Introduction
Mesenchymal chondrosarcomas are rare tumors that
account for 2–10% of primary chondrosarcomas (1). Their typical histological appearance
includes a biphasic pattern with areas of round, primitive
mesenchymal cells and interspersed islands of well differentiated
hyaline cartilage (2). They are two
to three times more common in bone than in soft tissue and are
mostly found in the head and neck area, particularly the orbit, the
cranial and spinal dura mater, and the lower extremities,
especially the thigh (2). However,
rare cases of mesenchymal chondrosarcoma have been described in
virtually every anatomic site. Unlike other types of
chondrosarcoma, mesenchymal chondrosarcomas grow fast and often
give rise to local recurrences and metastases. The majority of the
cases are diagnosed in the second and third decade of life and the
prognosis is highly variable with published 10-year overall
survival rates ranging from 21% to 67% (1). Moreover, some patients live for long
periods with metastatic disease, whereas others die shortly after
diagnosis (1). Adequate surgery is
the treatment of choice for localized disease (3). The role of chemotherapy and
radiotherapy remains poorly defined (4,5).
To date, only 12 mesenchymal chondrosarcomas have
been karyotyped (6–15) and no consistent aberration pattern
has been established. Recently, however, two fusion genes were
reported in mesenchymal chondrosarcomas. Wang et al used a
genome-wide exon-resolution expression screen to identify a fusion
between the hairy/enhancer-of-split related with YRPW motif 1
(HEY1; on 8q21.13) gene and the nuclear receptor coactivator
2 (NCOA2; on 8q13.3) gene; no karyotypic data were available
on the tumors thus examined (16).
Nyquist et al used karyotyping followed by RNA-Seq to
identify an IRF2BP2-CDX1 fusion gene in a case of
mesenchymal chondrosarcoma carrying a solitary t(1;5)(q42;q32)
chromosomal translocation (12).
Here, we present a mesenchymal chondrosarcoma which proved to have
an informative karyotype and which, by RT-PCR, was found to carry a
HEY1-NCOA2 fusion gene.
Materials and methods
Ethics statement
The study was approved by the regional ethics
committee (Regional komité for medisinsk forskningsetikk Sør-Øst,
Norge, http://helseforskning.etikkom.no) and written informed
consent was obtained from the patient.
Case report - pathology
The patient was a 26-year-old woman who had noticed
a tumor in the right thigh three months prior to diagnosis.
Radiologic evaluation revealed a 9.0×8.0×5.4 cm large tumor in the
large adductor muscle. A soft tissue lesion, presumed to be a
metastasis, was detected in the neck, and another lesion of unknown
origin was found in the left iliac bone. The patient received four
cycles of chemotherapy; two cycles of vincristine, doxorubicin and
cyclophosphamide, one cycle of vincristine, ifosfamide and
actinomycin D, and one cycle of etoposide and ifosfamide.
Radiologic evaluation after chemotherapy revealed no significant
change in size of any of the tumors. A wide resection of the
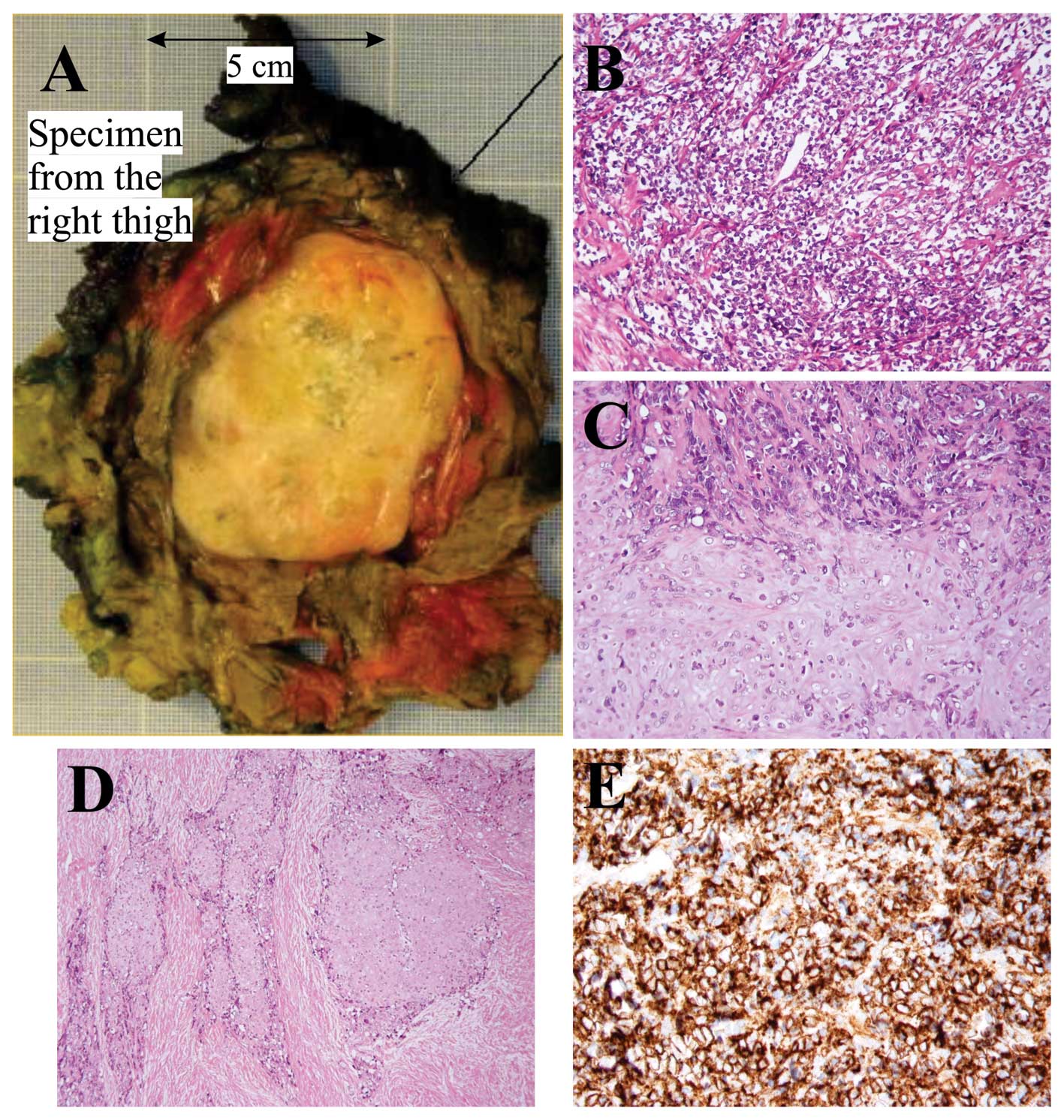
primary tumor (thigh) was performed (Fig. 1A). Radiotherapy and subsequent
surgery of the metastasis in the neck is planned.
Microscopic examination of the specimen from the
thigh showed a biphasic tumor with cellular areas with high-grade,
malignant-looking, small undifferentiated round cells and some more
pleomorphic cells (Fig. 1B)
alternating with cartilage of hyaline type consistent with a
low-grade malignant chondrosarcoma (Fig. 1C). The transition between the two
components was mostly abrupt. There was bone formation close to the
chondroid areas. A desmoplastic stroma was seen both around the
small round cells and the chondroid areas (Fig. 1D). Immunohistochemical analysis
showed a strong positive reaction to the antibody CD99 in the small
round cells (Fig. 1E). Microscopic
examination of the lesions in the neck and iliac bone showed tumor
tissue consistent with mesenchymal chondrosarcoma.
Karyotyping
Tumor samples from both the neck and thigh were
removed by core needle biopsy. The samples were mechanically and
enzymatically disaggregated and then short-term cultured as
described elsewhere (17). The
cultures were harvested and the chromosomes G-banded using Wright
stain. The subsequent cytogenetic analysis and karyotype
description followed the recommendations of the ISCN (18).
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was extracted using TRIzol reagent from
the core needle biopsy of the tumor of the thigh. No material was
available from the tumor of the neck. Then, 1 μg of total RNA was
reverse-transcribed in a 20 μl reaction volume using iScript
Advanced cDNA Synthesis Kit for RT-qPCR according to the
manufacturer’s instructions (Bio-Rad). The cDNA was diluted to 50
μl and 2 μl (corresponding to 40 ng of total RNA) were used as
template in subsequent PCR assays. As a positive control, a
mesenchymal chondrosarcoma known to carry the HEY1-NCOA2
fusion was used (12). The 25 μl
PCR-volume contained 12.5 μl of Premix Taq (Takara Bio Europe/SAS,
Saint-Germain-en-Laye, France), 2 μl of diluted cDNA, and 0.2 μM of
each of the forward HEY1-F1 (CGAGGTGGAGAAGGAGAGTG) and reverse
NCOA2-E13-R3 (AGTTGGGCTTTGCAATGTGA) primers. Both samples were
tested for expression of the HEY1 gene to assess the quality
of RNA and cDNA synthesis. The PCR components were the same as
above except that the primer combination HEY1-F1 and HEY1-551R
(CTCCGATAGTCCATAGCAAGG) was used. The PCRs were run on a C-1000
Thermal Cycler (Bio-Rad) using the following cycling conditions: an
initial denaturation at 94°C for 30 sec followed by 35 cycles of 7
sec at 98°C, 30 sec at 55°C and 2 min at 68°C, and a final
extension for 5 min at 68°C.
Four microliters of the PCR products were stained
with GelRed (Biotium, Hayward, CA, USA), analyzed by
electrophoresis through 1.0% agarose gel, and photographed. The
amplified fragment was purified using the Qiagen gel extraction kit
(Qiagen). Direct (Sanger) sequencing was performed using the light
run sequencing service of GATC Biotech (http://www.gatc-biotech.com/en/sanger-services/lightrun-sequencing.html).
The BLAST software (http://www.ncbi.nlm.nih.gov/BLAST/) was used for
computer analysis of sequence data.
Results
The G-banding analysis of cells cultured from the
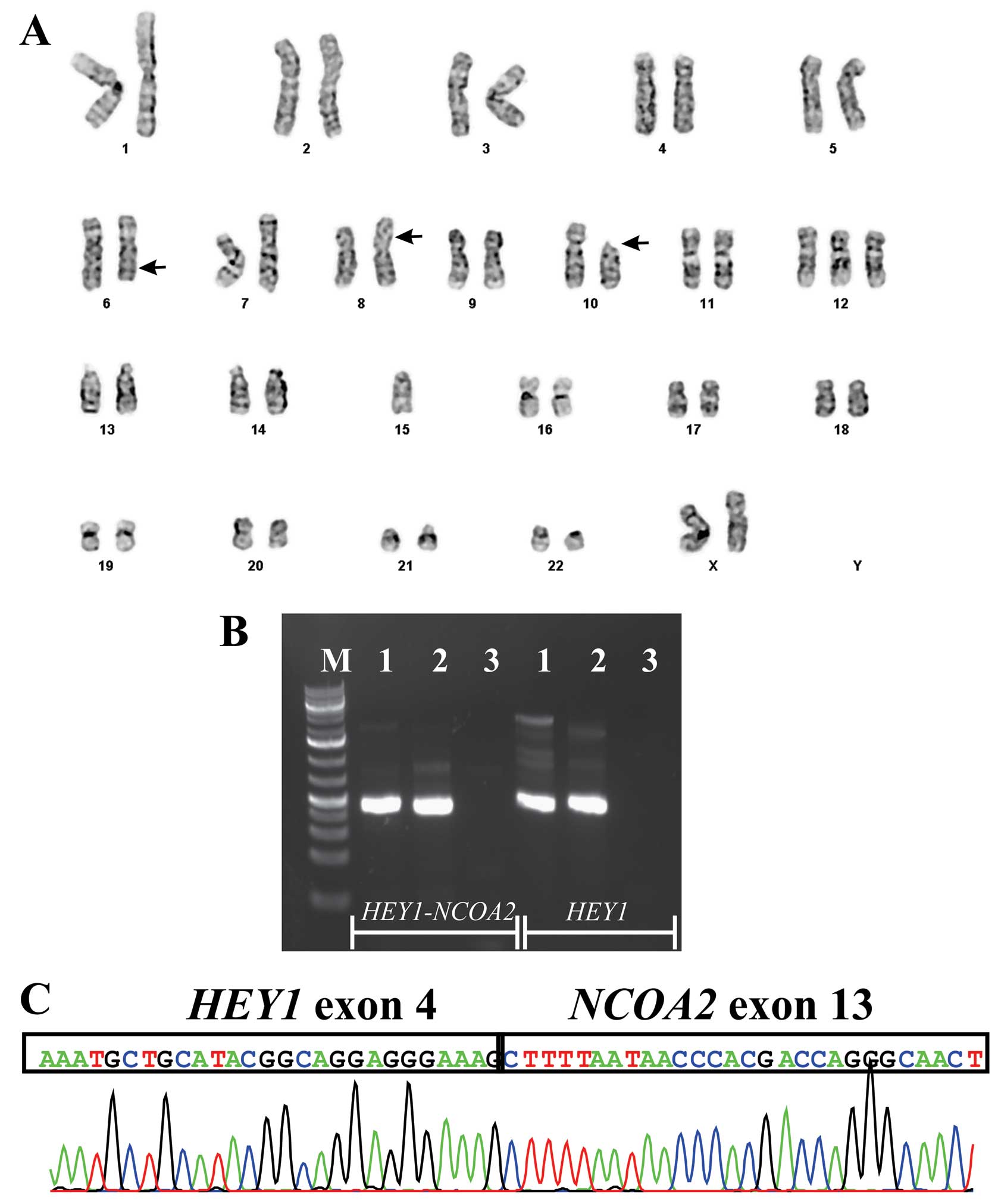
tumor of the neck yielded the karyotype 46,XX,add(6)(q23),add(8)(p23),del(10)(p11),+12, −15[6]/46,XX[5] (Fig. 2A), whereas a normal karyotype,
46,XX, was found in the tumor of the thigh.
RT-PCR with the HEY1-F1/NCOA2-E13-R3 primer
combination amplified a single cDNA fragment in both the tumor of
the thigh and the positive control (Fig. 2B). Sequencing of the amplified
fragment showed that exon 4 of HEY1(nt 531 in sequence with
accession number NM_012258 version 3) was fused to exon 13 of NCOA2
(nt 2768 in sequence with accession number NM_006540 version 2)
(Fig. 2C). Normal HEY1cDNA
fragments were amplified in both cases (Fig. 2B).
Discussion
Despite being a recognized entity for more than 50
years, mesenchymal chondrosarcoma continues to present substantial
diagnostic, prognostic and management challenges, due, in large
part, to its rarity (1). The
cytogenetic information is restricted to 12 cases with variable
karyotypes and no consistent aberration pattern has been
established. Dobin et al reported an intrathoracal
mesenchymal chondrosarcoma with a near-tetraploid karyotype which
included structural chromosomal abnormalities such as add(7)(p13), add(22)(q13), markers and double minutes
(7). Gatter et al reported
trisomy 8 as the sole cytogenetic abnormality (9), while three other reports described
different chromosomal translocations, t(4;19)(q35;q13),
t(6;10)(p21;q22), and t(1;5)(q42;q32), as the sole cytogenetic
abnormality (6,12,13).
Nevertheless, examining the karyotypes as part of this study we
observed that involvement of chromosome 8 was found in as many as 7
out of the 12 karyotyped mesenchymal chondrosarcomas with
aberrations: +8 was found in three cases (7,9,14), −8
was found in two cases (8,10), whereas structural aberrations of 8q
were found in three cases (in one case together with −8) (8,11).
Chromosome 8 was also structurally affected in the present study.
The pathogenetic mechanisms behind this nonrandom involvement are
unknown, but the presence on 8q of two genes, HEY1 and
NCOA2, now known to be involved in mesenchymal
chondrosarcoma tumorigenesis is, of course, suggestive.
Recently, Wang et al identified a
HEY1-NCOA2 fusion in mesenchymal chondrosarcomas. Using a
combination of FISH and RT-PCR methodologies they found that 10 out
of 15 examined mesenchymal chondrosarcomas carried the
HEY1-NCOA2 (16). The
findings were verified in two other studies: Nyquist et al
(12) showed by RT-PCR the presence
of HEY1-NCOA2 in 3 of 4 mesenchymal chondrosarcomas, and
Nakayama et al (19) found
HEY1-NCOA2 in 8 of 10 mesenchymal chondrosarcomas using FISH
on formalin-fixed and paraffin-embedded samples. The absence of
HEY1-NCOA2 fusion in some cases has been explained as being
due to methodological inadequacy (16,19)
but the possibility of other disease-specific fusion gene(s), and
thus pathogenetic heterogeneity in this diagnostic entity, should
not be ruled out. As demonstrated by Nyquist et al, the
chromosomal translocation t(1;5)(q42;q32) resulted in fusion of
IRF2BP2 (located on 1q42) with CDX1 (on 5q32) to
generate an IRF2BP2-CDX1 fusion gene in the mesenchymal
chondrosarcoma they studied (12).
In spite of this, the HEY1-NCOA2 fusion does seem to be
common as well as specific for mesenchymal chondrosarcomas since it
was not found in conventional and dedifferentiated chondrosarcomas
(16). Both genes are located on
the long arm of chromosome 8, HEY1 in 8q21.13 and
NCOA2 in 8q13.3, therefore the fusion may result from an
interstitial deletion (16) or a
t(8;8)(q13;q21) chromosomal translocation.
The involvement of the NCOA2 gene in
neoplasia was first reported by Carapeti et al who showed
that in acute myeloid leukemia the cytogenetic aberration
inv(8)(p11q13) resulted in a
KAT6A-NCOA2, also known as MOZ-TIF2, fusion gene
(20,21). Since then, the gene has also been
implicated in various other malignancies. Strehl et al
identified a novel recurrent t(8;12)(q13;p13) resulting in a fusion
between the transcriptional repressor ETV6 (TEL) and
NCOA2 in six cases of childhood leukemia expressing both
T-lymphoid and myeloid antigens (22). A PAX3-NCOA2 gene was found as
a rare variant fusion in alveolar rhabdomyosarcoma; it was brought
about by a t(2;8)(q35;q13) translocation (23). The AHRR-NCOA2 and
GTF2I-NCOA2 fusion genes were described in soft tissue
angiofibroma through translocations t(5;8)(p15;q13) and
t(7;8)(q11;q13), respectively, emphasizing the role of NCOA2
in soft tissue angiofibroma development (24,25).
Recently, SRF-NCOA2 and TEAD1-NCOA2 fusions were
reported in rhabdomyosarcomas (26). In all the above mentioned fusions,
NCOA2 is the 3′-partner gene and all fusion proteins contain
the two C-terminal activation domains AD1/CID (activation domain
1/CREB binding protein interacting domain) and AD2 (20–26).
The transforming activities of KAT6A-NCOA2 and
PAX3-NCOA2 have been demonstrated experimentally (23,27).
In addition, KAT6A-NCOA2 was shown to induce acute myeloid leukemia
in transgenic fish (28). Deguchi
et al (27) showed that the
KAT6A-NCOA2 interaction with CREBBP through AD1/CID is essential
for transformation. Similarly, Sumegi et al (23) showed that while deletion of the AD2
portion of PAX3-NCOA2 fusion protein reduced transforming activity,
deletion of the AD1/CID domain fully abrogated the transforming
activity of the chimeric protein. Thus, the presence of the AD1/CID
and AD2 domains of NCOA2 seems to be essential for the
transformation capacity of the various cancer fusion genes.
HEY1 encodes a nuclear protein belonging to
the hairy and enhancer of split-related (HESR) family of basic
helix-loop-helix (bHLH)-type transcriptional repressors (29). Expression of this gene is induced by
the Notch and c-Jun signal transduction pathways (30). HEY1 protein binds to specific DNA
sequences in the promoter regions of target genes as a dimer,
recruiting co-repressors to repress the target genes of Notch
signaling. The HEY1-NCOA2 fusion replaces the C-terminal portion of
HEY1 by the NCOA2 AD1/CID and AD2 domains, while retaining the HEY1
bHLH DNA-binding/dimerization domain. Therefore, the HEY1-NCOA2
fusion protein, instead of recruiting co-repressors, may recruit
co-activators through its NCOA2 part to some Notch/HEY1 target
genes (16). Additional experiments
are required to confirm or falsify the validity of this
hypothesis.
Acknowledgements
The authors thank Saeedeh Shahmohammadi for the
technical help. This work was supported by grants from the
Norwegian Cancer Society and The South-East Norway Regional Health
Authority.
References
|
1
|
Shakked RJ, Geller DS, Gorlick R and
Dorfman HD: Mesenchymal chondrosarcoma: clinicopathologic study of
20 cases. Arch Pathol Lab Med. 136:61–75. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weiss SW and Goldblum JR: Enzinger and
Weiss’s Soft Tissue Tumors. Mosby; St Louis, MO: 2001
|
|
3
|
Riedel RF, Larrier N, Dodd L, Kirsch D,
Martinez S and Brigman BE: The clinical management of
chondrosarcoma. Curr Treat Options Oncol. 10:94–106. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cesari M, Bertoni F, Bacchini P, Mercuri
M, Palmerini E and Ferrari S: Mesenchymal chondrosarcoma. An
analysis of patients treated at a single institution. Tumori.
93:423–427. 2007.PubMed/NCBI
|
|
5
|
Dantonello TM, Int-Veen C, Leuschner I, et
al: Mesenchymal chondrosarcoma of soft tissues and bone in
children, adolescents, and young adults: experiences of the CWS and
COSS study groups. Cancer. 112:2424–2431. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Crosswell H, Buchino JJ, Sweetman R and
Reisner A: Intracranial mesenchymal chondrosarcoma in an infant.
Med Pediatr Oncol. 34:370–374. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dobin SM, Donner LR and Speights VO Jr:
Mesenchymal chondrosarcoma. A cytogenetic, immunohistochemical and
ultrastructural study. Cancer Genet Cytogenet. 83:56–60. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fletcher CD, Dal Cin P, de Wever I, et al:
Correlation between clinicopathological features and karyotype in
spindle cell sarcomas. A report of 130 cases from the CHAMP study
group. Am J Pathol. 154:1841–1847. 1999. View Article : Google Scholar
|
|
9
|
Gatter KM, Olson S, Lawce H and Rader AE:
Trisomy 8 as the sole cytogenetic abnormality in a case of
extraskeletal mesenchymal chondrosarcoma. Cancer Genet Cytogenet.
159:151–154. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mandahl N, Gustafson P, Mertens F, et al:
Cytogenetic aberrations and their prognostic impact in
chondrosarcoma. Genes Chromosomes Cancer. 33:188–200. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Naumann S, Krallman PA, Unni KK, Fidler
ME, Neff JR and Bridge JA: Translocation der(13;21)(q10;q10) in
skeletal and extraskeletal mesenchymal chondrosarcoma. Mod Pathol.
15:572–576. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nyquist KB, Panagopoulos I, Thorsen J, et
al: Whole-transcriptome sequencing identifies novel IRF2BP2-CDX1
fusion gene brought about by translocation t(1;5)(q42;q32) in
mesenchymal chondrosarcoma. PLoS One. 7:e497052012. View Article : Google Scholar
|
|
13
|
Richkind KE, Romansky SG and Finklestein
JZ: t(4;19)(q35;q13.1): a recurrent change in primitive mesenchymal
tumors? Cancer Genet Cytogenet. 87:71–74. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sainati L, Scapinello A, Montaldi A, et
al: A mesenchymal chondrosarcoma of a child with the reciprocal
translocation (11;22)(q24;q12). Cancer Genet Cytogenet. 71:144–147.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Szymanska J, Tarkkanen M, Wiklund T, et
al: Cytogenetic study of extraskeletal mesenchymal chondrosarcoma.
A case report. Cancer Genet Cytogenet. 86:170–173. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang L, Motoi T, Khanin R, et al:
Identification of a novel, recurrent HEY1-NCOA2 fusion in
mesenchymal chondrosarcoma based on a genome-wide screen of
exon-level expression data. Genes Chromosomes Cancer. 51:127–139.
2012. View Article : Google Scholar
|
|
17
|
Mandahl N: Methods in solid tumour
cytogenetics. Human Cytogenetics: Malignancy and Acquired
Abnormalities. Rooney DE: Oxford University Press; New York: pp.
165–203. 2001
|
|
18
|
Schaffer LG, Slovak ML and Campbell LJ:
ISCN 2009: An International System for Human Cytogenetic
Nomenclature. Karger; Basel: 2009
|
|
19
|
Nakayama R, Miura Y, Ogino J, et al:
Detection of HEY1-NCOA2 fusion by fluorescence in-situ
hybridization in formalin-fixed paraffin-embedded tissues as a
possible diagnostic tool for mesenchymal chondrosarcoma. Pathol
Int. 62:823–826. 2012. View Article : Google Scholar
|
|
20
|
Carapeti M, Aguiar RC, Goldman JM and
Cross NC: A novel fusion between MOZ and the nuclear receptor
coactivator TIF2 in acute myeloid leukemia. Blood. 91:3127–3133.
1998.PubMed/NCBI
|
|
21
|
Carapeti M, Aguiar RC, Watmore AE, Goldman
JM and Cross NC: Consistent fusion of MOZ and TIF2 in AML with
inv(8)(p11q13). Cancer Genet Cytogenet. 113:70–72. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Strehl S, Nebral K, Konig M, et al:
ETV6-NCOA2: a novel fusion gene in acute leukemia associated with
coexpression of T-lymphoid and myeloid markers and frequent NOTCH1
mutations. Clin Cancer Res. 14:977–983. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sumegi J, Streblow R, Frayer RW, et al:
Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma
without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear
receptor transcriptional coactivator family. Genes Chromosomes
Cancer. 49:224–236. 2010.
|
|
24
|
Arbajian E, Magnusson L, Mertens F,
Domanski HA, Vult von Steyern F and Nord KH: A novel GTF2I/NCOA2
fusion gene emphasizes the role of NCOA2 in soft tissue
angiofibroma development. Genes Chromosomes Cancer. 52:330–331.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jin Y, Möller E, Nord KH, et al: Fusion of
the AHRR and NCOA2 genes through a recurrent translocation
t(5;8)(p15;q13) in soft tissue angiofibroma results in upregulation
of aryl hydrocarbon receptor target genes. Genes Chromosomes
Cancer. 51:510–520. 2012. View Article : Google Scholar
|
|
26
|
Mosquera JM, Sboner A, Zhang L, et al:
Recurrent NCOA2 gene rearrangements in congenital/infantile spindle
cell rhabdomyosarcoma. Genes Chromosomes Cancer. 52:538–550. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Deguchi K, Ayton PM, Carapeti M, et al:
MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome
binding motif and TIF2-mediated recruitment of CBP. Cancer Cell.
3:259–271. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhuravleva J, Paggetti J, Martin L, et al:
MOZ/TIF2-induced acute myeloid leukaemia in transgenic fish. Br J
Haematol. 143:378–382. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Steidl C, Leimeister C, Klamt B, et al:
Characterization of the human and mouse HEY1, HEY2, and HEYL genes:
cloning, mapping, and mutation screening of a new bHLH gene family.
Genomics. 66:195–203. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Maier MM and Gessler M: Comparative
analysis of the human and mouse Hey1 promoter: Hey genes are new
Notch target genes. Biochem Biophys Res Commun. 275:652–660. 2000.
View Article : Google Scholar : PubMed/NCBI
|