Introduction
Extracellular matrix (ECM) proteins provide
structural support for lung cells involved in gas exchange and may
also actively regulate various aspects of lung function in both
homeostatic and pathological conditions. Persistent exposure to
cigarette smoke or airborne pollutants leads to the recruitment and
activation of monocytes and alveolar macrophages (1,2).
These, in turn release proteases that destroy ECM components in the
alveolar septa, resulting in the development of emphysema, a common
type of chronic obstructive pulmonary disease (3). On the other hand, increased
degradation of ECM may promote aberrant remodeling of the
connective tissues with resultant lung fibrosis. Lung fibrosis and
emphysema may coexist and indeed, expression of a large number of
ECM-related genes are upregulated in the lungs of smokers with
severe emphysema (4,5). Among increased ECM proteins, type VI
collagen (COL6) is deposited early in lung fibrosis (6). Both emphysema and lung fibrosis have a
strong association with the occurrence of lung cancers (7–9).
However, the expression patterns of COL6 in human lung cancers are
largely unknown.
Collagen, an ECM protein, is important in
maintaining the integrity of the lung tissues (10). The pulmonary interstitial
compartment provides the scaffold and connection to the alveolar
network via the contents of matrix proteins. Although COL6 is not
the major structural element of the lung, it is present in the
interstitium of the lung (10), as
fine microfibrils with 3–10 nm in diameter that exhibit a
double-beaded period of ~100 nm (11). COL6 consists of monomers containing
at least 3 major polypeptides, α1, α2 and α3. The monomers
aggregate intracellularly into dimers and then tetramers (12). Three additional polypeptides (α4, α5
and α6) have been identified to have high homology to the α3 chain
(13,14).
The multi-domain structure of COL6 allows its
interaction with components of matrix, including biglycan, decorin
(15,16), hyaluronic acid (17), heparin sulphate (18) and type I (COL1) and type IV (COL4)
collagen (6,19,20).
Such interaction can stabilize ECM architecture, which is important
in maintaining the integrity of tissues in the lung (10). In addition to the
matrix-interaction, COL6 binds to integrins (21) of various cell types, initiating the
matrix-cell interaction and affecting signaling pathways that
regulate cellular functions, such as proliferation (22), migration (23), differentiation (24) and survival (25). COL6 has potent growth-stimulatory
effects, which is associated with aggressive tumor growth (22). Increased expression of COL6 has also
been positively correlated with tumorigenesis (26–29).
In this report, we revealed increased levels of COL6
in the lung tissues of patients with non-small cell lung cancers.
Since COL6 is located in the pulmonary interstitial compartment
where it encounters various cell types, including infiltrated
monocytes and epithelial cells, we hypothesized that excessive COL6
may have effects on its surrounding cells. The objective of this
study was to understand the role of excessive COL6 in the lung
tissues with aforementioned lung diseases. Our results demonstrated
the stimulatory properties of COL6 on various cell types, which in
turn may have impacts on the development of lung neoplasia from
emphysematous or fibrotic lungs where COL6 is upregulated.
Materials and methods
Collagens, antibodies and other
reagents
Human collagen type IV (COL4) and VI (COL6) purified
chromatographically and immunologically from human placenta were
obtained from Rockland Immunochemicals Inc. (Gilbertsville, PA, USA
). Human anti-COL6A1 polyclonal antibody for immunohistochemical
staining was obtained from Santa Cruz Biotechnology (Santa Cruz,
CA, USA ). Lipopolysaccharide (LPS from Escherichia coli
0111:B4) was from Sigma-Aldrich (St. Louis, MO, USA).
Cell lines, human blood samples and
primary cells
The human TH P-1 monocytic cell line (AT CC;
American Type Culture Collection, Manassas, VA, USA ; provided by
Dr Michael Klemsz from Indiana University School of Medicine) was
cultured in complete RPMI-1640 medium. Human blood samples of
healthy donors were obtained from the Indiana Blood Center.
Peripheral blood mononuclear cells (PBMCs) were separated using
Ficoll centrifugation and aliquots of PBMCs were cryopreserved in
liquid nitrogen. Human primary monocytes were isolated from PBMCs
using CD14 magnetic beads with ~93% of purity (Miltenyi Biotec
Inc., Auburn, CA, USA ). Human bronchial epithelial cells (HBEpCs)
derived from the surface epithelium of normal human bronchi were
obtained from Cell Applications (San Diego, CA, USA ) and were
cultured in Bronchial/Tracheal Epithelial Cell Growth Medium
provided by the same supplier.
Gene expression in normal and neoplastic
lung tissues
TissueScan Lung Cancer Tissue qPCR Panel II
containing 48 tissues covering 4 disease stages (IA, IB, IIA, IIB,
IIIA, IIIB and IV) of NS CLC and normal controls were obtained from
OriGene Technologies (Rockville, MD, USA). The gene expression of
COL6A1 and IL23A was analyzed using the TaqMan assay
primers with ACTB (β-actin) as endogenous control in the ABI
7300 system (Applied Biosystems by Life Technologies, Carlsbad, CA,
USA ).
Analysis of COL6 protein in human lung
tissues using immunohistochemistry staining
Lung disease tissue array slides (LUD481 and LUC962)
containing normal controls, adenocarcinoma and squamous cell
carcinoma were obtained from US Biomax (Rockville, MD, USA ). The
levels of COL6 protein were analyzed using immunohistochemical
staining with an anti-COL6A1 polyclonal antibody (Santa Cruz
Biotechnology).
Cytokine production by monocytes
Monocytes (2–4×106 cells/ml) freshly
purified from normal PBMCs were treated with medium only, LPS (1
μg/ml) and COL6 at the concentrations of 10 or 30 μg/ml. Eight
hours following treatment, the supernatants were collected and the
cell pellets were resuspended in TRIzol reagents (Invitrogen,
Carlsbad, CA, USA ) for total RNA extraction, first-strand cDNA
synthesis (Invitrogen) followed by real-time qPCR with TaqMan assay
primers for IL1B (IL-1β), IL6 (IL-6), TNFA
(TNFα), IL23A (p19) and IL12B (p40). The levels of
cytokine secretion in the supernatants, including IL-23 and TNFα
were measured using ELISA as previously described (30).
Analysis of focal adhension kinase (FAK)
and extracellular signal-regulated kinase (ERK) activation
following stimulation
TH P-1 or HBEpC cells were stimulated as indicated
at 37°C in a 5% CO2 incubator for 1 h. Cells were lysed
using the RIPA protein lysis buffer consisting of 10% glycerol, 1%
Igepal, 50 mM Tris-pH 7.4, 150 mM NaCl, 1 mM EDTA- pH 8.0, 1%
Na-deoxycholate, 0.1% SDS and protease inhibitors (31). Activation of FAK was evaluated using
western blot analysis with antibodies against phospho-FAK (P-FAK)
at Tyr 397 (Y397) and Y925 followed by total FAK (Cell Signaling
Technology, Danvers, MA, USA). Activation of ERK1/2 was evaluated
using western blot analysis with phospho-P44/42 ERK1/2
(Thr202/Tyr204) followed by total ERK antibodies (Cell Signaling
Technology). The band intensity was determined using Image J
program (NIH) and the ratios were calculated as the intensity of
phosphorylated proteins divided by the intensity of total
proteins.
Statistical analysis
PAS W Statistics (IBM-SPSS, Chicago, IL, USA) was
used to analyze the data. P-value was determined with an
independent Student’s t-test.
Results
COL6 expression is upregulated in
neoplastic lung tissues
In this study, we evaluated the levels of COL6 in
neoplastic lung tissues. The gene expression of COL6A1 was
significantly higher in lung tissues with NSCLC at various stages,
as compared to that from normal controls (Fig. 1A). In addition, the levels of COL6
protein were elevated in lung tissues from NS CLC patients with
adenocarcinoma and squamous cell carcinoma (Fig. 1B and C). Positive staining for
COL6A1 was more intense in the cytoplasm of stromal cells and in
the immediately adjacent extracellular space. Uninvolved healthy
lung tissue sections displayed the normal structure of alveoli,
whereas lung adenocarcinoma sections exhibited various stages of
neoplastic differentiation with loss of normal alveolar structures
(Fig. 1B). A marked staining of
COL6 was noted in 65% (15/23) of the lung tissues from the NS CLC
patients analyzed (Fig. 1C). We
found no significant difference in COL6 staining from the lung
tissues among the three different grades (grade 1–3) of lung cancer
(Fig. 1B and C).
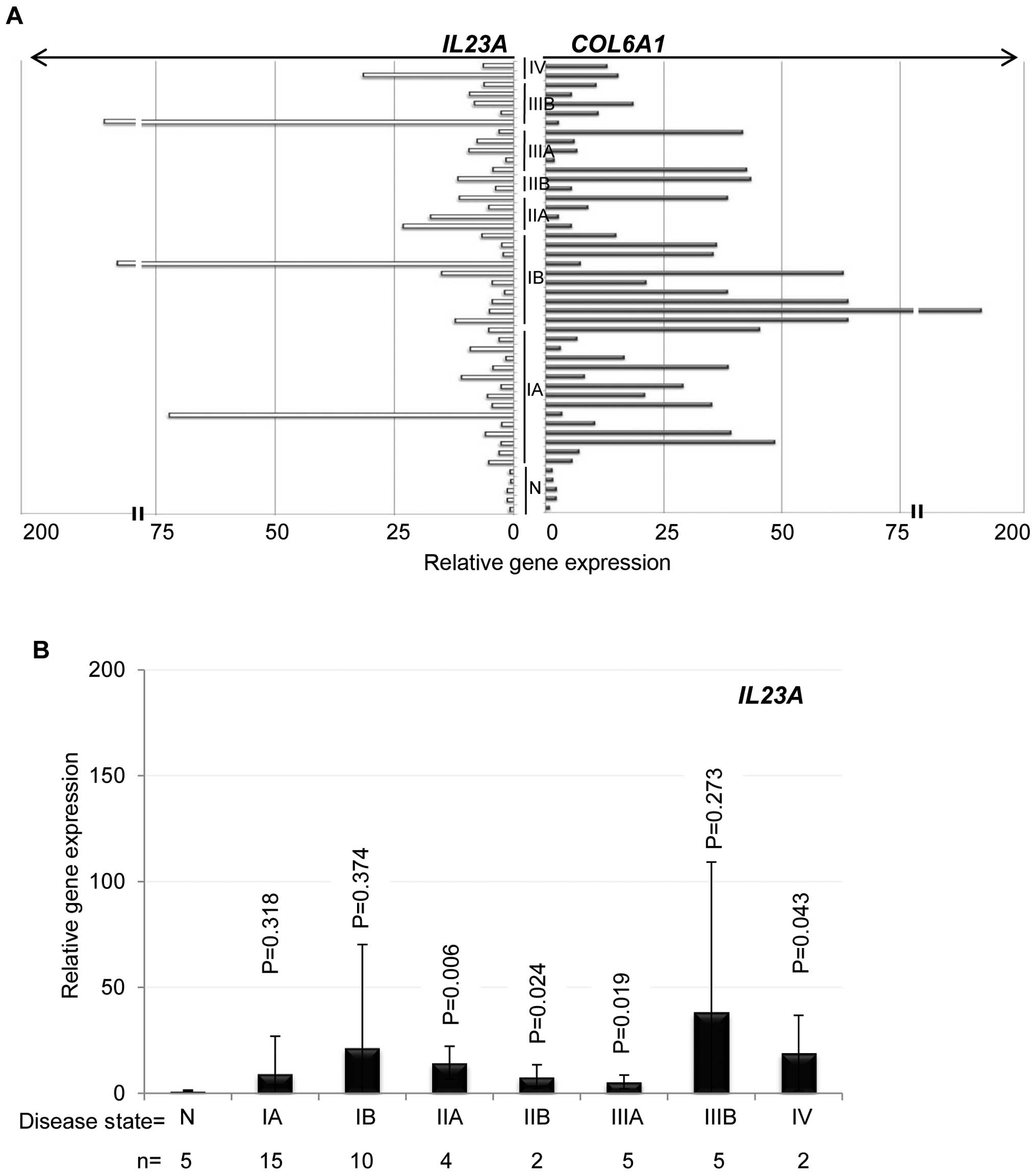
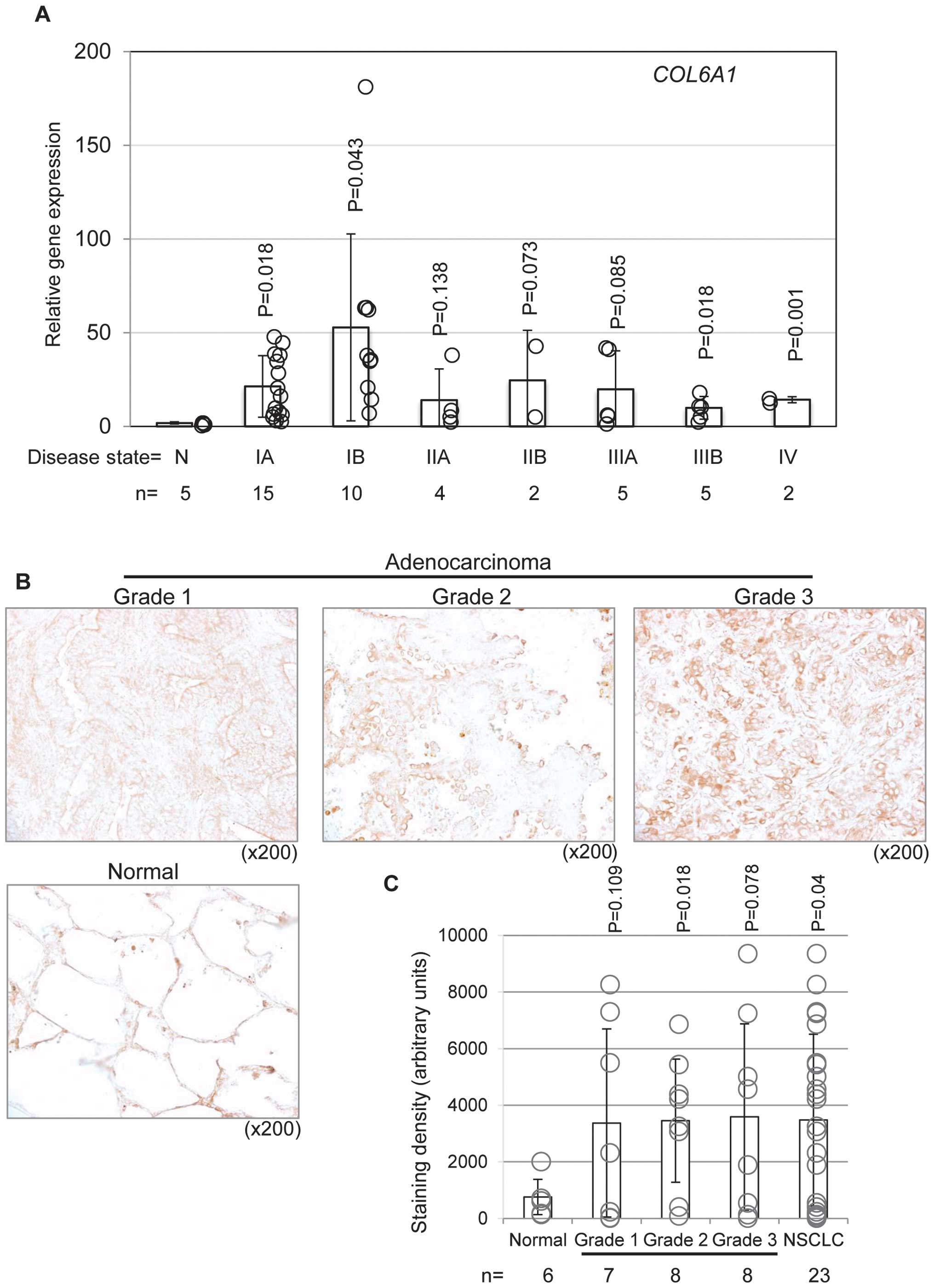 | Figure 1Expression of COL6 in normal and
neoplastic lung tissues. (A) COL6A1 gene expression in human
lung tissues. Tissue Scan Lung Cancer Tissue qPCR Panel II
containing 48 tissues covering 4 disease stages (IA, IB, IIA, IIB,
IIIA, IIIB and IV) of NS CLC and normal tissues (N) was used. The
gene expression level of COL6A1 was analyzed using TaqMan
assay primers with ACTB (β-actin) as endogenous control.
Data are presented as mean ± SD in a bar graph from each group.
Open circle (○) indicates the expression level of individual tissue
from the group. P-value is compared to the normal tissues. (B)
Immunohistochemical staining of COL6 protein from human lung
tissues. Lung disease tissue array slides (LUD481 and LUC962)
containing normal controls and NSCLC (grade 1–3) were used. The
levels of COL6 protein in the slides were analyzed using
immunohistochemical staining with anti-COL6A1 polyclonal antibody.
Representative image of adenocarcinoma (grade 1–3) and normal lung
at higher magnification (x200) is shown from slide LUD481. (C)
Intensity of COL6A1 staining in the whole tissues in the slides was
analyzed using Image J program and presented as the mean intensity
± SD from normal (n=6) and NS CLC (n=23; total grade 1–3),
including grade 1 (n=7; 3 adenocarcinoma and 4 squamous cell
carcinoma), grade 2 (n=8; 3 adenocarcinoma and 5 squamous cell
carcinoma), grade 3 (n=8; 3 adenocarcinoma and 5 squamous cell
carcinoma). P-value is compared to the normal tissues. COL6, type
VI collagen; NS CLC, non-small cell lung cancer. |
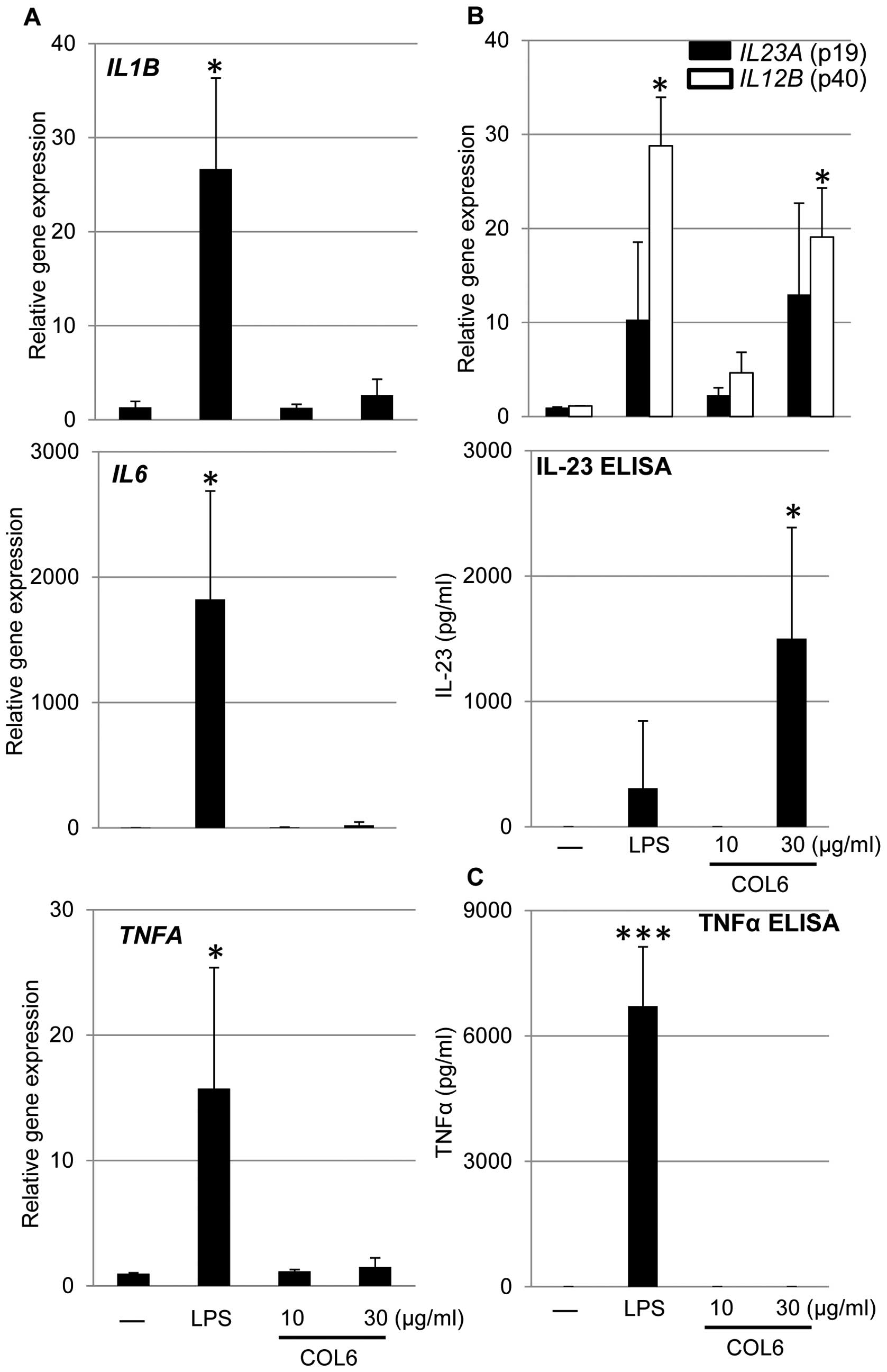
COL6 stimulates monocytes to produce
IL-23
During lung inflammation, lung epithelial cells are
activated upon exposure to irritants and produce chemokines that
attract immune cells from the periphery to the lungs. Among these
chemokines, CCL2 attracts CCR2-expressing monocytes (32). Given the fact that elevation of COL6
is present in the pulmonary interstitium, an area shared with
recruited monocytes during lung inflammation, we first tested the
hypothesis that excessive COL6 has stimulatory effects on
monocytes. To investigate whether monocytes respond to COL6, human
primary monocytes were purified from PBMCs of normal control donors
using CD14+ magnetic microbeads with a purity of ~93%.
Isolated monocytes were stimulated with different concentrations of
COL6 followed by analysis of cytokine production.
Human monocytes express toll-like receptor 4 that
responds to LPS stimulation, resulting in induction of
pro-inflammatory cytokines (33).
LPS was therefore used as a positive control in the in vitro
study. As expected, LPS stimulation in human monocytes resulted in
upregulation of pro-inflammatory cytokines such as IL1B,
IL6 and TNFA (Fig.
2A). We found that COL6 stimulation had no significant effects
on expression of IL1B, IL6 and TNFA (Fig. 2A). However, increased gene
expression of each subunit (IL23A for p19 and IL12B
for p40) of IL-23 was noted following LPS or COL6 stimulation in
the monocytes (Fig. 2B, upper
panel). In addition, production of IL-23 was dependent on the dose
of COL6 as no detectable level of secreted IL-23 was noticed using
a lower concentration of COL6 (Fig.
2B, lower panel). Similar to TNFA gene expression,
secreted TNFα was only detected following stimulation with LPS, but
not with medium or COL6 (Fig.
2C).
IL23A expression is upregulated in
neoplastic lung tissues
IL-23 has been implicated in the development of
cancers (34). The induction of
IL-23 by monocytes following COL6 stimulation prompted us to
examine whether the levels of IL-23 were elevated in the lung
tissues of patients with NSCLC. The expression of IL23A
encoding the p19 subunit of IL-23 was examined from another
duplicate lung cancer tissue panel as in Fig. 1A. Elevated gene expression of
IL23A was found in lung tissues of NSCLC as compared to that
from normal controls (Fig. 3A, left
panel). This profile was similar to that of COL6A1
expression (Fig. 3A, right panel).
However, the tissue with higher COL6A1 expression was not
always correlated with higher IL23A levels (Fig. 3A). Despite the individual variation
and sample sizes, IL23A expression was significantly higher
at various stages (IIA, IIB, IIIA and IV) of NS CLC than that of
normal controls (Fig. 3B).
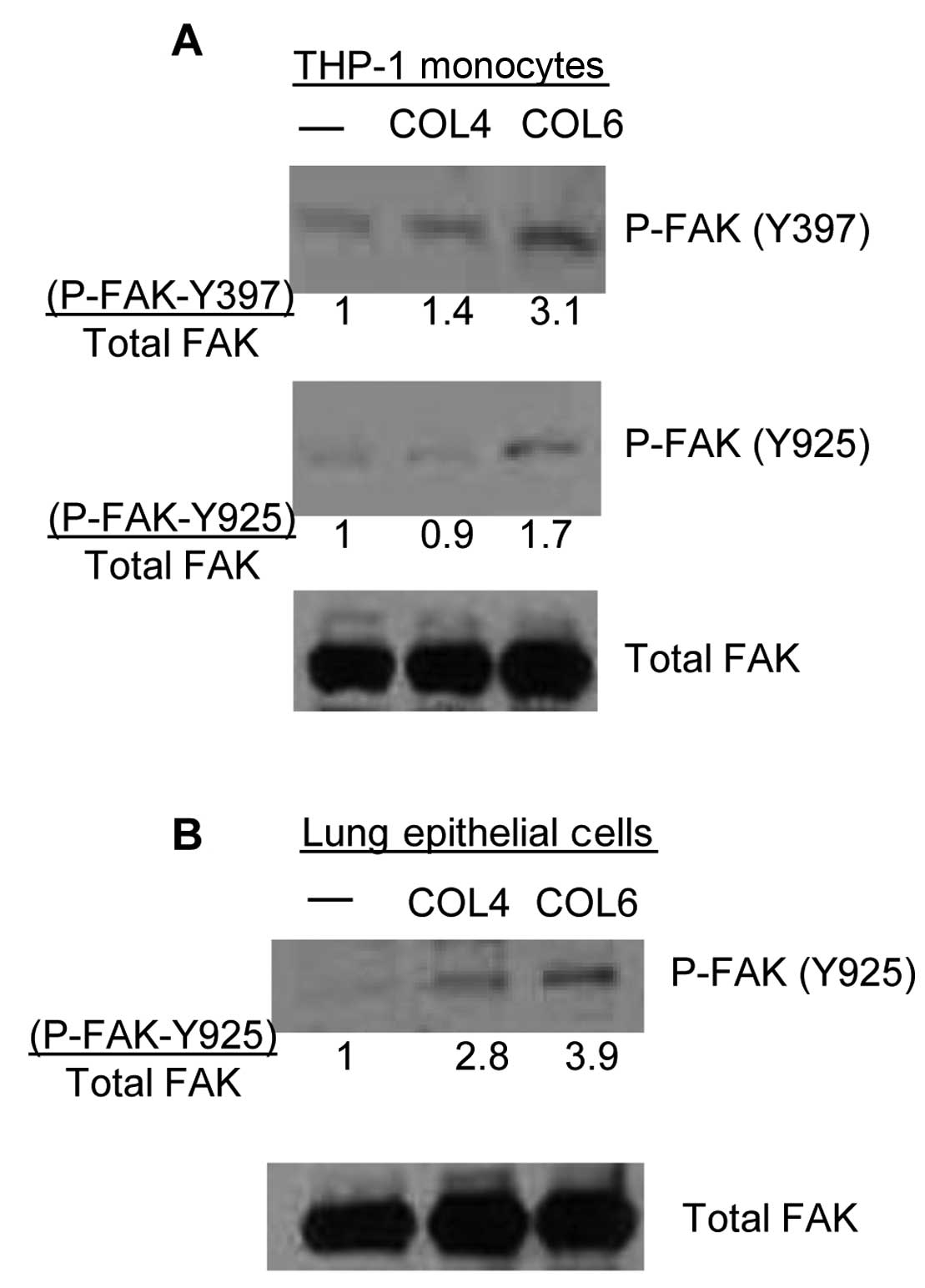
COL6 activates the FAK signaling pathway
in monocytes
Monocytes express integrins of the β1, β2, β3 and β5
subfamilies (35). Upon binding to
its ligands, integrin stimulates the activation of FAK outside-in
signaling, which contributes to various biological functions
including cytokine production, cell proliferation, survival and
differentiation (36). FAK acts as
a scaffolding molecule for assembly of the other kinases upon
integrin ligation. Activation of FAK by integrin clustering leads
to autophosphorylation at tyrosine (Y)397, which is a binding site
for the Src homology 2 (SH2) domain of the Src-protein tyrosine
kinases. Recruitment of Src family kinases results in the
phosphorylation of Y925 in the C-terminal region of FAK (37,38).
Integrin is one of the receptors for COL6 (21). To test whether the integrin-FAK
signaling pathway was activated in monocytes upon COL6 stimulation,
human THP-1 monocytes were stimulated followed by analysis of FAK
activation using western blot analysis (Fig. 4A). COL4 known to activate FAK in
intestinal epithelial cells (39)
was included in the stimulation for comparison. We found that FAK
was phosphorylated at Y397 and Y925 in THP-1 monocytes stimulated
with COL6, suggesting the activation of FAK and Src upon COL6
stimulation. In contrast, COL4 had little if any effects on P-FAK
in the monocytes.
COL6 activates FAK signaling in lung
epithelial cells
Signaling through FAK activation has been implicated
in the tumorigenic properties of lung cancer cells (40,41).
Having shown the activation of FAK in monocytes, we next tested if
COL6 was able to stimulate lung epithelial cells that also express
integrin. Phosphorylation of FAK at Y925 was not detected from
human primary epithelial cells cultured in medium only, while
stimulation with COL6 in human primary epithelial cells resulted in
an ~4-fold increase in phosphorylation of FAK at Y925 relative to
those cultured in medium only. COL4 is known to activate FAK in
intestinal epithelial cells (39)
and we also observed an intermediate increase (~3-fold) in P-FAK
(Y925) upon stimulation with COL4 in lung epithelial cells relative
to the control (Fig. 4B).
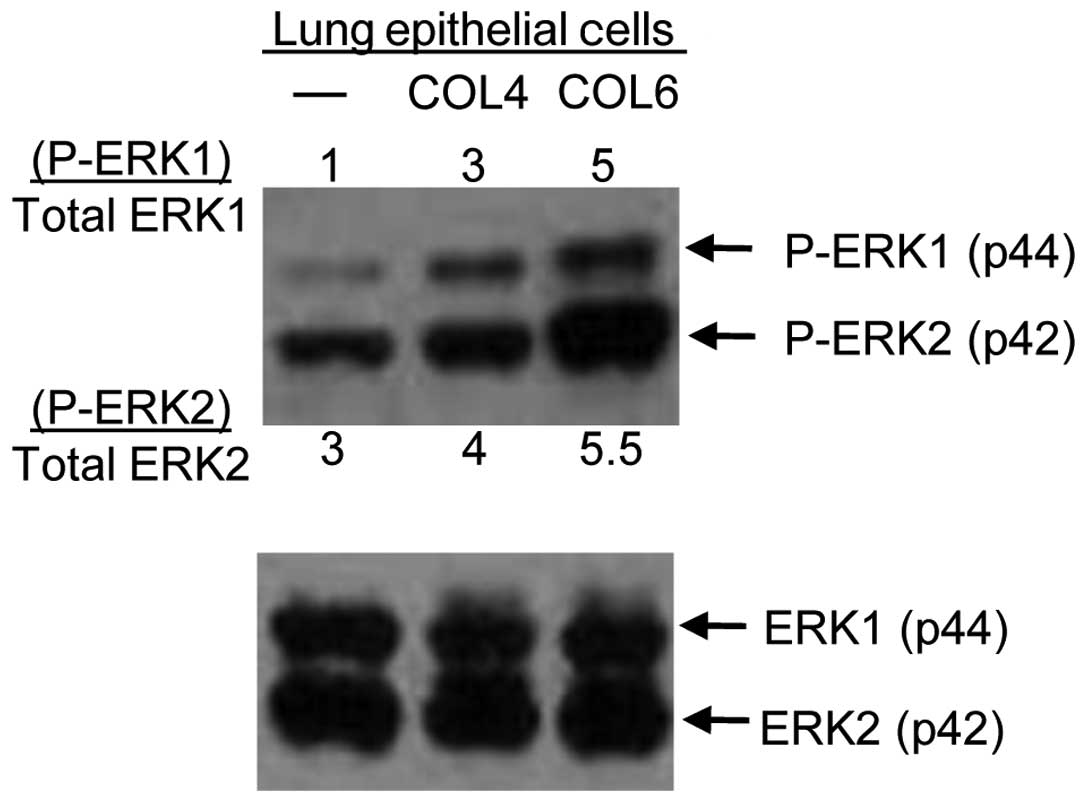
COL6 activates MAPK/ERK signaling in lung
epithelial cells
It is known that phosphorylation of FAK at Y925
creates a binding site for the growth factor-receptor-bound protein
2 (GRB2) and activates a small G protein, RAS (37). Activated RAS recruits
mitogen-activated protein kinase kinase kinase (MAPKKK or RAF),
which leads to activation of mitogen-activated protein kinase
kinase (MAPKK or MEK), which then activates the mitogen-activated
protein kinase (MAPK) extracellular signal-regulated kinase
(ERK)1/2 (42). To further
investigate the involvement of ERK1/2 in FAK signaling following
COL6 stimulation, we examined the activation of ERK1/2 using
western blot analysis. We found that human primary epithelial cells
increased the levels of phosphorylation of ERK1/2 upon treatment of
COL6 and an intermediate increase in ERK1/2 phosphorylation was
also induced by COL4 (Fig. 5).
Discussion
In the present study, we demonstrated that elevated
gene expression of COL6A1 was present in the lungs with
NSCLC at various stages. Lung tissues with adenocarcinoma or
squamous cell carcinoma were associated with deposition of more
COL6 as compared to normal lungs. COL6 is known for its potent
growth-stimulating effects on promoting tumor progression (22,26–29).
Our results from in vitro cultures suggest a novel function
of excess COL6 on stimulating IL-23 production by monocytes. We
also found upregulation of IL23A expression in neoplastic
lung tissues at various stages, consistent with the reports that
overexpression of IL23A was present in human tumor samples
(34). IL-23 has been implicated in
the development of cancers by increasing neutrophil and inhibiting
CD8+ T cell infiltration, thereby promoting tumor
incidence and growth (34). Our
results suggest a potential role of COL6 in promoting inflammation
that is favorable for tumor development.
Although IL-23 was induced by monocytes using a
soluble form of COL6 in a dose-dependent manner, it was less
efficiently induced using plate-bound COL6 (data not shown).
Instead, we found that plate-bound COL6 had effects on endothelial
cells, which are also present in the pulmonary interstitium.
Primary lung endothelial cells treated with plate-bound COL6 had
increased levels of cleaved caspase-3 (data not shown), suggesting
the induction of apoptosis. In this study, we did not identify the
specific fragments of COL6, which are required for induction of
IL-23 expression by monocytes. It is likely that different forms of
COL6 are able to stimulate various cell types in physiological
conditions.
In order to exhibit stimulatory effects in the
lungs, COL6 needs to be secreted and exposed to its surrounding
target cells. It is known that COL6 is mainly produced by
fibroblasts (43,44) and other cell types such as
adipocytes (29) or macrophages are
capable of expressing COL6 (45).
It has been shown that TGF-β1 and TGF-β3 are responsible for
increasing extracellular matrix expression by human lung
fibroblasts (46). In addition,
COL6 expression is induced by human macrophages following
stimulation with IL-4, IL-10, or IL-13, which are involved in T
helper type 2 (Th2) allergic inflammation (45). Therefore, the existing inflammatory
environment especially mediated by Th2 immunity may increase the
expression of COL6 in the lung tissues.
Another possible mechanism for upregulation of
COL6A1 gene expression is due to DNA hypomethylation in
stromal cells under hypoxic conditions. It has been shown that a
global genomic hypomethylation occurs in synovial fibroblasts from
rheumatoid arthritis patients (47). Our preliminary data also found a
greatly reduced DNA methylation in the COL6A1 gene from lung
tissue with both emphysema and adenocarcinoma as compared to that
of adjacent normal section from the same patient (data not shown).
A hypoxic lung environment as in emphysema or lung fibrosis is
likely to modulate expression of an array of genes such as ECM
component COL6.
To determine if COL6 was increased during the early
phases of emphysema, a mouse model of cigarette smoke-induced
emphysema was established. Destruction of alveolar septa was
evident in mice exposed to cigarette smoke for 4 months (data not
shown). However, there was no evidence of increased Col6a1
transcription or protein deposition in the lung tissues during this
phase of cigarette-induced lung injury characterized by lung tissue
loss, but no neoplasia (data not shown). These results suggest
destruction of ECM alone may not be sufficient to enhance COL6
expression and transform the cells. It is possible this occurs in
the faulty repair phase of lung injury, since increased degradation
of the ECM leads to aberrant remodeling of the connective tissues
and excess matrix protein deposition leads to pulmonary fibrosis,
which is sometimes present in the emphysematous lungs. Combined
pulmonary fibrosis and emphysema (CPFE) is a recently recognized
distinct condition in which the lung is prominently affected by
both emphysema and lung fibrosis (48). CPFE may be even more strongly
associated with lung cancer than either emphysema or lung fibrosis
alone (49). Future investigations
will have to mechanistically evaluate whether or not overexpression
of COL6 is critical to or it is merely associated with the
progression of emphysema or lung fibrosis to lung neoplasm.
In this study, we showed that COL6 exerted its
stimulatory effects involving the signaling pathway that activated
FAK molecules in monocytes and epithelial cells. MAPK/ERK, the
downstream molecule of FAK, was also activated in lung epithelial
cells following COL6 stimulation. ERK is known to promote cell
proliferation, angiogenesis, cell differentiation and cell
survival, which contributes to the development of various types of
cancers including NSCLC (31). In
addition to promote IL-23-mediated lung inflammation by targeting
monocytes, COL6 was capable of activating signaling molecules
(e.g., FAK and ERK) in normal lung epithelial cells, which may
promote the development of lung cancer. Inhibition of FAK has been
beneficial on retardation of tumor progression (38). Studies using small-molecule FAK
inhibitor are currently in clinical trials for patients with
various types of tumors (50). FAK
is a relevant target in the inhibition of tumor progression as well
as abrogation of COL6-mediated activity during tissue
remodeling.
Destruction of ECM in the lung is regarded as the
pathological hallmark of emphysema. Although upregulation of COL6
is important for repairing the wounded tissues during the healing
process, excess matrix protein deposition may lead to unwanted
consequences, such as increased fibrosis, and/or as we postulate,
increased tumorigenesis potential. In this report, we defined that
COL6 exerted stimulatory effects on inducing IL-23 production by
monocytes, supporting its role in activating immune cells to
promote pro-tumor lung inflammation during the process of tissue
remodeling. Amelioration of the progression or abnormal reparative
phases of emphysema or lung fibrosis by blocking signaling pathways
exerted by excessive ECM components such as COL6, may be an
important step in inhibiting tumorigenesis.
Acknowledgements
This study was supported in part by the Lung Cancer
Working Group (to H-C.C.), Research Support Fund Grant (to H-C.C.)
and National Institutes of Health Grants R01CA118118 (to M.J.R.)
and R01HL077328 (to I.P.). The authors thank Dr Michael Klemsz for
generously providing the human THP-1 monocytic cell line, Dr
Matthias Clauss for primary human lung microvascular endothelial
cells and Mrs. Liying Yan (EpigenDx Inc.) for CpG methylation
analysis. We thank members of Dr Petrache’s laboratory for
preparing the lung tissue samples from the smoked mice. We thank Dr
Mark Kaplan for his critical review of the manuscript. We also
thank Dr John Turchi and other members in the Lung Cancer Working
Group for their continuous support and invaluable suggestions.
References
|
1
|
Barnes PJ, Shapiro SD and Pauwels RA:
Chronic obstructive pulmonary disease: molecular and cellular
mechanisms. Eur Respir J. 22:672–688. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shapiro SD: The macrophage in chronic
obstructive pulmonary disease. Am J Respir Crit Care Med.
160:S29–S32. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Barnes PJ: Immunology of asthma and
chronic obstructive pulmonary disease. Nat Rev Immunol. 8:183–192.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lang MR, Fiaux GW, Gillooly M, Stewart JA,
Hulmes DJ and Lamb D: Collagen content of alveolar wall tissue in
emphysematous and non-emphysematous lungs. Thorax. 49:319–326.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Spira A, Beane J, Pinto-Plata V, Kadar A,
Liu G, et al: Gene expression profiling of human lung tissue from
smokers with severe emphysema. Am J Respir Cell Mol Biol.
31:601–610. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Specks U, Nerlich A, Colby TV, Wiest I and
Timpl R: Increased expression of type VI collagen in lung fibrosis.
Am J Respir Crit Care Med. 151:1956–1964. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wilson DO, Weissfeld JL, Balkan A,
Schragin JG, Fuhrman CR, et al: Association of radiographic
emphysema and airflow obstruction with lung cancer. Am J Respir
Crit Care Med. 178:738–744. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li Y, Swensen SJ, Karabekmez LG, Marks RS,
Stoddard SM, et al: Effect of emphysema on lung cancer risk in
smokers: a computed tomography-based assessment. Cancer Prev Res.
4:43–50. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Usui K, Tanai C, Tanaka Y, Noda H and
Ishihara T: The prevalence of pulmonary fibrosis combined with
emphysema in patients with lung cancer. Respirology. 16:326–331.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baldock C, Sherratt MJ, Shuttleworth CA
and Kielty CM: The supramolecular organization of collagen VI
microfibrils. J Mol Biol. 330:297–307. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Engel J, Furthmayr H, Odermatt E, von der
Mark H, Aumailley M, et al: Structure and macromolecular
organization of type VI collagen. Ann NY Acad Sci. 460:25–37. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Engvall E, Hessle H and Klier G: Molecular
assembly, secretion, and matrix deposition of type VI collagen. J
Cell Biol. 102:703–710. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fitzgerald J, Rich C, Zhou FH and Hansen
U: Three novel collagen VI chains, α4(VI), α5(VI), and α6(VI). J
Biol Chem. 283:20170–20180. 2008.
|
|
14
|
Gara SK, Grumati P, Urciuolo A, Bonaldo P,
Kobbe B, et al: Three novel collagen VI chains with high homology
to the α3 chain. J Biol Chem. 283:10658–10670. 2008.PubMed/NCBI
|
|
15
|
Bidanset DJ, Guidry C, Rosenberg LC, Choi
HU, Timpl R and Hook M: Binding of the proteoglycan decorin to
collagen type VI. J Biol Chem. 267:5250–5256. 1992.PubMed/NCBI
|
|
16
|
Wiberg C, Hedbom E, Khairullina A, Lamande
SR, Oldberg A, et al: Biglycan and decorin bind close to the
n-terminal region of the collagen VI triple helix. J Biol Chem.
276:18947–18952. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
McDevitt CA, Marcelino J and Tucker L:
Interaction of intact type VI collagen with hyaluronan. FEBS Lett.
294:167–170. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Specks U, Mayer U, Nischt R, Spissinger T,
Mann K, et al: Structure of recombinant N-terminal globule of type
VI collagen alpha 3 chain and its binding to heparin and
hyaluronan. EMBO J. 11:4281–4290. 1992.PubMed/NCBI
|
|
19
|
Bonaldo P, Russo V, Bucciotti F, Doliana R
and Colombatti A: Structural and functional features of the alpha 3
chain indicate a bridging role for chicken collagen VI in
connective tissues. Biochemistry. 29:1245–1254. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kuo HJ, Maslen CL, Keene DR and Glanville
RW: Type VI collagen anchors endothelial basement membranes by
interacting with type IV collagen. J Biol Chem. 272:26522–26529.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Loeser RF, Sadiev S, Tan L and Goldring
MB: Integrin expression by primary and immortalized human
chondrocytes: evidence of a differential role for α1β1 and α2β1
integrins in mediating chondrocyte adhesion to types II and VI
collagen. Osteoarthritis Cartilage. 8:96–105. 2000.PubMed/NCBI
|
|
22
|
Sherman-Baust CA, Weeraratna AT, Rangel
LB, Pizer ES, Cho KR, et al: Remodeling of the extracellular matrix
through overexpression of collagen VI contributes to cisplatin
resistance in ovarian cancer cells. Cancer Cell. 3:377–386. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tillet E, Gential B, Garrone R and
Stallcup WB: NG2 proteoglycan mediates beta1 integrin-independent
cell adhesion and spreading on collagen VI. J Cell Biochem.
86:726–736. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Naugle JE, Olson ER, Zhang X, Mase SE,
Pilati CF, et al: Type VI collagen induces cardiac myofibroblast
differentiation: implications for postinfarction remodeling. Am J
Physiol Heart Circ Physiol. 290:H323–H330. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ruhl M, Sahin E, Johannsen M, Somasundaram
R, Manski D, et al: Soluble collagen VI drives serum-starved
fibroblasts through S phase and prevents apoptosis via
down-regulation of Bax. J Biol Chem. 274:34361–34368. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Han J, Daniel JC and Pappas GD: Expression
of type VI collagen during glioblastoma cell invasion in brain
tissue cultures. Cancer Lett. 88:127–132. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
McComb RD, Moul JM and Bigner DD:
Distribution of type VI collagen in human gliomas: comparison with
fibronectin and glioma-mesenchymal matrix glycoprotein. J
Neuropathol Exp Neurol. 46:623–633. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Daniels KJ, Boldt HC, Martin JA, Gardner
LM, Meyer M and Folberg R: Expression of type VI collagen in uveal
melanoma: its role in pattern formation and tumor progression. Lab
Invest. 75:55–66. 1996.PubMed/NCBI
|
|
29
|
Iyengar P, Espina V, Williams TW, Lin Y,
Berry D, et al: Adipocyte-derived collagen VI affects early mammary
tumor progression in vivo, demonstrating a critical interaction in
the tumor/stroma microenvironment. J Clin Invest. 115:1163–1176.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mathur AN, Chang HC, Zisoulis DG,
Stritesky GL, Yu Q, et al: Stat3 and Stat4 direct development of
IL-17-secreting Th cells. J Immunol. 178:4901–4907. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Meng XN, Jin Y, Yu Y, Bai J, Liu GY, et
al: Characterisation of fibronectin-mediated FAK signalling
pathways in lung cancer cell migration and invasion. Br J Cancer.
101:327–334. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Antoniades HN, Neville-Golden J,
Galanopoulos T, Kradin RL, Valente AJ and Graves DT: Expression of
monocyte chemoat-tractant protein 1 mRNA in human idiopathic
pulmonary fibrosis. Proc Natl Acad Sci USA. 89:5371–5375. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guha M and Mackman N: LPS induction of
gene expression in human monocytes. Cell Signal. 13:85–94. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Langowski JL, Zhang X, Wu L, Mattson JD,
Chen T, et al: IL-23 promotes tumour incidence and growth. Nature.
442:461–465. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Haskill S, Yurochko AD and Isaacs KL:
Regulation of macrophage infiltration and activation in sites of
chronic inflammation. Ann NY Acad Sci. 664:93–102. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Abram CL and Lowell CA: The ins and outs
of leukocyte integrin signaling. Annu Rev Immunol. 27:339–362.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schlaepfer DD, Hanks SK, Hunter T and van
der Geer P: Integrin-mediated signal transduction linked to Ras
pathway by GRB2 binding to focal adhesion kinase. Nature.
372:786–791. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schlaepfer DD and Mitra SK: Multiple
connections link FAK to cell motility and invasion. Curr Opin Genet
Dev. 14:92–101. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sanders MA and Basson MD: Collagen
IV-dependent ERK activation in human Caco-2 intestinal epithelial
cells requires focal adhesion kinase. J Biol Chem. 275:38040–38047.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang C, Yang R, Yue D and Zhang Z:
Expression of FAK and PTEN in bronchioloalveolar carcinoma and lung
adenocarcinoma. Lung. 187:104–109. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dy GK: The role of focal adhesion kinase
in lung cancer. Anticancer Agents Med Chem. 13:581–583. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hood JD and Cheresh DA: Role of integrins
in cell invasion and migration. Nat Rev Cancer. 2:91–100. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hatamochi A, Aumailley M, Mauch C, Chu ML,
Timpl R and Krieg T: Regulation of collagen VI expression in
fibroblasts. Effects of cell density, cell-matrix interactions, and
chemical transformation. J Biol Chem. 264:3494–3499.
1989.PubMed/NCBI
|
|
44
|
Zou Y, Zhang RZ, Sabatelli P, Chu ML and
Bonnemann CG: Muscle interstitial fibroblasts are the main source
of collagen VI synthesis in skeletal muscle: implications for
congenital muscular dystrophy types Ullrich and Bethlem. J
Neuropathol Exp Neurol. 67:144–154. 2008.PubMed/NCBI
|
|
45
|
Schnoor M, Cullen P, Lorkowski J, Stolle
K, Robenek H, et al: Production of type VI collagen by human
macrophages: a new dimension in macrophage functional
heterogeneity. J Immunol. 180:5707–5719. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Eickelberg O, Kohler E, Reichenberger F,
Bertschin S, Woodtli T, et al: Extracellular matrix deposition by
primary human lung fibroblasts in response to TGF-β1 and TGF-β3. Am
J Physiol. 276:L814–L824. 1999.
|
|
47
|
Karouzakis E, Gay RE, Michel BA, Gay S and
Neidhart M: DNA hypomethylation in rheumatoid arthritis synovial
fibroblasts. Arthritis Rheum. 60:3613–3622. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jankowich MD and Rounds SI: Combined
pulmonary fibrosis and emphysema syndrome: a review. Chest.
141:222–231. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kitaguchi Y, Fujimoto K, Hanaoka M,
Kawakami S, Honda T and Kubo K: Clinical characteristics of
combined pulmonary fibrosis and emphysema. Respirology. 15:265–271.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Parsons JT, Slack-Davis J, Tilghman R and
Roberts WG: Focal adhesion kinase: targeting adhesion signaling
pathways for therapeutic intervention. Clin Cancer Res. 14:627–632.
2008. View Article : Google Scholar : PubMed/NCBI
|