
strategies are needed for ICC.
In the present study, we showed that metformin
inhibited the growth of ICC cells by inducing apoptosis and cell
cycle arrest and inhibiting colony formation. Metformin inhibited
growth by activation of the AMPK/mTOR complex 1 (mTORC1) pathway.
Furthermore, metformin amplified the effect of chemotherapeutic
agents such as gemcitabine, 5-fluorouracil, arsenic trioxide
(As2O3) and sorafenib. Metformin might target
the mTOR/hypoxia inducible factor 1α (HIF1α)/multidrug resistance
proteins 1 (MRP1) signaling pathway and extracellular signal
regulated kinases 1/2 (ERK) to sensitize the ICC cells to
chemotherapy.
Materials and methods
Cell cultures
The ICC cell lines RBE and HCCC-9810 were purchased
from the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). The cell lines were cultured in RPMI-1640
(Gibco, USA) supplemented with 10% fetal bovine serum (FBS; Gibco)
and 100 μg/ml each of penicillin and streptomycin (Invitrogen, USA)
in 5% CO2 at 37°C.
Reagents
Metformin (1,1-dimethylbiguanide hydrochloride,
#D150959-5G), gemcitabine (2′-Deoxy-2′,2′-difluorocytidine,
#G6423-10MG) and 5-fluorouracil (2,4-dihydroxy-5-fluoropyrimidine,
#F6627-1G) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Sorafenib
(C21H16ClF3N4O3C7H8O3S,
CAS 475207-59-1) was purchased from Selleck Chemicals (Houston, TX,
USA). The Cell Counting Kit-8 (CCK-8, KGA317), the Annexin V-FITC
Apoptosis Detection Kit (KGA108) and the Cell Cycle Detection Kit
(KGA512) were purchased from KeyGen Biotech (Nanjing, China).
Antibodies
The following antibodies were used in western blot
analysis: β-actin (sc-47778, diluted 1:1,000) was from Santa Cruz
Biotechnology, Inc., (Santa Cruz, CA, USA). HIF-1α (N-term)
(AP4776a, diluted 1:1,000), active Caspase-3 (AJ1131b, diluted
1:1,000), Bcl-2 (AJ1082a, diluted 1:1,000), CDK4 (AP7520b, diluted
1:1,000) and Cyclin D1 (AP2612c, diluted 1:1,000) were from Abgent
(San Diego, CA, USA). AMPKα (Ab-172, #B0003, diluted 1:500) and
phosphorylated AMPKα (Phospho-Thr172, #A0003, diluted
1:500), mTOR (#B7156, diluted 1:500) and phosphorylated mTOR
(Phospho-Ser2448, #A7156, diluted 1:500) were from Assay
Biotech (Sunnyvale, CA, USA). MRP1 (PA5-30594, diluted 1:500) was
from Thermo Fisher Scientific Inc., (Rockford, IL, USA).
Phosphorylated Raptor (Phospho-Ser792, #2083, diluted
1:1,000), phosphorylated p70 S6 Kinase (Phospho-Thr389,
#9234, diluted 1:1,000), phosphorylated 4E-BP1
(Phospho-Thr37/46, #2855, diluted 1:1,000), ERK (#4696,
diluted 1:2,000) and phosphorylated ERK
(Phospho-Thr202/Tyr204, #4370, diluted
1:2,000) were from Cell Signaling Technology, Inc. (Danvers, MA,
USA). Goat anti-rabbit and goat anti-mouse IgG,
peroxidase-conjugated secondary antibodies (31460 and 31430, both
diluted 1:10,000) were from Thermo-Pierce (Rockford, IL, USA).
Cell viability assay
Cell viability was determined using the CCK-8 assay
according to the manufacturer’s instructions. Cells
(5×103) were seeded into a well of a 96-well plate and
cultured in 100 μl of RPMI-1640 supplemented with 10% FBS, 100
μg/ml penicillin and 100 μg/ml streptomycin. After 24 h, metformin
(0, 1, 5, 10, 20, 40 mmol/l) was added into the culture medium.
Then, after the cells were incubated at 37°C for different times
(24, 48, 72 h), the medium was exchanged for 100 μl of RPMI-1640
and 10 μl of CCK-8 reagent. The cells were incubated for 2 h at
37°C. Finally, the optical density was measured using an
EnSpireTM 2300 Multilabel Reader (PerkinElmer, Waltham,
MA, USA) at 450 nm. Five replicates were prepared for each
condition. The mean values were calculated and growth cures were
drawn.
Clonogenic assay
The inhibitory effect of metformin on ICC cell
proliferation was also determined by clonogenic assay.
Logarithmic-phase ICC cells were trypsinized and collected, and
then resuspended cells were seeded into 6-well plates in
triplicates at a density of 500 cells/well in 2 ml of medium
containing 10% FBS. After 48 h incubation, cultures were replaced
with fresh medium contain 0, 1, and 2.5 mM metformin and 2% FBS,
and grown for 2 weeks. The cell clones were stained by a solution
containing 1% crystal violet and 25% methanol for 2 min. The excess
dye was removed by gently rinsing with tap water for 15 min. The
clones were visually counted and the percentage of cells that
initiated a clone was determined as clonogenicity (number of
clones/500×100%).
Western blot analysis
Cells after different treatments were harvested and
lysed in RIPA buffer (KGP702), supplemented with 1 mM
phenylmethylsulfonyl fluoride (PMSF; KGP610) and 1 mM phosphatase
inhibitor cocktails (KGP602; all from KeyGen Biotech). The mixture
was centrifuged at 12,000 × g for 20 min, and the supernatant was
collected. The protein concentration was determined using the BCA
assay kit (KGPBCA), and each sample contained 30 μg protein per 10
μl. The protein samples were mixed with loading buffer (KGP101) and
the proteins were separated using 6 or 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad
Laboratories, Hercules, CA, USA). After soaking in blocking buffer
for 2 h, the blots were incubated at 4°C overnight with the primary
antibody and were subsequently incubated at 37°C for 1 h with the
HRP-conjugated secondary antibody. The bands were visualized by
chemiluminescence, they were imaged using a ChemiDoc XRS and were
analyzed using Image Lab (both from Bio-Rad).
Flow cytometric analysis
To evaluate the effects on cell cycle arrest and
induction of apoptosis by metformin, the cells were examined using
the Annexin V-FITC Apoptosis Detection Kit and the Cell Cycle
Detection Kit according to the manufacturer’s protocols. RBE and
HCCC-9810 cells were seeded into 6-well plates (1×105
and 2×105 cells/dish for analysis of cell cycle arrest
and apoptosis, respectively). For cell cycle analysis, after
treatment with metformin (0, 1, 5, 10, 20, 40 mmol/l) for 48 h, a
total of 1×106 cells was pelleted by centrifugation and
washed twice with PBS. Then, the cell pellets were resuspended in
500 μl of ice-cold 70% ethanol and incubated at 4°C overnight. The
fixed cells were centrifuged and the pellets were washed with PBS.
After incubation with 100 μl RNase A (10 μg/ml) for 30 min at 37°C
in the dark, the cells were resuspended in 400 μl PI (50 μg/ml) and
placed at 4°C in the dark for 30 min. The stained cells were
analyzed using an Accuri C6 flow cytometer (Accuri Cytometers Inc.,
Ann Arbor, MI, USA). For the apoptosis analysis, the cells were
trypsinized, washed with cold PBS and suspended in PBS. Then, the
cells were stained using the Annexin V-FITC reaction reagent (5 μl
of Annexin V-FITC, 5 μl of propidium iodide) at 37°C for 30 min in
the dark. The stained cells were analyzed using an Accuri C6 flow
cytometer (Accuri Cytometers Inc).
Statistical analysis
SPSS 13.0 statistical software was used for the
statistical analysis. Values are presented as the mean ± SD.
Statistical analyses were performed using Student’s t-test. The
analysis of multiple groups was performed with ANOVA with an
appropriate post hoc test.
Results
Metformin inhibits the proliferation of
ICC cells
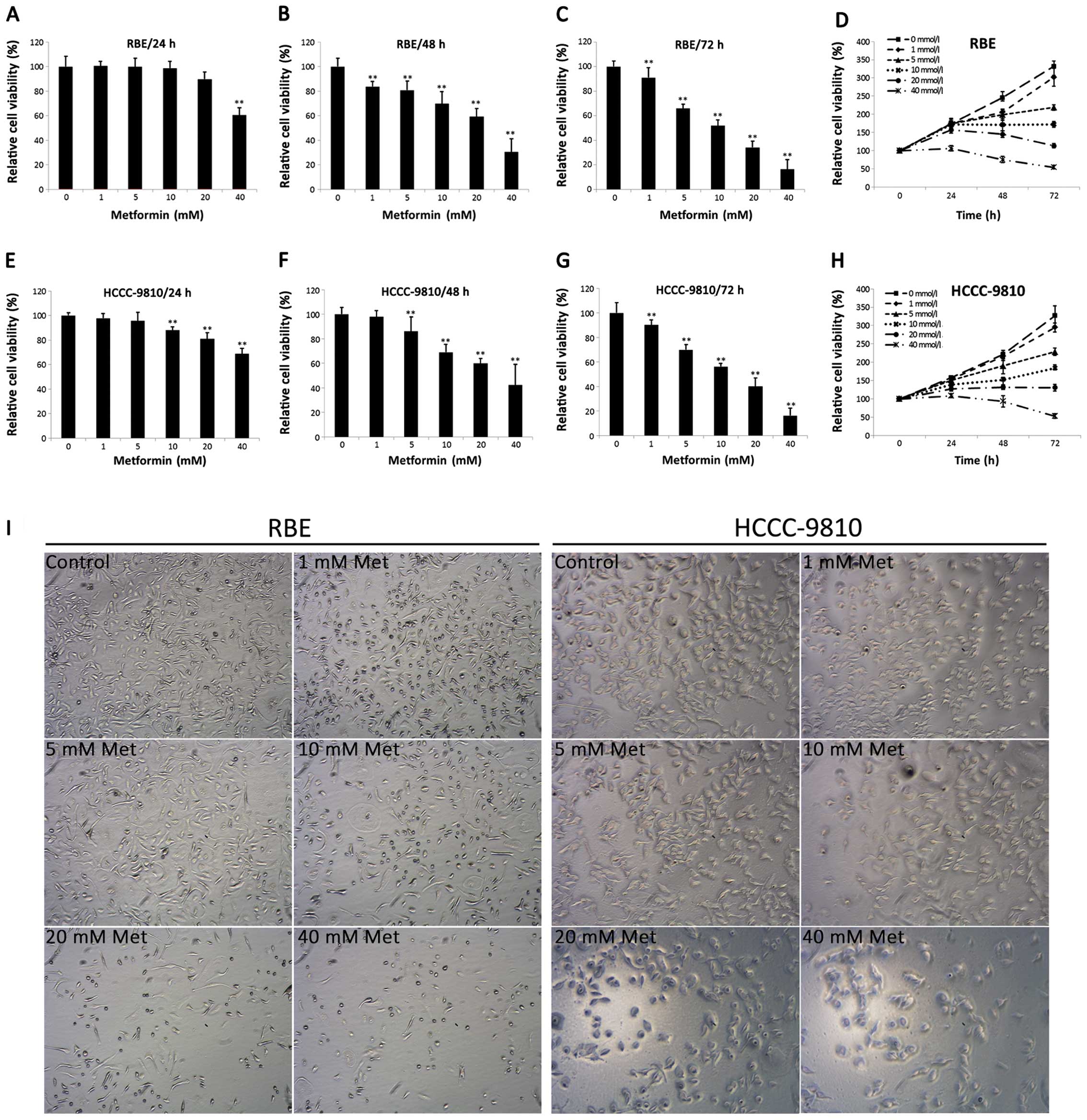
The effects of metformin on the proliferation of ICC
cells were investigated in RBE and HCCC-9810 cell lines using CCK-8
assay. As shown in Fig. 1A–H,
metformin significantly reduced cell viability in a dose- (0–40 mM)
and time-dependent (24, 48, 72 h) manner in both RBE and HCCC-9810
cell lines. With the 5 mM metformin and 48-h incubation, the two
cell lines showed statistical differences in relative cell
viability compared to the control cells. The microscopic
examination showed a significant decrease of cell density and
change of cell morphology, which displayed a smaller and granulated
shape in metformin treated cells (Fig.
1I). Taken together, the results show that metformin inhibits
the proliferation of ICC cells.
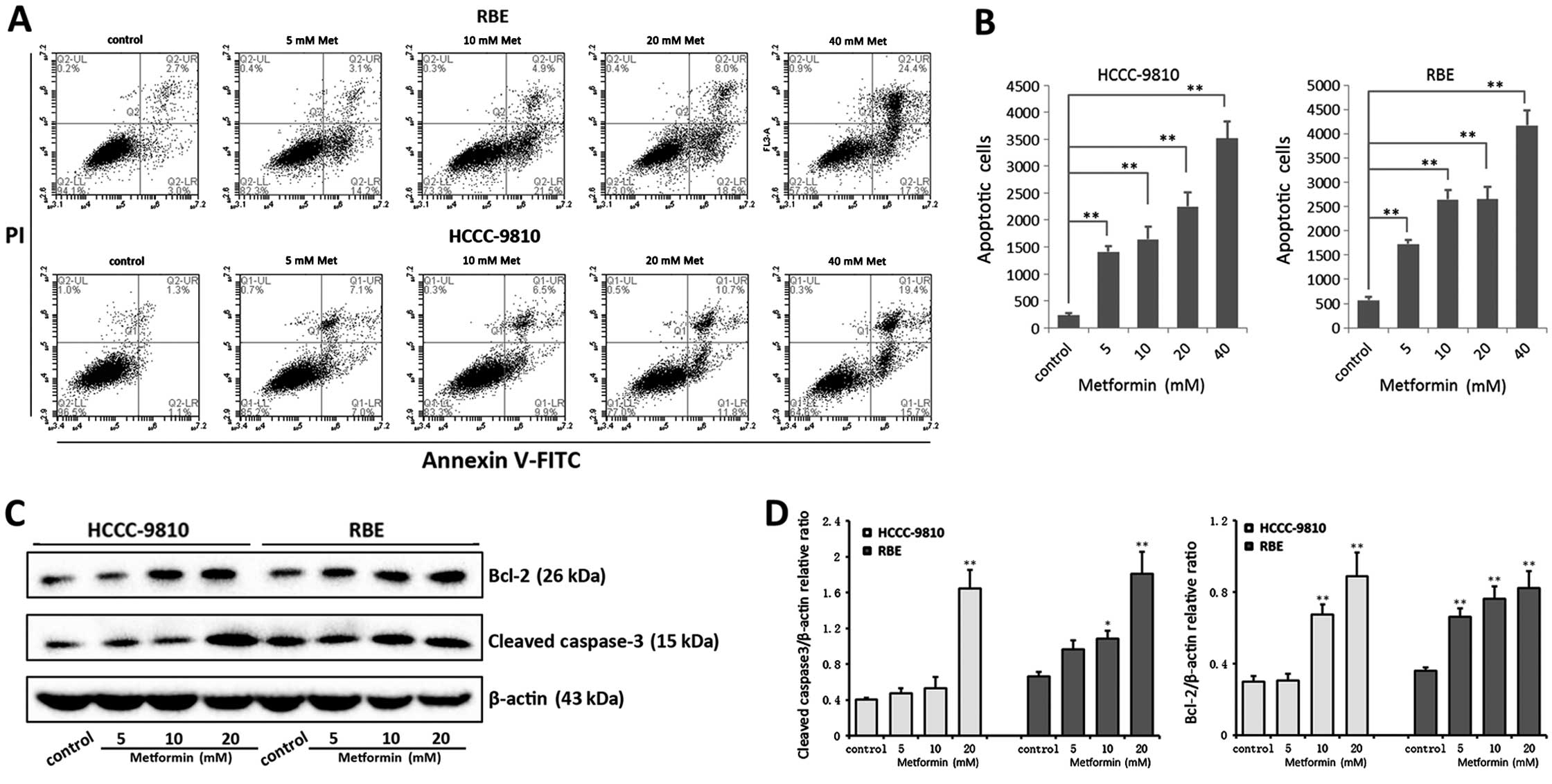
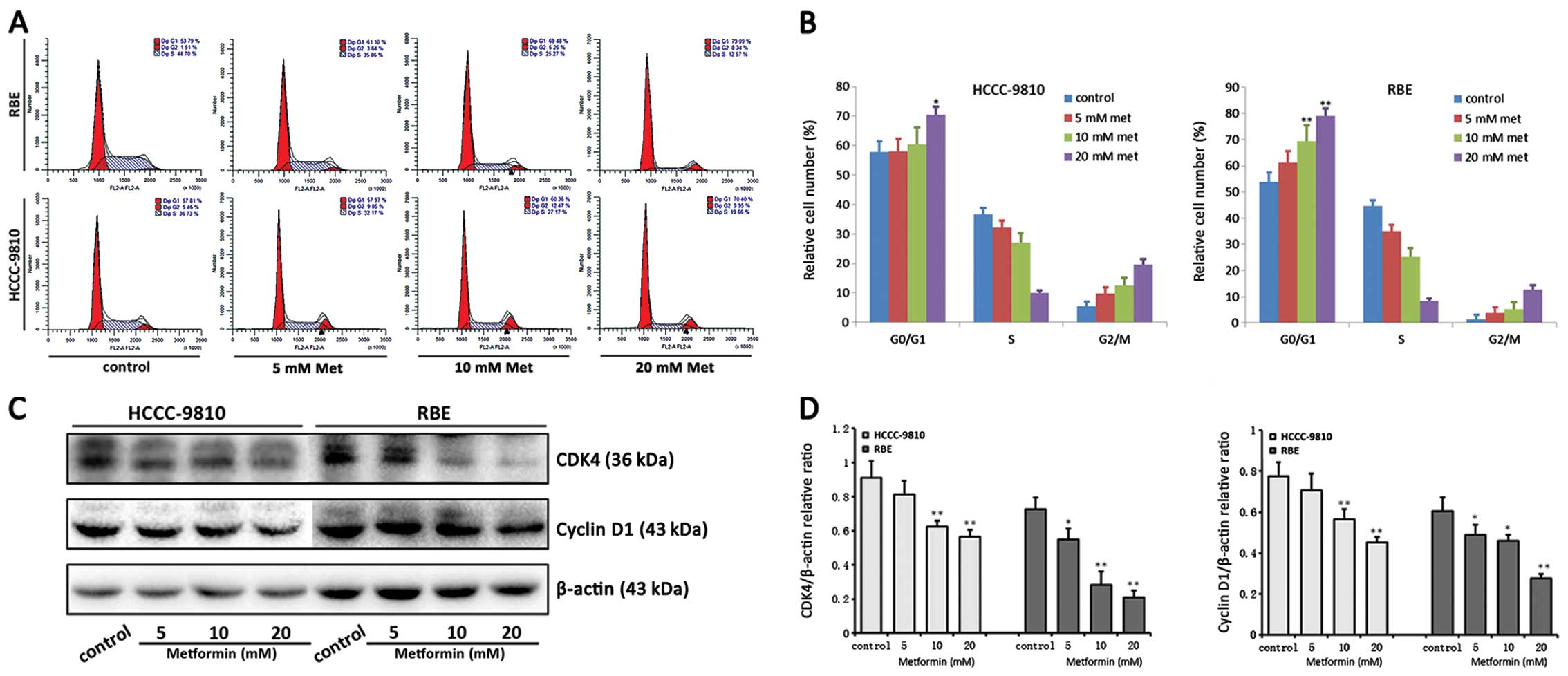
Metformin promotes apoptosis and induces
G0/G1 cell cycle arrest in ICC cells
The number of apoptotic cells and percentages of
cell cycle distribution were examined after 48 h metformin
incubation by flow cytometric analysis. A significant increase in
the number of apoptotic cells and cells undergoing G0/G1 cell cycle
arrest was observed in RBE and HCCC-9810 cells with metformin
treatment compared with the control cells (Figs. 2A and B; 3A and B). With 48 h metformin treatment,
the percentages of early apoptotic and late apoptotic/necrotic were
increased from 5.7±0.7% to 41.7±3.1% in RBE cells and 2.4±0.4% to
35.1±3.21% in HCCC-9810 cells, and the G0/G1 phase cells were
increased from 53.79±3.59% to 79.09±2.79% in RBE cells and
57.81±3.56% to 70.40±2.81% in HCCC-9810 cells, depending on the
metformin doses.
Furthermore, to confirm the metformin-induced
apoptosis and G0/G1 cell cycle arrest in ICC cells, cleaved
caspase-3, Bcl-2, cyclin D1 and CDK4 expression were monitored
using western blot analysis in RBE and HCCC-9810 cells (Figs. 2C and D; 3C and D). Activation of the apoptosis
promoter caspase-3 was related to the metformin-induced apoptosis.
However, Bcl-2, known as an apoptosis inhibitor, was upregulated by
treatment of metformin in a dose-dependent manner. Cyclin D1 and
CDK4, which are responsible for the transition from G0/G1 to S
phase, were downregulated after metformin treatment. Collectively,
these results suggest that metformin promotes apoptosis and induces
G0/G1 cell cycle arrest in ICC cells.
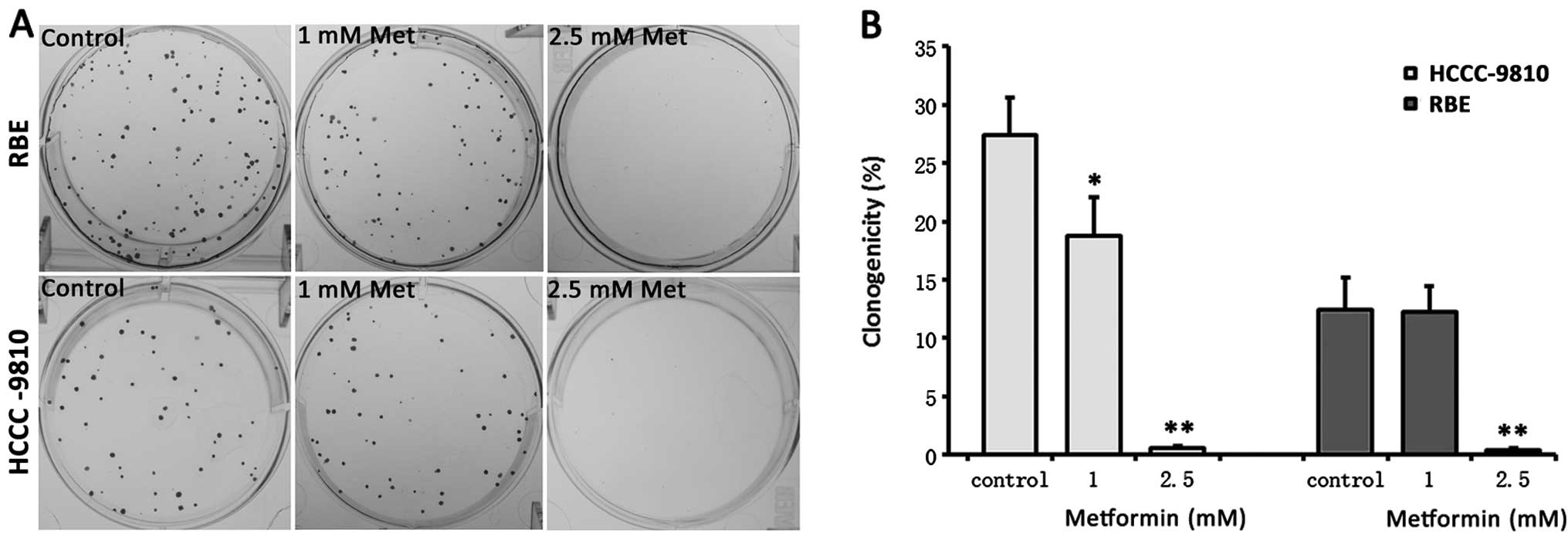
Metformin inhibits colony formation of
ICC cells
We studied the ability of these two ICC cell lines
to form colonies in 6-well plates in the presence or absence of
metformin. The RBE cells were more sensitive to 1 mM metformin than
HCCC-9810 cells (Fig. 4). At the
concentration of 2.5 mM metformin, almost no colonies formed in
either cell line.
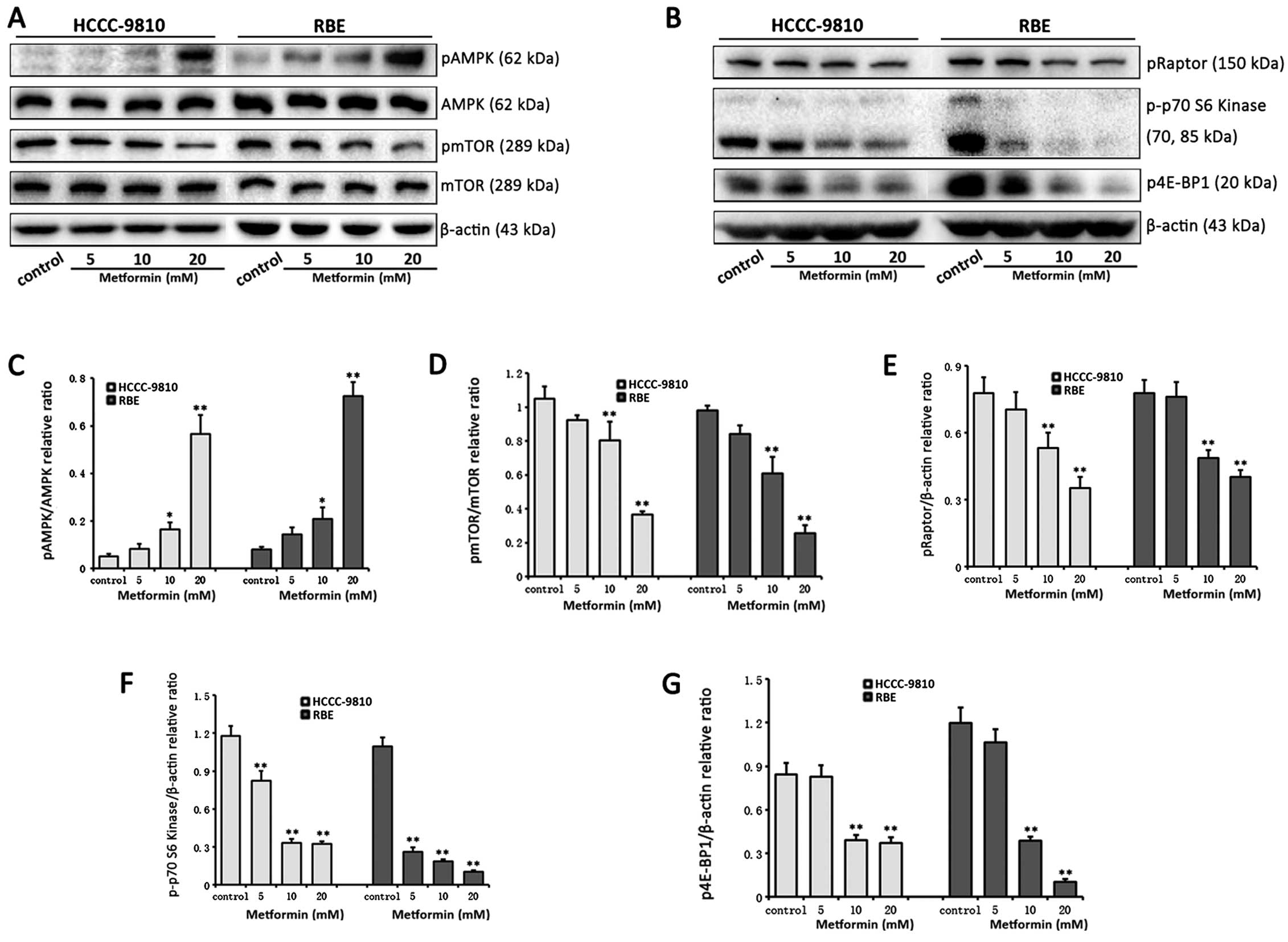
Metformin targets the AMPK/mTORC1 pathway
in ICC cells
To evaluate the specific effect of metformin on the
AMPK/mTORC1 pathway, which is widely believed to be the most common
target of metformin, western blot analysis was used to evaluate the
AMPK/mTORC1 pathway in HCCC-9810 and RBE cells. Metformin treatment
resulted in enhanced AMPK phosphorylation and reduced mTOR
phosphorylation in a dose-dependent manner in ICC cells.
Furthermore, the regulatory protein of mTOR (Raptor), which is
identified as an mTOR binding partner, mediates mTOR signaling to
downstream targets through binding to mTOR substrates, including
eIF4E-binding protein 1 (4E-BP1) and p70 S6 kinase. Metformin
dose-dependently inhibited the phosphorylation of Raptor, 4E-BP1
and p70 S6 kinase. Taken together, these results suggest metformin
targets the AMPK/mTORC1 pathway in ICC cells (Fig. 5).
Metformin targets the HIF-1α/MRP1 pathway
and ERK and sensitizes ICC cells to certain chemotherapeutic
agents
The combination of metformin with one of the
following chemotherapeutic agents, sorafenib, gemcitabine,
5-fluorouracil and arsenic trioxide (As2O3)
was more effective than certain agents alone (Fig. 6F and G). Markedly, metformin plus
sorafenib or As2O3 significantly enhanced the
inhibitory effect on RBE and HCCC-9810 cells induced by a single
agent. However, metformin did not significantly sensitize ICC cells
to gemcitabine, and alternatively sensitized ICC cells to
5-fluorouracil.
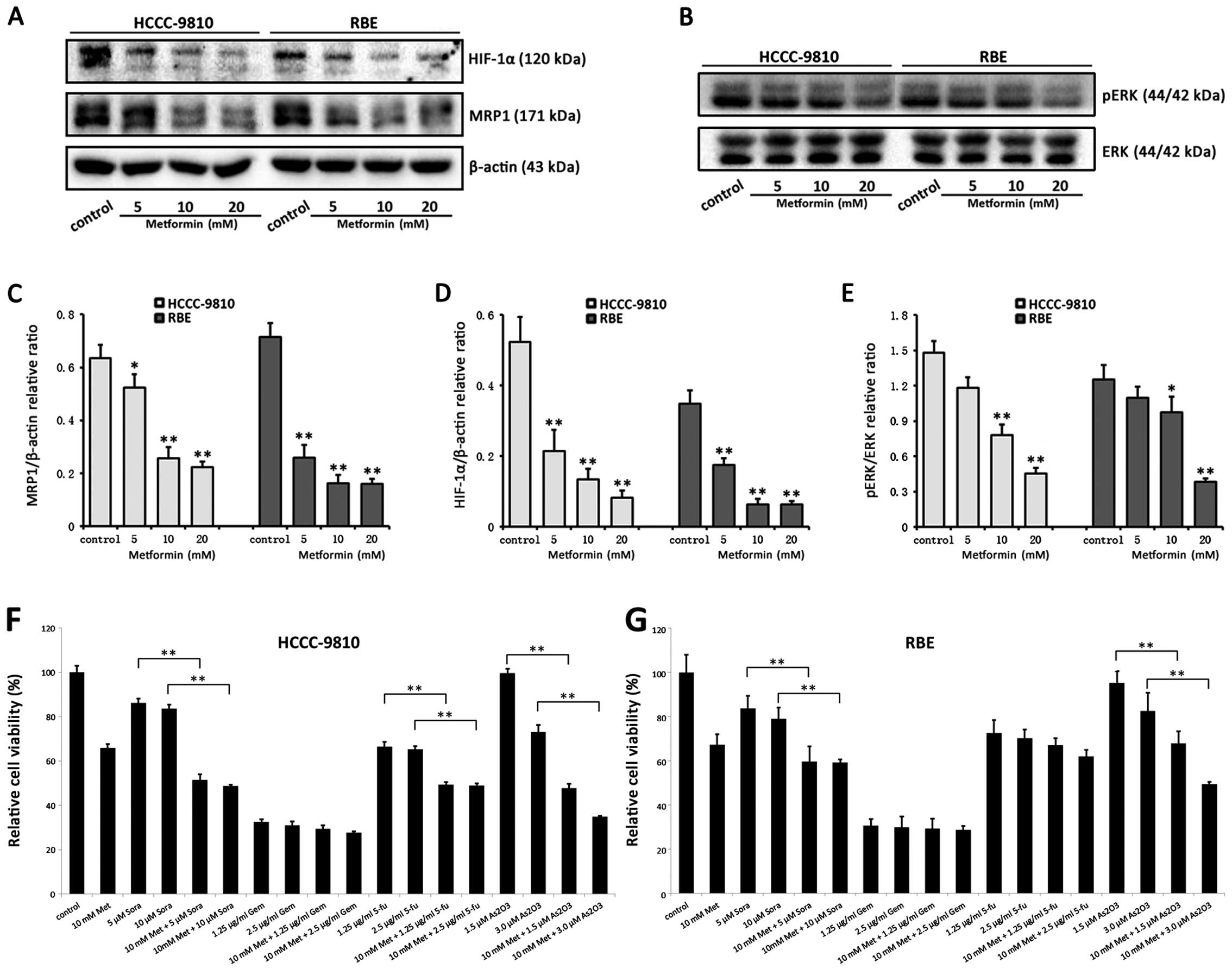 | Figure 6Metformin sensitizes ICC cells to
certain chemotherapeutic agents and the potential mechanisms. RBE
and HCCC-9810 cells were treated with a combination of metformin
and one of the chemotherapeutic agents, sorafenib (sora),
gemcitabine (gem), 5-fluorouracil (5-fu) and arsenic trioxide
(As2O3). (A) The expression of HIF-1α protein
and MRP1 protein was downregulated depending on the dose of
metformin determined using western blotting. (B) The
phosphorylation of ERK was downregulated depending on the dose of
metformin. (C–E) All the band intensities were shown after
semi-quantification using Image Lab 5.0 software and normalized
with β-actin or total EKR and are represented as the means ± SD
from 3 independent experiments (*P<0.05,
**P<0.01).(F and G) RBE and HCCC-9810 cells were
treated with sora (5 or 10 μM), gem (1.25 or 2.5 μg/ml), 5-fu (1.25
or 2.5 μg/ml) and As2O3 (1.5 or 3.0 μM),
respectively, for 48 h. Then, the CCK-8 assay was used to determine
cell viability. The cell viability with a combination of agent
treatments was compared to the cell viability with a single
chemotherapeutic agent treatment, respectively
(**P<0.01). |
To explore the potential mechanisms related to the
chemotherapy sensitization effect of metformin, HIF-1α/MRP1 and
phosphorylation of ERK were analyzed in RBE and HCCC-9810 cells.
Western immunoblot analysis revealed that metformin markedly
suppressed the expression of HIF-1α protein and MRP1 protein and
decreased the phosphorylation of ERK in a dose-dependent manner
(Fig. 6A–E).
Discussion
ICC is an aggressive malignancy, the incidence and
mortality rates of which are increasing worldwide. ICC is difficult
to diagnose at an early stage and is associated with a low surgical
resection rate. Management of ICC has not achieved significant
improvements in recent decades. Metformin is an oral
anti-hyperglycemic agent of the biguanide family, which is used
first-line for type II diabetes with few side-effects. Recent data
also described the anti-proliferation effect of metformin in
numerous cancer cells. A recent epidemiological study that included
1,828 potential ICC patients described that metformin use was
significantly associated with a 60% reduction in ICC risk in
diabetic patients, demonstrating the potential value of metformin
in ICC management. Thus, we investigated the anti-proliferation
effect and the mechanisms by which metformin affects the ICC cell
lines RBE and HCCC-9810. In addition, we evaluated the
pro-sensitive effect of metformin in chemotherapy in ICC cells, the
mechanisms of which were also explored.
In the present study, we showed that metformin
exhibited a dose- and time-dependent anti-proliferation effect on
ICC cell lines RBE and HCCC-9810. Metformin exerted inhibitory
effects on the clonogenicity and promoted apoptosis and induced
G0/G1 cell cycle arrest in RBE and HCCC-9810 cells, which were
consistent with the cell viability variation. The two ICC cell
lines showed similar sensitivity to metformin. Notably, expression
of Bcl-2, which is known to block apoptosis, was upregulated by
treatment of metformin in a dose-dependent manner in our study. To
the best of our knowledge, most of the previous studies reported
that metformin reduced expression of Bcl-2 (4,16,17).
We hypothesize that the ICC cells resist the metformin-induced
apoptosis via, paradoxically, upregulation of Bcl-2 protein and, on
the other hand, Bcl-2 conversely acts as an apoptosis inhibitor or
a promoter by certain mechanisms (18). Our future studies will focus on the
exact mechanism by which metformin affects expression of Bcl-2 in
ICC cells.
The AMPK/mTORC1 pathway is the most widely believed
target of metformin. Metformin inhibited the activation of the
mTORC1 by activating AMPK in RBE and HCCC-9810 cells in our study.
Although it remains controversial whether activation of mTOR
pathway promotes development of cholangiocarcinoma or predicts poor
prognosis in patients with cholangiocarcinoma (19,20),
our results are in line with numerous other reports describing the
inhibitory effect of metformin on cancer cells (6,10,21).
In addition, we detected the inhibitory effect of
combination treatment of metformin with sorafenib, gemcitabine,
5-fluorouracil or As2O3, which have been
clinically used for cholangiocarcinoma treatment. Metformin
significantly sensitized RBE and HCCC-9810 cells to sorafenib and
As2O3, while metformin alternatively
sensitized ICC cells to 5-fluorouracil and did not statistically
sensitize ICC cells to gemcitabine. This phenomenon may be
explained by the fact that RBE and HCCC-9810 cells were highly
sensitive to gemcitabine in our study. In particular, metformin
more significantly amplified the inhibitory effect of
As2O3 than other agents in ICC cells, the
mechanism of which we will explore in future studies. To approach
the potential mechanisms related to the chemotherapy sensitization
effect of metformin, we further investigated how metformin
suppressed the expression of HIF-1α and MRP1, which are associated
with the multidrug resistance of cancer cells, and decreased the
phosphorylation of ERK, which is critical in regulating therapy
response of cancer cells (22–24).
Recent studies suggested that metformin could improve oxygenation
and suppress HIF-1α accumulation in tumor- or diabetic-related
diseases through the activation of the AMPK/mTOR pathway and the
repression of oxygen consumption (25–28).
HIF-1, a basic helix-loop-helix transcription factor, plays a
significant role in regulating the transcription of various target
genes in response to hypoxia (29).
HIF-1α is an oxygen-regulated subunit that mediates the essential
function of HIF-1. The overexpression of HIF-1α may contribute to
the pathogenesis of tumor resistance to chemotherapy (30,31).
MRP1 is regarded as energy-dependent membrane efflux pumps and are
widely believed to be transcriptionally regulated by HIF-1α in
multiple human tumors (32–35). Consequently, our results indicated
that metformin might target the AMPK/mTOR/HIF-1α/MRP1 sensitizing
ICC cells to chemotherapeutic agents. Furthermore, decreasing the
phosphorylation of ERK, which is also the target of sorafenib and
As2O3 (36–38),
might compose the mechanisms by which metformin performed as a
chemosensitizer in our study.
In conclusion, our results revealed that metformin
inhibits ICC cell proliferation and sensitizes ICC cells to certain
chemotherapeutic agents, possibly by mechanisms including apoptosis
induction and cell cycle arrest, and targeting the AMPK/mTORC1,
AMPK/mTOR/HIF-1α/MRP1 pathway and ERK. Further studies are required
to investigate how metformin increases the expression of Bcl-2 and
the impact exerted by Bcl-2 on ICC cells. As it is an inexpensive
and widely used antidiabetic drug without severe adverse effects,
metformin may be a prospective chemotherapeutic agent or a
chemosensitizer in ICC treatment.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (81272368, 81102069) and the
Public Technology Research and Social Development Project of
Science and Technology Department of Zhejiang Province
(2013C33128).
References
|
1
|
Evans JM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee MS, Hsu CC, Wahlqvist ML, Tsai HN,
Chang YH and Huang YC: Type 2 diabetes increases and metformin
reduces total, colorectal, liver and pancreatic cancer incidences
in Taiwanese: a representative population prospective cohort study
of 800,000 individuals. BMC Cancer. 11:202011. View Article : Google Scholar
|
|
3
|
Dowling RJ, Niraula S, Stambolic V and
Goodwin PJ: Metformin in cancer: translational challenges. J Mol
Endocrinol. 48:R31–R43. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nair V, Pathi S, Jutooru I, et al:
Metformin inhibits pancreatic cancer cell and tumor growth and
downregulates Sp transcription factors. Carcinogenesis.
34:2870–2879. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Akinyeke T, Matsumura S, Wang X, et al:
Metformin targets c-MYC oncogene to prevent prostate cancer.
Carcinogenesis. 34:2823–2832. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alimova IN, Liu B, Fan Z, et al: Metformin
inhibits breast cancer cell growth, colony formation and induces
cell cycle arrest in vitro. Cell Cycle. 8:909–915. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kato K, Gong J, Iwama H, et al: The
antidiabetic drug metformin inhibits gastric cancer cell
proliferation in vitro and in vivo. Mol Cancer Ther. 11:549–560.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Petrushev B, Tomuleasa C, Soritau O, et
al: Metformin plus PIAF combination chemotherapy for hepatocellular
carcinoma. Exp Oncol. 34:17–24. 2012.PubMed/NCBI
|
|
9
|
Ashinuma H, Takiguchi Y, Kitazono S, et
al: Antiproliferative action of metformin in human lung cancer cell
lines. Oncol Rep. 28:8–14. 2012.
|
|
10
|
Chen G, Xu S, Renko K and Derwahl M:
Metformin inhibits growth of thyroid carcinoma cells, suppresses
self-renewal of derived cancer stem cells, and potentiates the
effect of chemotherapeutic agents. J Clin Endocrinol Metab.
97:E510–E520. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chaiteerakij R, Yang JD, Harmsen WS, et
al: Risk factors for intrahepatic cholangiocarcinoma: association
between metformin use and reduced cancer risk. Hepatology.
57:648–655. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Patel T: Increasing incidence and
mortality of primary intrahepatic cholangiocarcinoma in the United
States. Hepatology. 33:1353–1357. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tyson GL and El-Serag HB: Risk factors for
cholangiocarcinoma. Hepatology. 54:173–184. 2011. View Article : Google Scholar
|
|
14
|
Khan SA, Thomas HC, Davidson BR and
Taylor-Robinson SD: Cholangiocarcinoma. Lancet. 366:1303–1314.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shaib YH, Davila JA, McGlynn K and
El-Serag HB: Rising incidence of intrahepatic cholangiocarcinoma in
the United States: a true increase? J Hepatol. 40:472–477. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chaudhary SC, Kurundkar D, Elmets CA,
Kopelovich L and Athar M: Metformin, an antidiabetic agent reduces
growth of cutaneous squamous cell carcinoma by targeting mTOR
signaling pathway. Photochem Photobiol. 88:1149–1156. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Luo Q, Hu D, Hu S, Yan M, Sun Z and Chen
F: In vitro and in vivo anti-tumor effect of metformin as a novel
therapeutic agent in human oral squamous cell carcinoma. BMC
Cancer. 12:5172012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin B, Kolluri SK, Lin F, et al:
Conversion of Bcl-2 from protector to killer by interaction with
nuclear orphan receptor Nur77/TR3. Cell. 116:527–540. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee D, Do IG, Choi K, et al: The
expression of phospho-AKT1 and phospho-MTOR is associated with a
favorable prognosis independent of PTEN expression in intrahepatic
cholangiocarcinomas. Mod Pathol. 25:131–139. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chung JY, Hong SM, Choi BY, Cho H, Yu E
and Hewitt SM: The expression of phospho-AKT, phospho-mTOR, and
PTEN in extrahepatic cholangiocarcinoma. Clin Cancer Res.
15:660–667. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu B, Fan Z, Edgerton SM, et al:
Metformin induces unique biological and molecular responses in
triple negative breast cancer cells. Cell Cycle. 8:2031–2040. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McCubrey JA, Steelman LS, Chappell WH, et
al: Roles of the Raf/MEK/ERK pathway in cell growth, malignant
transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
McCubrey JA, Steelman LS, Chappell WH, et
al: Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how
mutations can result in therapy resistance and how to overcome
resistance. Oncotarget. 3:1068–1111. 2012.PubMed/NCBI
|
|
24
|
McCubrey JA, Steelman LS, Chappell WH, et
al: Mutations and deregulation of Ras/Raf/MEK/ERK and
PI3K/PTEN/Akt/mTOR cascades which alter therapy response.
Oncotarget. 3:954–987. 2012.PubMed/NCBI
|
|
25
|
Takiyama Y, Harumi T, Watanabe J, et al:
Tubular injury in a rat model of type 2 diabetes is prevented by
metformin: a possible role of HIF-1alpha expression and oxygen
metabolism. Diabetes. 60:981–992. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ece H, Cigdem E, Yuksel K, Ahmet D, Hakan
E and Oktay TM: Use of oral antidiabetic drugs (metformin and
pioglitazone) in diabetic patients with breast cancer: how does it
affect serum Hif-1 alpha and 8Ohdg levels? Asian Pac J Cancer Prev.
13:5143–5148. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zannella VE, Pra AD, Muaddi H, et al:
Reprogramming metabolism with metformin improves tumor oxygenation
and radiotherapy response. Clin Cancer Res. 19:6741–6750. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sheng B, Liu J and Li GH: Metformin
preconditioning protects Daphnia pulex from lethal hypoxic
insult involving AMPK, HIF and mTOR signaling. Comp Biochem Physiol
B Biochem Mol Biol. 163:51–58. 2012.PubMed/NCBI
|
|
29
|
Li Y and Ye D: Cancer therapy by targeting
hypoxia-inducible factor-1. Curr Cancer Drug Targets. 10:782–796.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rho JK, Choi YJ, Lee JK, et al: Gefitinib
circumvents hypoxia-induced drug resistance by the modulation of
HIF-1α. Oncol Rep. 21:801–807. 2009.PubMed/NCBI
|
|
31
|
Huang C, Xu D, Xia Q, Wang P, Rong C and
Su Y: Reversal of P-glycoprotein-mediated multidrug resistance of
human hepatic cancer cells by Astragaloside II. J Pharm Pharmacol.
64:1741–1750. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu H, Luo SF, Wang J, et al: Effect of
environmental factors on chemoresistance of HepG2 cells by
regulating hypoxia-inducible factor-1alpha. Chin Med J (Engl).
125:1095–1103. 2012.PubMed/NCBI
|
|
33
|
Ding Z, Yang L, Xie X, et al: Expression
and significance of hypoxia-inducible factor-1 alpha and
MDR1/P-glycoprotein in human colon carcinoma tissue and cells. J
Cancer Res Clin Oncol. 136:1697–1707. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li DW, Dong P, Wang F, Chen XW, Xu CZ and
Zhou L: Hypoxia induced multidrug resistance of laryngeal cancer
cells via hypoxia-inducible factor-1alpha. Asian Pac J Cancer Prev.
14:4853–4858. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen M, Huang SL, Zhang XQ, et al:
Reversal effects of pantoprazole on multidrug resistance in human
gastric adenocarcinoma cells by down-regulating the
V-ATPases/mTOR/HIF-1alpha/P-gp and MRP1 signaling pathway in vitro
and in vivo. J Cell Biochem. 113:2474–2487. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Woo HY and Heo J: Sorafenib in liver
cancer. Expert Opin Pharmacother. 13:1059–1067. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Eguchi R, Fujimori Y, Takeda H, et al:
Arsenic trioxide induces apoptosis through JNK and ERK in human
mesothelioma cells. J Cell Physiol. 226:762–768. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Petit A, Delaune A, Falluel-Morel A, et
al: Importance of ERK activation in
As2O3-induced differentiation and
promyelocytic leukemia nuclear bodies formation in neuroblastoma
cells. Pharmacol Res. 77:11–21. 2013.
|