Introduction
Chronic inflammation and inflammatory responses are
considered to play crucial roles in the development of cancer
(1). Inflammatory bowel diseases
(IBDs), characterized by immune deregulation and autoimmunity, are
recognized to have a high risk for the development of
colitis-associated colorectal cancer (CAC) (2,3).
Animal models of IBDs have presented important clues to the
decisive roles of inflammatory mediators and molecular events
leading to the development of CAC (4). Within this population, the variability
in the cancer risk is complex. Although genetic variability plays
an important role in the malignant transformation of IBDs (5), the long duration of the disease, as
well as the severity and extent of the inflammation are considered
to be the most important risk factors, particularly the severity of
inflammation (6). However, changes
at the molecular level under different degrees of inflammation in
colitis and CAC still remain puzzling.
mTOR, a serine/threonine kinase that regulates cell
cycle progression, cellular proliferation and growth, autophagy and
angiogenesis, is often deregulated in human cancers (7,8).
Functioning as the protein kinase of the mammalian target of
rapamycin complex 1 (mTORC1), mTOR regulates the regulators of
eukaryotic translation initiation factor 4E binding protein 1
(4EBP1) and the p70 ribosomal protein S6 kinase 1 (S6K1) (9,10).
Activation of mTORC1 actuates protein synthesis via activation of
S6K1, which phosphorylates the ribosomal protein S6 and initiates
translation (11). S6K1, a member
of the AGC kinase family, plays important roles in cell growth,
proliferation and cell differentiation via a wide range of
extracellular signals including growth factors, hormones, nutrients
and stress (12,13). Rapamycin and its analogs are likely
to be the first mTOR-perturbing drugs for therapeutic use in human
cancer (14). Rapamycin is the
universal inhibitor of mTORC1 and S6K1 and is a cell-type specific
inhibitor of mTORC2 and Akt signaling (15). Various tumor cells have been
effectively treated with rapamycin, including rhabdomyosarcoma,
osteosarcoma, pancreatic carcinoma, RCC, Ewing sarcoma, brain,
lung, prostate and breast cancer (16). Intriguingly, Thiem et al
found that coactivation of mTOR and STAT3 signaling occurs in
gastric tumors in humans. They demonstrated that mTORC1 is
activated via GP130 in a STAT3- and STAT1-independent manner and
induces inflammation-associated gastrointestinal tumorigenesis
(17). Generally, excessive
activation of STAT3 has been identified as a hallmark of
inflammation-associated cancers (18). STAT3 activation commonly contributes
to an abundance of tumor and stromal cell-derived cytokines, such
as IL-6 and IL-11 (19). It has
been reported that IL-6-dependent persistent activity of STAT3 is
required to maintain the survival capacity of intestinal epithelial
cells and the development of colitis-associated cancer (19). However, the relationship between the
activation of STAT3 and mTORC1 in colitis and CAC is not yet
clear.
In the present study, we employed the colonotropic
mutagen azoxymethane (AOM) and different concentrations of dextran
sulfate sodium salt (DSS) to produce different degrees of
inflammation in the CAC mouse model. We determined the activity of
mTOR and STAT3 and compared the efficiency of tumor formation at
the different inflammation levels. We found that as the
inflammation was aggravated, the activity of STAT3 and mTOR was
elevated. Coactivation of STAT3 and mTOR signaling occurred with
increased inflammation and persisted until tumor development. We
also demonstrated that rapamycin did not only inhibit mTOR
signaling but also STAT3 signaling in colitis and suppressed
inflammation-associated colon cancer.
Materials and methods
Animals
All female mice (C57BL/6) used were 7–8 weeks of
age, with body weight 18–20 g. They were housed in pathogen-free
rooms in filter-topped cages at the Laboratory Animal Facility at
Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou.
All mice were quarantined for 3 days after arriving. They were
maintained in a room with controlled temperature (22±1°C), humidity
(50–70%) and a 12-h light/dark cycle. All animal experiments were
approved by the Ethics Committee for Animal Care and Use of
Zhongshan School of Medicine, Sun Yat-sen University according to
an approved protocol.
AOM/DSS experimental inflammation-induced
CAC model
We followed a modified protocol outlined recently by
Chumanevich et al (20). The
CAC model was induced by AOM (Sigma-Aldrich, St. Louis, MO, USA)
and DSS (MP Biomedicals, Solon, OH, USA). Briefly, female mice were
injected intraperitoneally with a single dose of AOM (10 mg/kg),
and the mice received a course of 1–2% DSS in sterile drinking
water for 7 days followed by pure drinking water for 14 days in a
total of 3 cycles. Assessment of AOM/DSS-treated mice was performed
daily for general appearance, food uptake, body weight, stool
consistency and rectal bleeding. In the control group, the same
procedure was performed with an intraperitoneal injection of AOM
(10 mg/kg) and drinking pure water instead of DSS. A scheme of the
treatment protocol is shown in Fig.
1.
Before AOM injection, the mice were randomized into
control and experimental groups (20 animals per group). Rapamycin
(LC Laboratories, Woburn, MA, USA) was dissolved in ethanol and
stored at −80°C. For dosing of animals, the stock solution was
diluted with aqueous solution to give final concentrations of 4%
ethanol, 5% polyethylene glycol 400 and 5% Tween-80 immediately
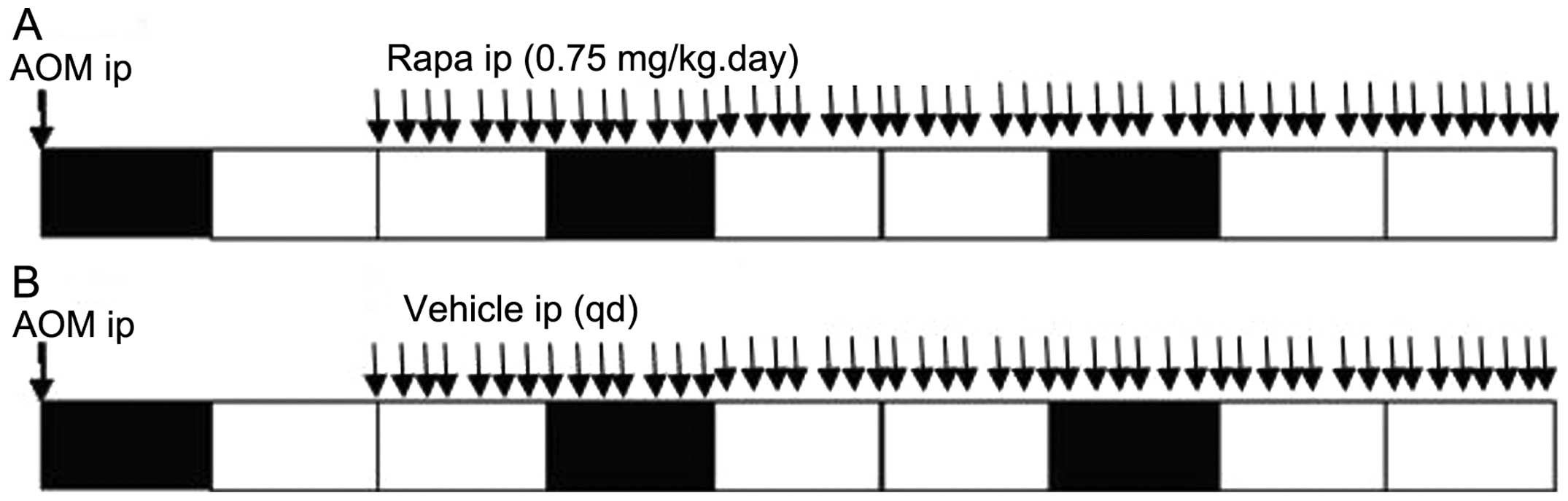
before intraperitoneal injections (21). After 14 days of AOM injection, mice
in the experimental group received 0.75 mg/kg rapamycin every day
and the control animals received the drug vehicle only according to
the same dosing schedule. The mice were sacrificed 63 days after
AOM injection. A scheme of the treatment protocol is shown in
Fig. 2. The mouse colons were
isolated, slit open longitudinally for tumor count and size
determination using a dissecting microscope. Tumors were excised
for RNA and protein isolation for gene expression analysis using
real-time PCR and western blot analysis, respectively. Non-tumorous
colonic epithelia were scraped from the middle and proximal
portions of the colon. Colons adjacent to tumors were fixed in 10%
fresh formalin, and sections were cut out to embed in paraffin for
immunohistochemical analysis.
mRNA isolation and quantitative
expression studies
Total RNA was isolated from the AOM/DSS + rapamycin
and AOM/DSS + vehicle groups using the AxyPrep™ Multisource Total
RNA Miniprep kit (Axygen, Union City, CA, USA) according to the
manufacturer’s suggested protocol. Total RNA (0.5 μg) was reverse
transcribed using ReverTra Ace® qPCR RT kit (Toyobo,
Osaka, Japan) in a 20-μl reaction volume with the GeneAmp PCR
System 9700 (Applied Biosystems, Foster City, CA, USA) and from
that, 2 μl of the reaction volume was added to Thunderbird SYBR
qPCR Mix (Toyobo) for quantitative real-time PCR using the 7500
Real-Time PCR System (Applied Biosystems). Primers used were as
follows: forward (GCCAGCCGATGGGTTGTAC) and reverse
(TTGACGGCAGAGAGGAGGTT) primers for the detection of TNF-α
expression; forward (TTACTGCCACGGCACAGTCA) and reverse
(TGCAGGATTTTCATGTCACCAT) primers for IFN-γ expression; forward
(AACCACGGCCTTCCCTACTT) and reverse (TCTTTTCTCATTTCCACGATTTCC)
primers for IL-6 expression; forward (CAGGGCCCTTTGCTATGGT) and
reverse (TCTGAGCTGCTGCAGGAATG) primers for IL-10 expression;
forward (GCCATCAACGCAGCACTTC) and reverse (TGCTTCTCCCACAGGAGGTT)
primers for IL-12α expression and actin was chosen as a
housekeeping gene with forward (ATGACCCAAGCCGAGAAGG) and reverse
(CGGCCAAGTCTTAGAGTTGTTG) primers. Expression of various genes was
normalized to the expression of actin.
Histological analysis of the colonic
tumors in the AOM/DSS-treated mice
Mouse colon tissues were fixed in 4%
paraformaldehyde, paraffin-embedded and sectioned at a 4-μm
thickness. The sections were stained with hematoxylin and eosin
(H&E) and microscopically examined for histopathologic and
inflammatory changes as described by Chumanevich et al
(20).
Immunohistochemistry and western blot
analysis
Standard immunohistochemistry and western blotting
techniques were used. The antibodies used were as follows: mTOR was
detected with polyclonal anti-rabbit mTOR antibody [Abcam (Hong
Kong) Ltd., HKSP, New Territories, Hong Kong]; phosphorylated
(p)-mTOR (S2448) was detected with polyclonal anti-rabbit p-mTOR
(S2448) antibody (Abcam); S6K was detected with monoclonal
anti-rabbit S6K antibody (Abcam); p-S6K (T389) was detected with
monoclonal anti-rabbit p-S6K (T389) antibody (Abcam); STAT3 was
detected with monoclonal anti-rabbit STAT3 antibody (Cell Signaling
Technology, Danvers, MA, USA); p-STAT3 (Tyr705) was detected with
monoclonal anti-rabbit p-STAT3 (Tyr705) antibody (Cell Signaling
Technology); GAPDH was detected with polyclonal anti-GAPDH (Santa
Cruz Biotechnology, Santa Cruz, CA, USA).
Statistical analysis
Data are expressed as the mean ± standard error of
the mean (SEM). All statistical analyses were carried out using an
unpaired t-test analysis in GraphPad Prism 4 software (GraphPad
Software, San Diego, CA, USA). Significance was defined by
P<0.05.
Results
Inflammation promotes the malignant
transformation of colitis in an experimental colitis-associated
colorectal cancer (CAC) model
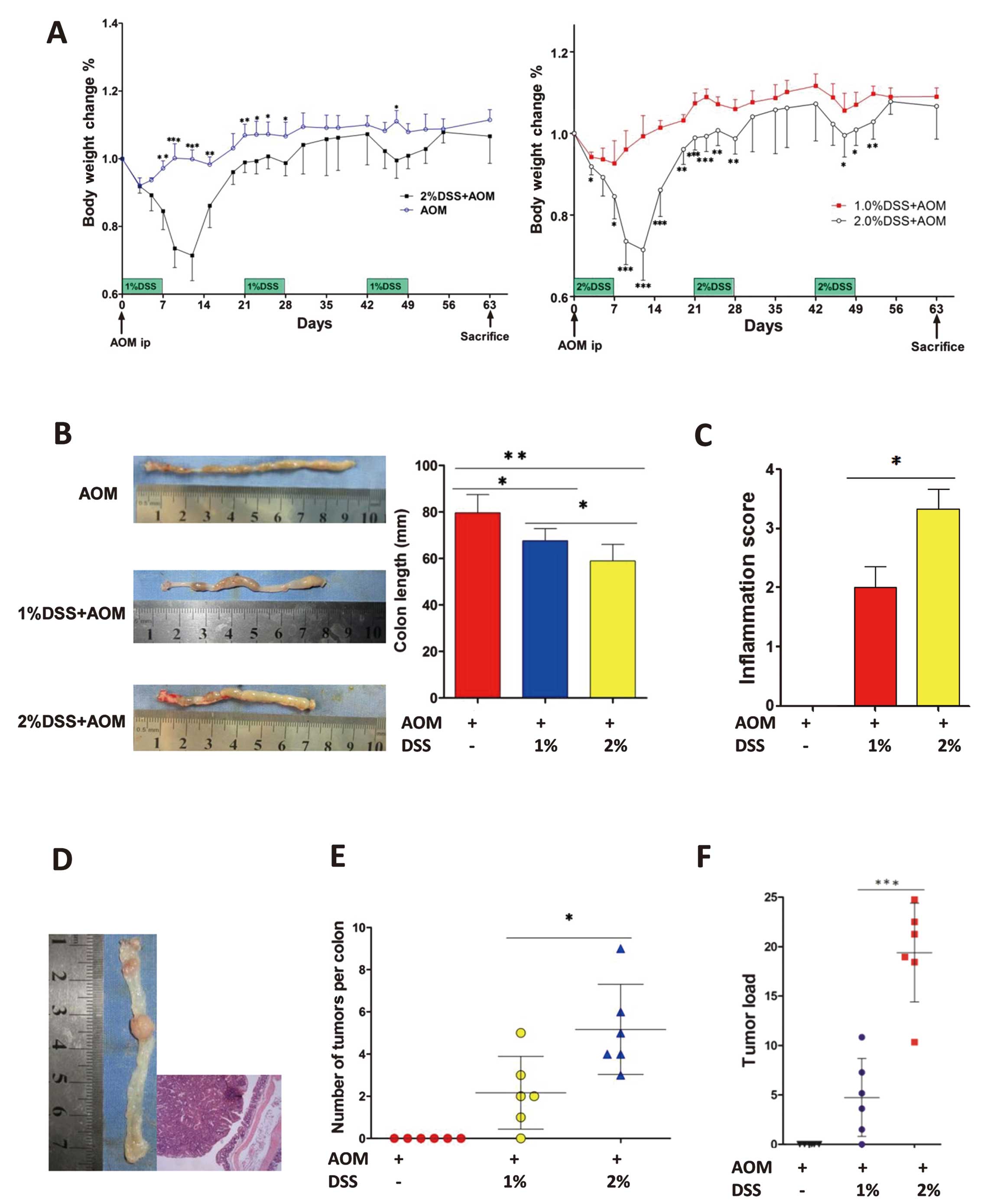
To imitate different levels of colonic inflammation,
the concentration of dextran sulfate sodium (DSS) in drinking water
was changed. We randomized mice into 3 groups (6 animals per group)
and administered axozymethane (AOM) only to the control group, 1%
DSS + AOM to the low inflammation group and 2% DSS + AOM to the
high inflammation group. This showed that a higher concentration of
DSS in drinking water could induce higher colonic inflammation. A
lower concentration of DSS (1% DSS + AOM) had less effect on body
weight loss when compared with the control group (AOM only); mice
in the high inflammation group (2% DSS + AOM) showed a higher body
weight loss when compared with the mice in the control group (AOM
only) and the low inflammation group (1% DSS + AOM) (Fig. 3A). The colon length was
significantly reduced in the high inflammation group than that in
the low inflammation group (Fig.
3B). Specifically, high inflammation shrank the colon to
58.9±7.1 mm (P=0.001 vs. AOM only) and lower inflammation shrank
the colon to 67.6±5.2 mm (P=0.01 vs. AOM only). The high
inflammation group had shorter colons than those of the low
inflammation group (P=0.044). In addition, the histopathological
inflammation score in the high inflammation group was significantly
higher than that in the low inflammation group (3.3±1.0 vs.
2.0±1.2, P=0.016) (Fig. 3C). Single
intraperitoneal injection of the mutagenic agent AOM and repeated
oral administration of the pro-inflammatory agent DSS could induce
the CAC model (Fig. 3D). The tumor
multiplicity of the high inflammation group was higher than that of
the low inflammation group (5.2±0.9 vs. 2.2±0.7, P=0.0232)
(Fig. 3E), and the tumor load (sum
of tumor diameters per mouse) in these two groups was also
significant (19.4±2.0 vs. 4.7±1.6, P=0.0002) (Fig. 3F).
STAT3 and mTORC1 pathways are highly
activated in colitis-associated cancer
It has been shown that the STAT3 and mTORC1 pathways
may play an important role in inflammation and carcinogenesis
(7,22). To illuminate the function of the
STAT3 and mTORC1 pathways in CAC, we determined the expression of
STAT3 and mTORC1 pathways in colonic tumors and inflammatory
colonic tissues adjacent to tumors. Phosphorylated (p)-STAT3
(Tyr705), the active phase of STAT3, in the high inflammation colon
tissue was expressed at a higher level than that in the low
inflammation tissues while the expression of total STAT3 was not
obviously altered (Fig. 4A).
Moreover, the expression of p-STAT3 in the colitis-associated
cancer was higher than that in the high inflammation tissues
(Fig. 4B), indicating that with the
development of inflammation, the activation of the STAT3 signaling
pathway increased gradually. p-mTOR (S2448) is one of the active
phases of mTOR and as one of the downstream molecules of the mTORC1
pathway, the active phase of S6K is phosphorylated-S6K (T389).
Fig. 4C–E shows that the expression
levels of p-mTOR (S2448) and p-S6K (T389) in the high inflammation
tissues were higher than these levels in the low inflammation
tissues. As shown in Fig. 4F–H, the
expression levels of p-mTOR (S2448) and p-S6K (T389) in
colitis-associated cancer were higher than these levels in the high
inflammation tissues. This indicated that with the development of
inflammation, the activation of the mTORC1 pathway increased
gradually similar to the STAT3 pathway.
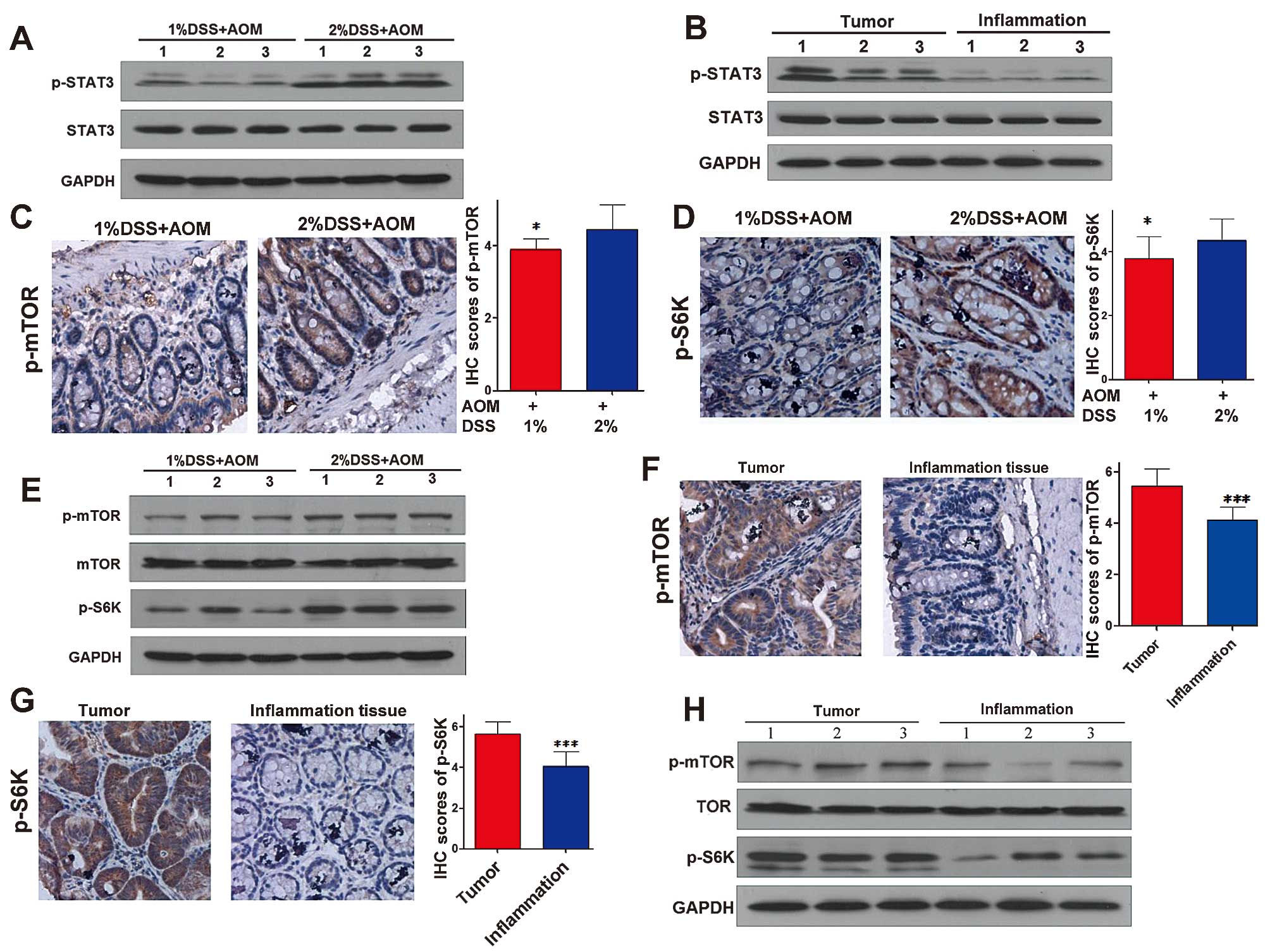 | Figure 4STAT3 and mTORC1 pathways are highly
activated in CAC. Immunohistochemical analysis of low inflammation
tissues, high inflammation tissues and tumors for phosphorylated
(p)-mTOR (S2448) and p-S6K (T389). Western blot analysis of low
inflammation tissues, high inflammation tissues and tumors for
STAT3, p-STAT3 (Tyr705), mTOR, p-mTOR (S2448), p-S6K (T389) and
glyceraldehyde 3-phosphate dehydrogenase (GAPDH) housekeeping gene.
(A and B) Expression of the STAT3 pathway in the low inflammation
tissues, high inflammation tissues and tumors. (C) Magnification,
×200, (D) magnification, ×200, and (E) expression of the mTORC1
pathway in the low inflammation tissue and higher inflammation
tissue. (F) Magnification, ×200, (G) magnification, ×200, and (H)
expression of the mTORC1 pathway in high inflammation tissues and
tumors. *P<0.05, **P<0.01,
***P<0.001. CAC, colitis-associated colorectal
cancer. |
mTORC1 inhibitor suppresses the malignant
transformation of colitis in mice
To further illuminate the role of the mTORC1 and
STAT3 pathways in CAC, we used an mTORC1 inhibitor, rapamycin, to
block the mTORC1 pathway in vivo. After 14 days of AOM
injection, mice in the experimental group were peritoneally
injected with 0.75 mg/kg rapamycin daily, and the control animals
received drug vehicle only according to the same dosing schedule
(20 animals per group) (Fig. 2).
After 63 days of AOM injection, all mice were sacrificed, and the
tumor incidence, multiplicity and tumor size were evaluated.
Rapamycin, the mTORC1 inhibitor, reduced the incidence of CAC to
40% (8/20), compared with 100% (20/20) in the control group
(vehicle + AOM/DSS) (P<0.001). The multiplicity of CAC in the
experimental group was significantly lower than that in the control
group (1.55±1.92 vs. 8.30±3.59, P<0.001) (Fig. 5A and B). In addition, the tumor load
(sum of tumor diameters per mouse) in these two groups was
significant (4.8±2.3 vs. 18.1±8.0, P<0.001) (Fig. 5C).
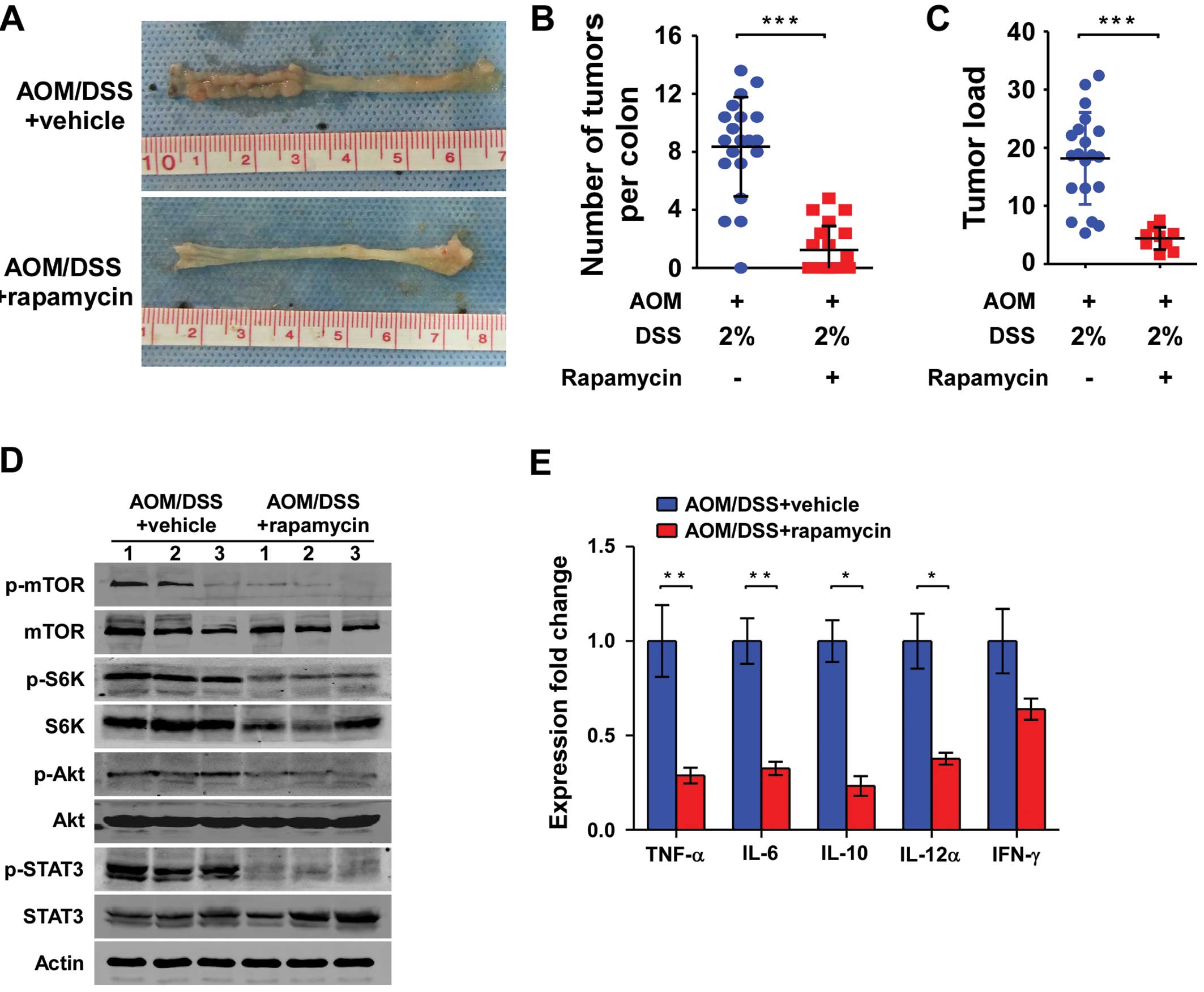 | Figure 5Rapamycin, an inhibitor of the mTORC1
pathway, inhibits the development of CAC and reduces the expression
of pro-inflammatory and anti-inflammatory chemokines. (A)
Macroscopic view of colons of CAC treated with rapamycin. (B and C)
Number of colonic tumors per animal (multiplicity) and tumor load
(sum of tumor diameters per mouse) per animal (n=20) following
treatment with rapamycin. (D) Western blot analysis of the colon
tissues for the STAT3 and mTORC1 pathways (n=3 per group). (E)
Effects of rapamycin on colonic TNF-α, IL-6, IL-12α, IL-10 and
INF-γ mRNA expression patterns. Both the experimental group
(AOM/DSS + rapamycin) and control group (AOM/DSS + vehicle) mice
were sacrificed on day 63 following 3 cycles of 2% DSS
administration. Following sacrifice, the expression levels of
TNF-α, IL-6, IL-12α, IL-10 and INF-γ were quantified by
TaqMan-qPCR, relative to the expression of the housekeeping gene
(actin). mRNA expression for each gene was quantified for each
individual mouse in triplets and averaged per mouse (n=7 per
group). Data represent mean ± SEM; *P<0.05,
**P<0.01, ***P<0.001. n=7 in each
group. CAC, colitis-associated colorectal cancer. |
Rapamycin suppresses the malignant
transformation through inhibition of the mTORC1 and STAT3
pathways
To confirm the effect of rapamycin on the mTORC1 and
STAT3 pathways, we detected the expression of mTORC1 and STAT3
pathways in colon tissues by western blotting. Previous research
showed that there exists crosstalk between the mTORC1 and STAT3
pathway in vitro (23). In
the present study, as shown in Fig.
5D, the mTORC1 inhibitor rapamycin reduced the expression of
p-STAT3 (Tyr705), which indicated that rapamycin inhibited the
activation of the STAT3 pathway in vivo. Moreover, rapamycin
also inhibited the activation of the mTORC1 pathway in vivo
(Fig. 5D).
Rapamycin reduces the expression of
pro-inflammatory and anti-inflammatory chemokines
As demonstrated above, inflammation is one of the
main processes driving tumor progression in CAC. Therefore, to
direct our mechanistic study of how rapamycin, an mTORC1 inhibitor,
reduced CAC, changes in colonic messenger RNA expression levels of
common pro-inflammatory and anti-inflammatory cytokines were
determined. Compared with mice in the control group, rapamycin
suppressed the colonic mRNA expression of pro-inflammatory
cytokines, such as tumor necrosis factor-α (TNF-α) (P=0.002), IL-6
(P<0.001) and IL-12α (P=0.01). Notably, rapamycin also
suppressed the colonic mRNA expression of anti-inflammatory
cytokine IL-10 (P<0.001). Furthermore, mRNA expression of
another anti-inflammatory cytokine, IFN-γ, in colonic tissues of
rapamycin-treated CAC mice tended to be reduced (P=0.179, Fig. 5E).
Discussion
Recent research has demonstrated that the risk
factors for the development of colorectal neoplasia in patients
with IBD include disease duration, anatomic extent of the disease,
age at onset of IBD, family history of sporadic CRC and severity of
inflammation (24). Among these
factors, severity of inflammation may be the most crucial and the
easiest to remedy. A cohort study showed that the severity of
microscopic inflammation over time is an independent risk factor
for developing advanced colorectal neoplasia among patients with
long-standing ulcerative colitis (25). In addition, in Crohn’s disease
patients, perianal disease, bypasses and strictures may be sites of
increased risk for neoplastic transformation (26,27).
These findings indicate that, in IBD patients, chronic intestinal
inflammation may be the leading cause of CAC. Moreover, a large
epidemiological study showed that patients regularly ingesting
anti-inflammation agent, aminosalicylates (5-ASA) for IBD treatment
had a lower CRC risk (28,29). These findings together strongly
indicate that inflammation plays an important role in CAC
development. In the present study, we varied the concentration of
pro-inflammatory agent, DSS, in drinking water to induce different
degrees of colonic inflammation. We found that the tumor
multiplicity in mice in the high inflammation group was higher than
that in the low inflammation group, which was consistent with the
colonic inflammatory activity. In addition, tumor size in the high
inflammation group tended to be larger. All of these findings
directly demonstrate that inflammation promotes the malignant
transformation of colitis.
STAT3 belongs to the STAT (signal transducer and
activator of transcription) family of signal responsive
transcription factors, which similar to NF-κB are kept in an
inactive form in the cytoplasm of non-stimulated cells (18). Activation of STAT3 is mediated by
phosphorylation of a tyrosine residue (Tyr705) that induces STAT3
dimerization through phosphotyrosine-SH2 domain interaction.
Activators of STAT3 include interleukins, epidermal growth factor
(EGF) family members and interferon (18). When activated in cancer or immune
cells, STAT3 can induce the expression of a wide variety of genes
including IL-6, IL-22, EGF, IL-23 and IL-10 as well as
proto-oncogenes such as K-Ras, Src and c-Abl, whose products are
capable of inducing STAT3 activation in return (22). Therefore, there is no doubt that
STAT3 is a crucial link between inflammation and cancer.
Grivennikov et al recently demonstrated that ablation of
IL-6 reduced the activation of STAT3, which was consistent with the
reduction in CAC tumorigenesis. When STAT3 in IL-6−/−
animals was conditionally knocked out, there was almost no
tumorigenesis in the CAC model (19). In the present study, the activation
of the STAT3 signaling pathway was more excessive in the high
inflammation tissues than that in the low inflammation tissues.
Moreover, the activation of the STAT3 signaling pathway was higher
in the tumors than that in the inflammation tissues. These findings
suggest that with the development of inflammation, the activation
of the STAT3 pathway increased gradually. Moreover, CAC
carcinogenesis was associated with the persistent activation of the
STAT3 pathway.
The mTORC1 pathway integrating both intracellular
and extracellular signals, serves as a central regulator of cell
metabolism, growth, proliferation and survival. mTORC1 is also a
crucial molecule in the processes of inflammation and cancer
development. Dysregulation of the mTORC1 pathway is commonly found
in human inflammation and cancers (30–33).
Rapamycin, an inhibitor of mTORC1, was originally developed as an
immunosuppressive agent, suggesting that the mTORC1 pathway plays
an important role in the regulation of the immune system. mTORC1 is
essential for survival, cytokine production and migration of immune
cells involved in innate immune response and adaptive immune
response, such as mast cells (34),
macrophages (35), natural killer
cells (36), B cells (37) and T cells (38), which play a crucial role in the
inflammatory response. Furthermore, the mTORC1 signaling pathway
also plays an important role in carcinogenesis. Dysregulation of
the mTORC1 pathway can lead to many types of cancers, such as lung
(39) and breast (31), hepatocarcinoma (33) and colorectal cancer (32). Foremost, there exists crosstalk
between the mTOR and the STAT3 signaling pathway which is a crucial
pathway in CAC as mentioned above. A recent study showed that when
human breast, prostate, lung, pancreatic and liver cancer cell
lines were treated with rapamycin, activation of the STAT3
signaling pathway decreased (40).
When the mTORC1 signaling pathway was overactive using siRNA for
PTEN, an inhibitor of the mTORC1 pathway, activation of the STAT3
pathway was increased (40).
Therefore, we hypothesized that the mTORC1 pathway may play an
important role in CAC. Farkas et al recently showed that
rapamycin, an mTORC1 inhibitor, ameliorated experimental murine
colitis (41). Collectively, these
findings demonstrate that mTORC1 may be a therapeutic target for
IBD and CAC. In the present study, we found that activation of the
mTORC1 pathway was gradually increased in inflammatory colic and
CAC tissues. These findings indicated that with the development of
inflammation, the activation of the mTORC1 signaling pathway
increased gradually. CAC carcinogenesis was associated with the
persistent activation of the mTORC1 pathway. When we treated the
mice with rapamycin, activation of the STAT3 signaling pathway was
decreased, which was consistent with previous research (42). Furthermore, the tumor incidence,
multiplicity and size decreased. These findings revealed that the
mTORC1 and STAT3 signaling pathways promoted the malignant
transformation of colitis.
TNF-α, IL-6 and IL-12 are crucial pro-inflammatory
cytokines in the pathogenesis of IBD. As essential switches between
inflammation and cancer, much evidence has pointed to a critical
role of TNF-α, IL-6 and IL-12 in tumor initiation, proliferation,
migration, invasion and metastasis (43–46).
In our study, we demonstrated that levels of mRNA expression of
TNF-α, IL-6, IL-12α in mice in the CAC model treated with rapamycin
were lower when compared with the levels in mice given vehicle
alone. These findings reveal that rapamycin can reduce a
pro-inflammatory response, which may lead to suppression of
carcinogenesis. Notably, rapamycin also inhibited the expression of
the anti-inflammatory cytokines, IFN-γ and IL-10. The most viable
explanation is that the mTORC1 pathway is an essential signal of
immune cells such as neutrophils, monocytes, dendritic cells, B and
T cells (10,38). Rapamycin, an inhibitor of the mTORC1
pathway can widely suppress the expression of not only
pro-inflammatory cytokines, but also anti-inflammatory
cytokines.
In conclusion, the results of this study suggest
that inflammation promotes colitis-associated cancer through
activation of the mTORC1 and STAT3 pathways. Our data also indicate
that rapamycin, an inhibitor of the mTORC1 pathway, can inhibit the
development of CAC through widely suppressing the expression of
pro-inflammatory and anti-inflammatory cytokines including TNF-α,
IL-6, IL-12α, IL-10 and INF-γ.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (nos. 81072046 and
91029702).
References
|
1
|
Balkwill F and Mantovani A: Inflammation
and cancer: back to Virchow? Lancet. 357:539–545. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Eaden JA, Abrams KR and Mayberry JF: The
risk of colorectal cancer in ulcerative colitis: a meta-analysis.
Gut. 48:526–535. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gillen CD, Walmsley RS, Prior P, Andrews
HA and Allan RN: Ulcerative colitis and Crohn’s disease: a
comparison of the colorectal cancer risk in extensive colitis. Gut.
35:1590–1592. 1994.
|
|
4
|
Saleh M and Trinchieri G: Innate immune
mechanisms of colitis and colitis-associated colorectal cancer. Nat
Rev Immunol. 11:9–20. 2011. View
Article : Google Scholar
|
|
5
|
Rhodes JM: Unifying hypothesis for
inflammatory bowel disease and associated colon cancer: sticking
the pieces together with sugar. Lancet. 347:40–44. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rutter M, Saunders B, Wilkinson K, et al:
Severity of inflammation is a risk factor for colorectal neoplasia
in ulcerative colitis. Gastroenterology. 126:451–459. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Strimpakos AS, Karapanagiotou EM, Saif MW
and Syrigos KN: The role of mTOR in the management of solid tumors:
an overview. Cancer Treat Rev. 35:148–159. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guertin DA and Sabatini DM: Defining the
role of mTOR in cancer. Cancer Cell. 12:9–22. 2007. View Article : Google Scholar
|
|
9
|
Hay N and Sonenberg N: Upstream and
downstream of mTOR. Genes Dev. 18:1926–1945. 2004. View Article : Google Scholar
|
|
10
|
Delgoffe GM and Powell JD: mTOR: taking
cues from the immune microenvironment. Immunology. 127:459–465.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Magnuson B, Ekim B and Fingar DC:
Regulation and function of ribosomal protein S6 kinase (S6K) within
mTOR signalling networks. Biochem J. 441:1–21. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jastrzebski K, Hannan KM, Tchoubrieva EB,
Hannan RD and Pearson RB: Coordinate regulation of ribosome
biogenesis and function by the ribosomal protein S6 kinase, a key
mediator of mTOR function. Growth Factors. 25:209–226. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Duvel K, Yecies JL, Menon S, et al:
Activation of a metabolic gene regulatory network downstream of
mTOR complex 1. Mol Cell. 39:171–183. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guertin DA and Sabatini DM: An expanding
role for mTOR in cancer. Trends Mol Med. 11:353–361. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sarbassov DD, Ali SM, Sengupta S, et al:
Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB.
Mol Cell. 22:159–168. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bjornsti MA and Houghton PJ: The TOR
pathway: a target for cancer therapy. Nat Rev Cancer. 4:335–348.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thiem S, Pierce TP, Palmieri M, et al:
mTORC1 inhibition restricts inflammation-associated
gastrointestinal tumorigenesis in mice. J Clin Invest. 123:767–781.
2013.PubMed/NCBI
|
|
18
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: a leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Grivennikov S, Karin E, Terzic J, et al:
IL-6 and Stat3 are required for survival of intestinal epithelial
cells and development of colitis-associated cancer. Cancer Cell.
15:103–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chumanevich AA, Poudyal D, Cui X, et al:
Suppression of colitis-driven colon cancer in mice by a novel small
molecule inhibitor of sphingosine kinase. Carcinogenesis.
31:1787–1793. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Namba R, Young LJ, Abbey CK, et al:
Rapamycin inhibits growth of premalignant and malignant mammary
lesions in a mouse model of ductal carcinoma in situ. Clin Cancer
Res. 12:2613–2621. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu H, Kortylewski M and Pardoll D:
Crosstalk between cancer and immune cells: role of STAT3 in the
tumour microenvironment. Nat Rev Immunol. 7:41–51. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Park E, Park J, Han SW, et al: NVP-BKM120,
a novel PI3K inhibitor, shows synergism with a STAT3 inhibitor in
human gastric cancer cells harboring KRAS mutations. Int J Oncol.
40:1259–1266. 2012.PubMed/NCBI
|
|
24
|
Farraye FA, Odze RD, Eaden J and Itzkowitz
SH: AGA technical review on the diagnosis and management of
colorectal neoplasia in inflammatory bowel disease.
Gastroenterology. 138:746–774.e4. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gupta RB, Harpaz N, Itzkowitz S, et al:
Histologic inflammation is a risk factor for progression to
colorectal neoplasia in ulcerative colitis: a cohort study.
Gastroenterology. 133:1099–1105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sobala A, Herbst F, Novacek G and
Vogelsang H: Colorectal carcinoma and preceding fistula in Crohn’s
disease. J Crohns Colitis. 4:189–193. 2010.
|
|
27
|
Laukoetter MG, Mennigen R, Hannig CM, et
al: Intestinal cancer risk in Crohn’s disease: a meta-analysis. J
Gastrointest Surg. 15:576–583. 2011.
|
|
28
|
van Staa TP, Card T, Logan RF and Leufkens
HG: 5-Aminosalicylate use and colorectal cancer risk in
inflammatory bowel disease: a large epidemiological study. Gut.
54:1573–1578. 2005.PubMed/NCBI
|
|
29
|
Rubin DT, LoSavio A, Yadron N, Huo D and
Hanauer SB: Aminosalicylate therapy in the prevention of dysplasia
and colorectal cancer in ulcerative colitis. Clin Gastroenterol
Hepatol. 4:1346–1350. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Slomovitz BM and Coleman RL: The
PI3K/AKT/mTOR pathway as a therapeutic target in endometrial
cancer. Clin Cancer Res. 18:5856–5864. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lauring J, Park BH and Wolff AC: The
phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target
in breast cancer. J Natl Compr Cancer Netw. 11:670–678.
2013.PubMed/NCBI
|
|
32
|
Lee YK, Park SY, Kim YM, et al:
Suppression of mTOR via Akt-dependent and -independent mechanisms
in selenium-treated colon cancer cells: involvement of AMPKα1.
Carcinogenesis. 31:1092–1099. 2010.PubMed/NCBI
|
|
33
|
Zhang DM, Liu JS, Deng LJ, et al:
Arenobufagin, a natural bufadienolide from toad venom, induces
apoptosis and autophagy in human hepatocellular carcinoma cells
through inhibition of PI3K/Akt/mTOR pathway. Carcinogenesis.
34:1331–1342. 2013. View Article : Google Scholar
|
|
34
|
Smrz D, Kim MS, Zhang S, et al: mTORC1 and
mTORC2 differentially regulate homeostasis of neoplastic and
non-neoplastic human mast cells. Blood. 118:6803–6813. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fox R, Nhan TQ, Law GL, Morris DR, Liles
WC and Schwartz SM: PSGL-1 and mTOR regulate translation of ROCK-1
and physiological functions of macrophages. EMBO J. 26:505–515.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kawauchi K, Ihjima K and Yamada O: IL-2
increases human telomerase reverse transcriptase activity
transcriptionally and posttranslationally through
phosphatidylinositol 3′-kinase/Akt, heat shock protein 90, and
mammalian target of rapamycin in transformed NK cells. J Immunol.
174:5261–5269. 2005.PubMed/NCBI
|
|
37
|
Donahue AC and Fruman DA: Proliferation
and survival of activated B cells requires sustained antigen
receptor engagement and phosphoinositide 3-kinase activation. J
Immunol. 170:5851–5860. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Powell JD and Delgoffe GM: The mammalian
target of rapamycin: linking T cell differentiation, function, and
metabolism. Immunity. 33:301–311. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Memmott RM and Dennis PA: The role of the
Akt/mTOR pathway in tobacco carcinogen-induced lung tumorigenesis.
Clin Cancer Res. 16:4–10. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ma J, Meng Y, Kwiatkowski DJ, et al:
Mammalian target of rapamycin regulates murine and human cell
differentiation through STAT3/p63/Jagged/Notch cascade. J Clin
Invest. 120:103–114. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
Farkas S, Hornung M, Sattler C, et al:
Rapamycin decreases leukocyte migration in vivo and
effectively reduces experimentally induced chronic colitis. Int J
Colorectal Dis. 21:747–753. 2006.PubMed/NCBI
|
|
42
|
Deng L, Zhou JF, Sellers RS, et al: A
novel mouse model of inflammatory bowel disease links mammalian
target of rapamycin-dependent hyperproliferation of colonic
epithelium to inflammation-associated tumorigenesis. Am J Pathol.
176:952–967. 2010. View Article : Google Scholar
|
|
43
|
Wu Y and Zhou BP: TNF-α/NF-κB/Snail
pathway in cancer cell migration and invasion. Br J Cancer.
102:639–644. 2010.
|
|
44
|
Hodge DR, Hurt EM and Farrar WL: The role
of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer.
41:2502–2512. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Grivennikov S, Karin E, Terzic J, et al:
IL-6 and Stat3 are required for survival of intestinal epithelial
cells and development of colitis-associated cancer. Cancer Cell.
15:103–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lippitz BE: Cytokine patterns in patients
with cancer: a systematic review. Lancet Oncol. 14:e218–e228. 2013.
View Article : Google Scholar : PubMed/NCBI
|