Introduction
Oral cancer is the sixth most common cancer in the
world and the incidence of new cases indicates a continuing rise in
developing countries (1). Several
genetic and epigenetic alterations underlie the progressive
acquisition of a malignant phenotype in head and neck squamous cell
carcinoma (HNSCC) (2). The
molecular dissection of aberrant signaling networks, including
EGFR, Ras, NF-κB, Wnt/β-catenin, TGF-β, and PI3K-AKT-mTOR signaling
pathways, has increased our understanding of the basic mechanisms
controlling HNSCC progression (2,3).
Signal transducer and activator of transcription
(STAT) proteins constitute a family of transcription factors, which
exist in the cell as latent cytoplasmic transcription factors and
become activated in response to stimulation by cytokines and growth
factors (4,5). Once bound to their receptors, STATs
become phosphorylated, they dissociate from the receptor and form
homo- or heterodimers (4–7). STAT dimers then translocate to the
nucleus, where they interact with the promoters of target genes,
thus regulating transcription (4–7).
Phosphorylation of STAT molecules on a tyrosine
residue is the first critical event for their activation and has
been convincingly shown to correlate with STAT DNA binding and
transcriptional activity (4–7). In
contrast, STAT serine phosphorylation, which may also occur in
response to growth factor and cytokine stimulation, has been
associated mainly with negative regulation of STAT activity
(8–11). Other authors have indicated that
STAT1 and STAT3 are often phosphorylated in serine residues,
resulting in further STAT activation (12).
STAT signaling has been found to be involved in
oncogenesis (13,14). STAT3 persistent activation has been
convincingly shown to contribute to a malignant phenotype (13). Constitutive activation of STAT3 has
been associated with tumorigenesis in various types of human cancer
by stimulating specific target genes that enhance cell
proliferation and prevent apoptosis (13–15).
There is compelling evidence that STAT3 constitutive
activation, mainly associated with aberrant TGF-α/EGFR signaling,
is linked to HNSCC development and growth (2,14–18).
Previous findings suggested the existence of EGFR-independent STAT
oncogenic properties in HNSCC, including autocrine/paracrine
cytokine (IL-6, IL-10 or IL-22) stimulation or signaling through 7
nicotinic and erythropoietin receptor pathways (17,19–23).
Cytokine receptors are constitutively associated with members of
the Janus kinase (JAK) family of protein tyrosine kinases, whereas
growth factor receptors have intrinsic tyrosine kinase activity
(5).
Mitogen-activated protein kinase (MAPK) pathways are
evolutionarily conserved kinase modules that link extracellular
signals to the machinery that controls fundamental cellular
processes such as growth, proliferation, differentiation, migration
and apoptosis (24). MAPKs
phosphorylate serine and threonine residues of specific target
proteins (25). MAPKs are
classified into three major subfamilies, including extracellular
signal-regulated kinases (ERKs), p38 MAPKs, and c-Jun
NH2-terminal kinases (JNKs) (25–27).
Previous studies supported an association between
activation of specific members of the MAPK family and negative
regulation of STAT3 signaling in various cell types (8,28–34).
Chung et al showed that ERKs phosphorylate STAT3 on serine
727 (Ser727) in vitro and in vivo, while inhibiting
STAT3 tyrosine phosphorylation (8).
Similarly, Jain et al confirmed that ERKs induce STAT3
serine phosphorylation and suppress STAT3 tyrosine phosphorylation,
DNA binding and transcriptional activity induced by Src or Jak-2
(28). Moreover, Sengupta et
al provided evidence that activated ERKs induce a rapid
downregulation of IL-6-mediated STAT3 signaling through inhibition
of the upstream JAK kinases in several cell lines (29). Recently, Gough et al found
that the MEK-ERK pathway is required for activated Ras-induced
phosphorylation of mitochondrial STAT3 on Ser727, an important
process during cellular transformation (35). Also, Xue et al suggested that
treatment of human lung adenocarcinoma cells with RY10-4 affected
Bcl-2 family members, caspases, MMPs, TIMPs expression and ROS
production by inhibiting STAT3 activities through ERK and p38
pathways (36). Finally, Lee et
al found that blockade of MEK1/2-Erk1/2-RSK2 signaling by
silybin resulted in a reduced activation of NF-κB, activator
protein-1, and STAT3 in melanoma cells (37). Collectively, there is compelling
evidence to suggest that MAPK, especially ERK, activation has an
inhibitory effect on STAT3 signaling, manifested by downregulation
of STAT3 tyrosine phosphorylation and induction of STAT3 serine
phosphorylation, in various cell types including cancer cells.
The aim of the present investigation was to evaluate
whether oncogenic constitutive STAT3 signaling in oral squamous
cell carcinoma (OSCC) cells can be modulated by regulation of
specific MAPKs. The expression and activation status of STAT3 and
Erk1/2 in HNSCC cell lines were recorded and the effects of
selective Erk1/2 inhibition or activation on STAT3 signaling and
cellular proliferation were monitored, in an effort to elucidate
important molecular aspects of oral cancer with potential
therapeutic implications.
Materials and methods
Cell lines and cell culture
Experiments were performed using established cell
lines of human HNSCC (SCC9 and SCC25) obtained from the American
Type Culture Collection (ATCC; Manassas, VA, USA). Cells were
cultured in a 1:1 mixture of Ham’s F12 and DMEM containing 10%
fetal bovine serum (FBS), 100 units of penicillin, and 400 ng/ml
hydrocortisone (Sigma Chemical Co., St. Louis, MO, USA) at 37°C in
a 5% CO2 air atmosphere. Cells were subcultured by
disaggregation with trypsin (0.1%)-EDTA (0.01%) in
phosphate-buffered saline (PBS) at pH 7.5.
Selective inhibition of Erk1/2
Cells were plated in 6-well plates at a density of
5×104 cells/well and were allowed to grow to 80%
confluency. Then, they were treated either with the vehicle alone
(DMSO at a maximum concentration of 0.1%) or with the selective
MAPK (Erk1/2) inhibitor U0126 (Calbiochem, San Diego, CA, USA) at
concentrations of 20 and 50 μM for 24 h.
Selective induction of Erk1/2 MAPK
Cells were plated in 6-well plates at a density of
2×105 cells/well and were allowed to grow to 80%
confluency. They were then treated either with the vehicle alone
(DMSO at a maximum concentration of 0.1%) or with the selective
MAPK (Mek1/2) inducer (ProSpec, Israel) at concentrations of 2 and
5 μM for 48 h.
siRNA transfection
The human Erk1 and Erk2 specific siRNAs were based
on NCBI Reference Sequences (GenBank: Erk1: NM_002746 and Erk2:
NM_002745). Erk1/2 siRNA and scrambled control siRNA (siControl)
were purchased from Qiagen. All siRNA transfections were performed
using Lipofectamine 2000 (Invitrogen), according to the
manufacturer’s protocol, with final siRNA concentrations of 1 and
2.5 μM. OSCC cells were grown to mid-log phase and were transiently
transfected (2×106 cells) with 50 μg of the empty vector
or siRNA using Nucleofector reagents (Amaxa Biosystems,
Gaithersburg, MD, USA). Cells were collected at 48 h and whole
lysates were analyzed by western blotting.
Western blot experiments
Cells were washed twice with ice-cold PBS, followed
by lysis with radioimmunoprecipitation assay buffer (50 mM Tris pH
7.4, 150 mM NaCl, 1% Triton X-100, 1% deoxycholic acid, sodium
salt, 0.1% sodium dodecyl sulfate, 100 mg/ml phenylmethylsulfonyl
fluoride, 1 mg/ml aprotinin, 1 mM dichlorodiphenyltrichloroethane
and 1 mM sodium orthovanadate) for 10 min at 4°C. The wells were
scraped, and recovered cell products were centrifuged at 40,000 × g
for 15 min at 4°C. Recovered proteins were measured and equalized
using Bio-Rad Protein Assay (Bio-Rad Laboratories, Richmond, CA,
USA) as per the manufacturer’s instructions.
Western blotting was performed using antibodies
against: total STAT3, phospho-STAT3 (Tyr705), phospho-STAT3
(Ser727), total p44/42(Erk1/2) (Cell Signaling, Beverly, MA, USA),
phospho-Erk1/2 MAPK, (Upstate, Charlottesville, VA, USA), cyclin D1
(Cell Signaling) and β-actin (Sigma Chemical).
Cell proliferation and viability
Cells were counted with a hemocytometer under
inverted microscope. Cell viability after treatment was determined
by the trypan blue dye exclusion test 24 h after each treatment.
All assays were performed in quadruplicate and results are reported
as the mean ± SD.
Results
Effects of U0126 inhibitor on Erk1/2,
STAT3 and cyclin D1 protein expression and activation
The expression and activation status of Erk1/2 in
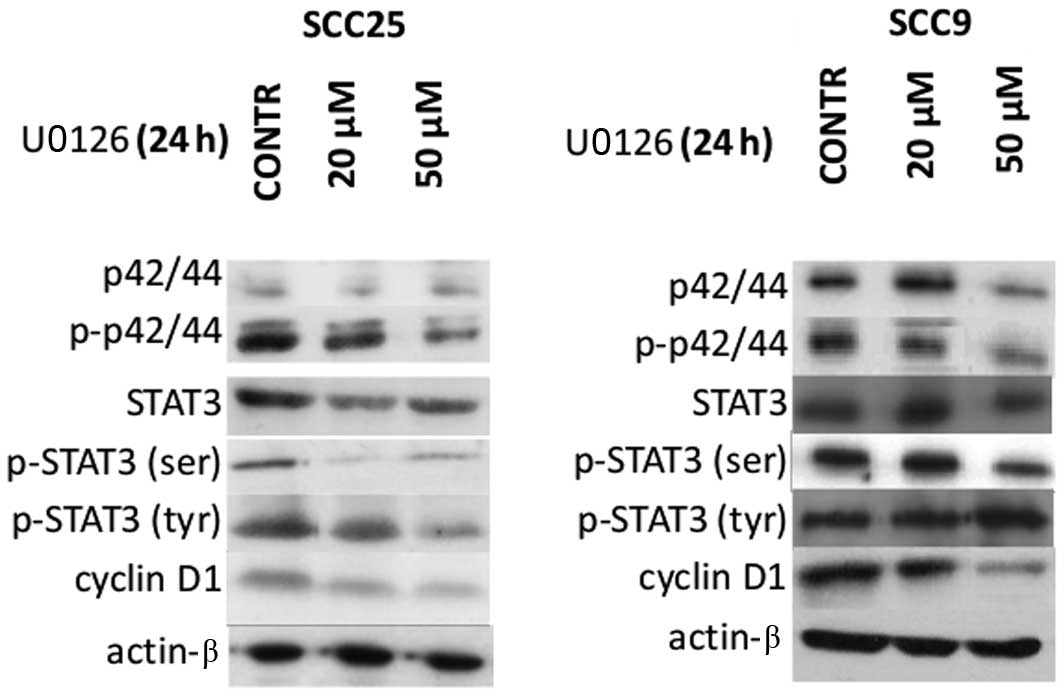
OSCC cells was examined first. According to western blotting
experiments, Erk1/2 (p42/44) total and phosphorylated (activated)
protein levels were detected in both OSCC cell lines tested (SCC9
and SCC25). Total, tyrosine phosphorylated (p-tyr) and serine
phosphorylated (p-ser) STAT3, as well cyclin D1 protein levels were
also observed in both cell lines (Fig.
1).
The effectiveness of Erk1/2 inhibition was then
assessed. Treatment of oral SCC25 cells with the Erk1/2 inhibitor
U0126 for 24 h resulted in inhibition of Erk1/2 phosphorylation,
which was more pronounced at the highest concentration used (50
μM). A decrease in total Erk1/2 protein expression levels following
20 or 50 μM of U0126 treatment was also noted in SCC25 cells. On
the other hand, U0126 treatment of oral SCC9 cells caused less
notable effects on Erk1/2 protein expression and phosphorylation
with decreases observed only at the highest concentration (50 μM)
(Fig. 1).
The effectiveness of Erk1/2 inhibition on STAT3
protein expression and activation levels was also examined. In oral
SCC25 cells, a significant reduction of p-ser STAT3 was detected
after 24 h of treatment with 20 or 50 μM of U0126; in contrast,
p-tyr STAT3 levels were not significantly affected. On the other
hand, treatment of SCC9 cells with U0126 led to a decrease of p-ser
STAT3 only when a concentration of 50 μM was used, along with a
moderate increase in p-tyr STAT3 levels. Total STAT3 levels were
not affected by U0126 treatment in either cell line (Fig. 1).
Moreover, western blot analysis demonstrated that
inhibition of Erk1/2 was associated with decreased levels of cyclin
D1 expression in a dose-dependent manner in both cell lines. In
contrast, the levels of actin remained stable throughout the
treatment, indicating that the observed effects on the
aforementioned proteins were not caused by a nonspecific reduction
of protein expression (Fig. 1).
Hence, Erk1/2 inhibition by U0126 treatment was more
potent in oral SCC25 cells and was associated with a decrease in
p-ser STAT3 and cyclin D1 levels without affecting p-tyr STAT3
levels. In contrast, U0126 treatment of oral SCC9 cells appeared to
be less effective in reducing Erk1/2 phosphorylation; nonetheless,
it induced decreases in p-ser STAT3 levels and cyclin D1 protein
expression as well as increases in p-tyr STAT3 levels at the
highest concentration used.
Effects of U0126 inhibitor on cell growth
and viability
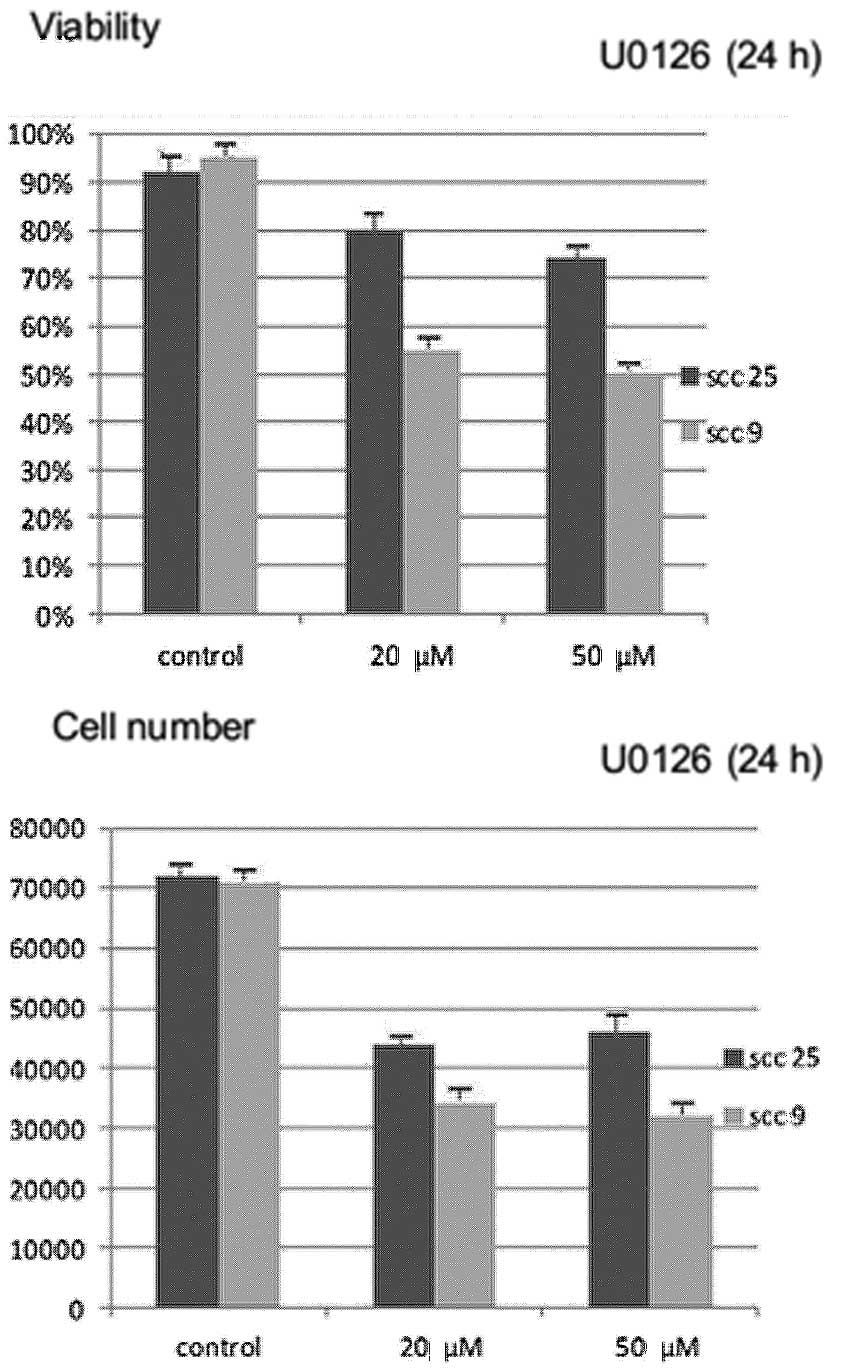
Treatment with U0126 for 24 h resulted in a
statistically significant (P<0.05) dose-dependent reduction in
cell growth (absolute number of cells) and cell viability (number
of viable cells) in both cell lines tested. The reduction appears
to be more prominent in the oral SCC9 cell line (Fig. 2).
Effects of Erk1/2 siRNA silencing on
STAT3 and cyclin D1 protein expression and activation
In order to corroborate the results from the
pharmacological inhibition of Erk1/2, specific inhibition was
performed by siRNA-targeting of Erk1/2 in both cell lines.
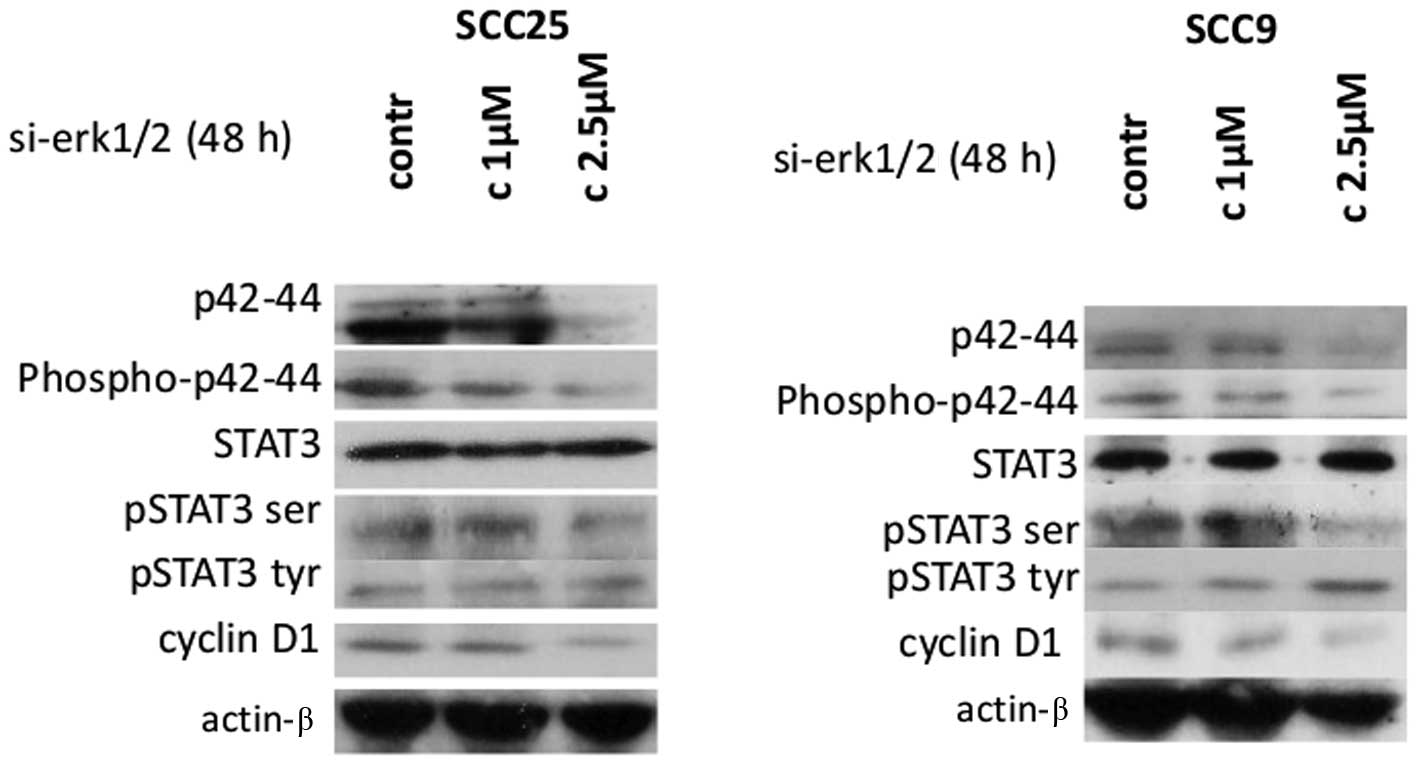
Following 48 h of transfection, western blotting revealed that
si-RNA against Erk1/2 efficiently silenced Erk1/2 causing
dose-dependent decreases in total and phosphorylated p42/44 Erk1/2
protein levels compared to control-transfected cells in both cell
lines (Fig. 3).
Decreases in Erk1/2 protein expression and
phosphorylation correlated with a decrease in p-ser STAT3 in both
cell lines, after 48 h of treatment with 2.5 μM of specific siRNA
against Erk1/2. Regarding STAT3 tyrosine phosphorylation, an
upregulation of p-tyr-STAT3 protein levels was detected,
particularly at the highest concentration, in oral SCC9 cells. On
the other hand, siRNA treatment against Erk1/2 did not appear to
cause any observable change in p-tyr-STAT3 levels in oral SCC25
cells. Total STAT3 protein levels were not affected in either cell
line (Fig. 3).
Furthermore, western blot analysis demonstrated that
silencing of Erk1/2 was associated with substantially decreased
levels of cyclin D1 protein expression in a dose-dependent manner
in both cell lines. Finally, the levels of actin remained stable
(Fig. 3). Therefore, specific
silencing of Erk1/2 resulted in a decrease in p-ser STAT3 and
cyclin D1 levels in both cell lines and an increase in p-tyr-STAT3,
particularly in oral SCC9 cells.
Effects of silencing Erk1/2 on cell
growth and viability
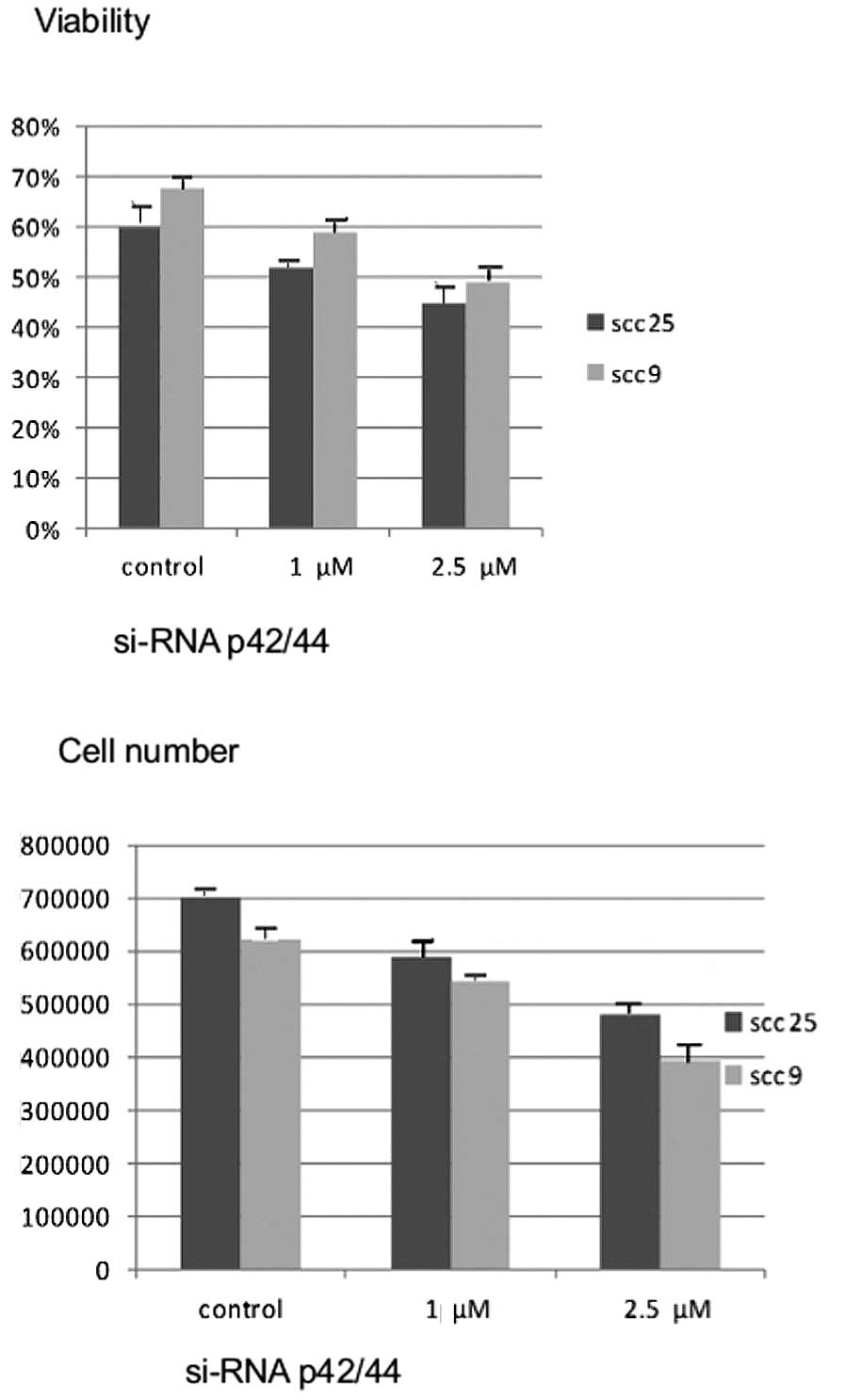
Similar to the effects of chemical inhibition with
U0126, 48 h of treatment with siRNA against Erk1/2 resulted in a
dose-dependent reduction in cell growth and cell viability in both
cell lines (P<0.05) (Fig.
4).
Effects of Erk1/2 induction on STAT3 and
cyclin D1 protein expression and activation
In order to further investigate the significance of
Erk1/2 for STAT3 and cyclin D1 modulation, pharmacological
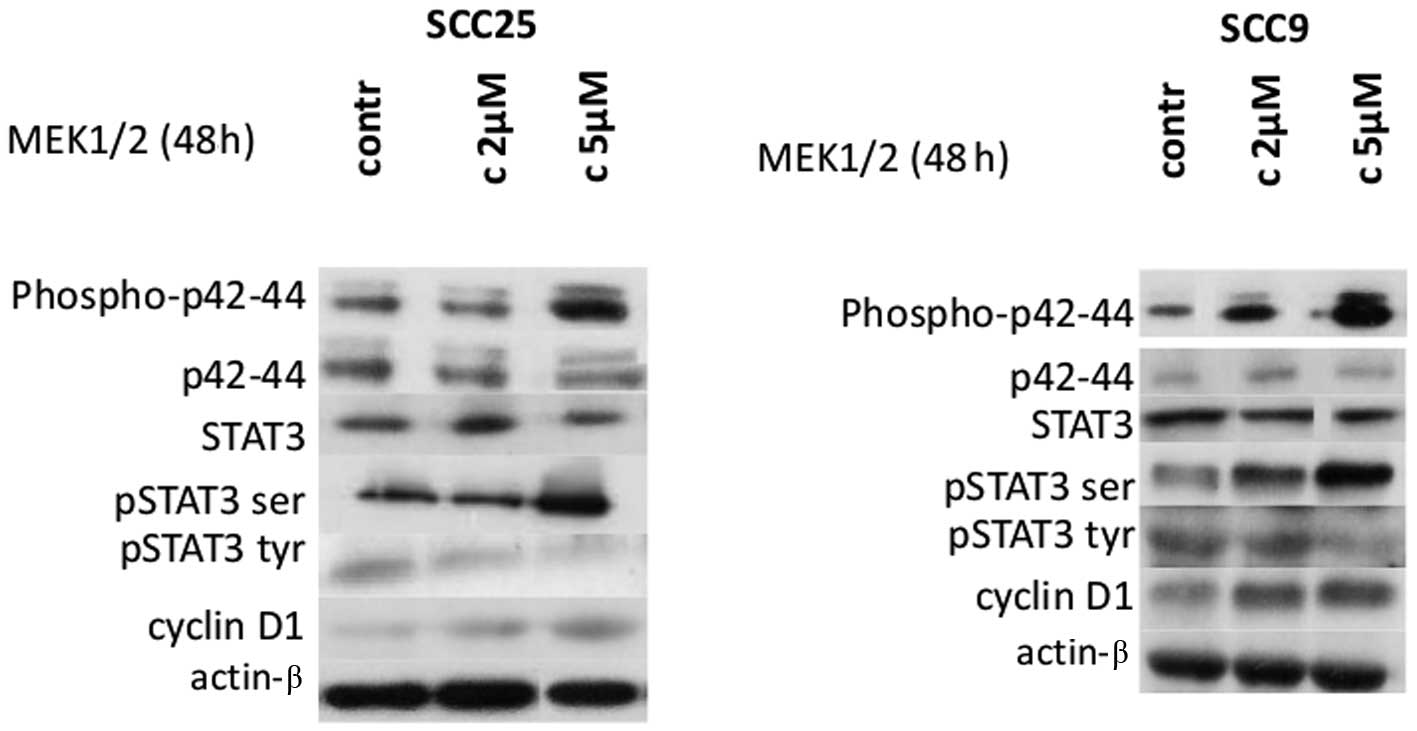
induction of Erk1/2 using active MEK1/2 was performed. Treatment of
cells with selective MEK1/2 inducer efficiently upregulated
phosphorylated Erk1/2 levels in a dose-dependent manner in both
cell lines without affecting total Erk1/2 levels, as expected
(Fig. 5).
Treatment of both cell lines with Erk1/2 inducer for
48 h resulted in significant induction of STAT3 phosphorylation on
Ser727, especially at a concentration of 5 μm/ml. In contrast,
p-tyr-STAT3 levels appeared to decrease after treatment with active
MEK1/2. Total STAT3 levels were not affected by Erk1/2 induction in
either cell line (Fig. 5).
With regards to cyclin D1, active MEK1/2 treatment
of both cell lines for 48 h caused an upregulation in cyclin D1
expression levels, particularly at the higher concentration.
Finally, actin protein levels remained stable throughout treatment
(Fig. 5). Thus, Erk1/2 induction
caused upregulation of p-ser STAT3 and cyclin D1 levels in both
cell lines and a decrease in p-tyr-STAT3.
Effects of Erk1/2 induction on cell
growth and viability
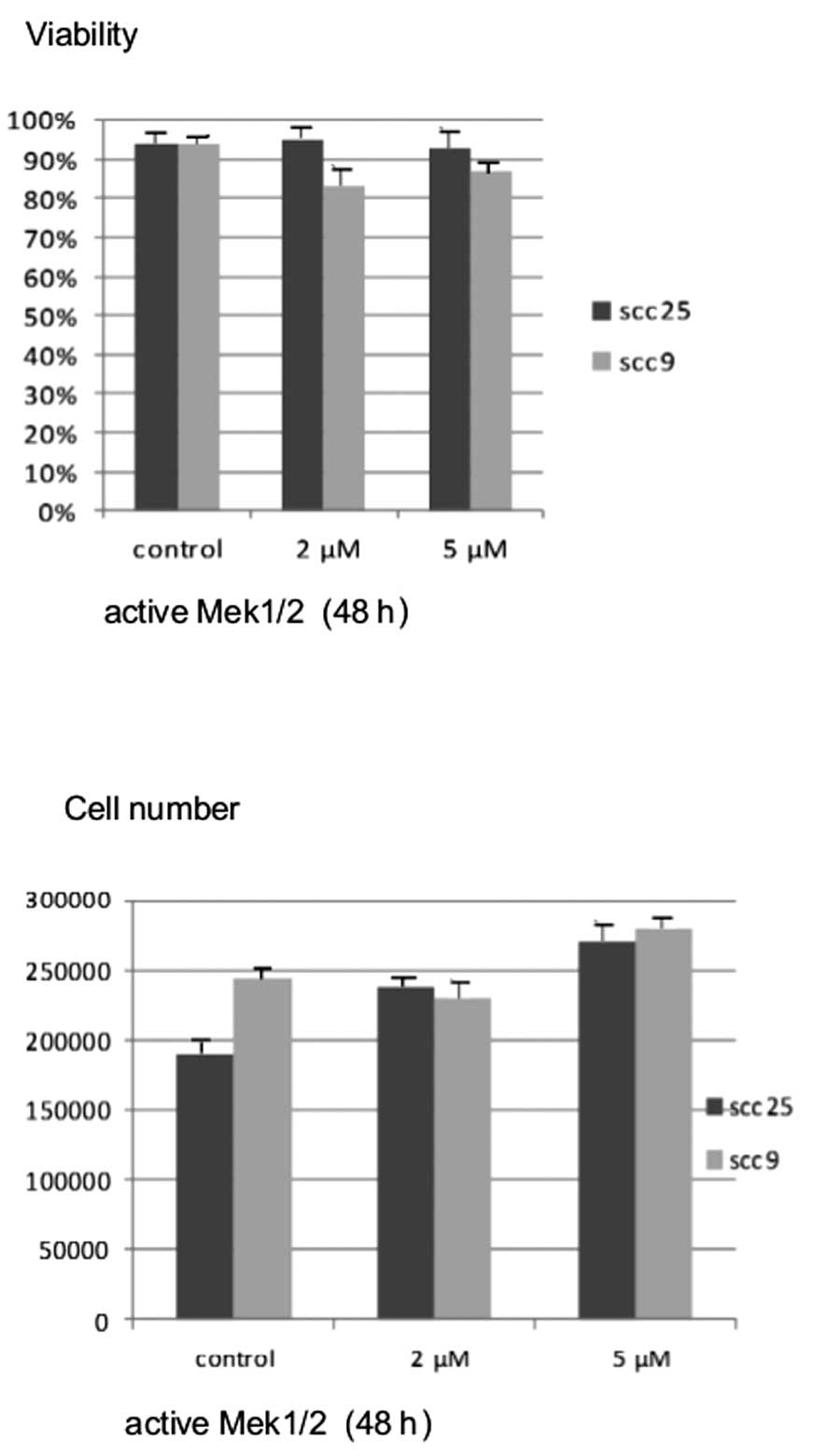
Active MEK1/2 treatment at the highest concentration
for 48 h resulted in a significant dose-dependent increase in cell
growth, which was more prominent in the oral SCC25 cell line
(P<0.05). On the contrary, treatment of cells with active MEK1/2
for 48 h did not appear to induce notable changes in cell viability
in either cell line (Fig. 6).
Discussion
The oncogenic role of MAPK signaling pathway has
been the focus of numerous studies indicating that aberrant MAPK
expression alters differentiation and deregulates proliferation and
apoptosis in several types of cancer (38,39).
In particular, ERK is a member of an oncogenic pathway activated by
the upstream oncoproteins Ras and Raf (24,40).
Upon activation, Erk1/2 phosphorylates either cytoplasmic
downstream targets, including p90 ribosomal S6 kinase, or nuclear
substrates (24,41). In the nucleus, ERK phosphorylates an
array of targets, including transcription factors and the family of
mitogen and stress-activated protein kinases (MSKs) (42). ERK nuclear targets also include the
Ternary Complex Factor (TCF), which plays a crucial role in
enhancing expression of the immediate early genes, such as c-Fos
and c-Myc (39,43).
The significance of the MEK/ERK pathway has been
shown in various types of cancer, such as liver, prostate and
malignant melanoma cancer cell growth in vitro and in
vivo (44–46). For example, knockdown of
serine/threonine kinase Mirk/Dyrk1B by siRNA in either ovarian
cancer or non-small cell lung cancer (NSCLC) cells led to
upregulated activation of c-Raf-MEK-ERK, followed by increased
growth rate (47). Induction of
anti-apoptotic proteins, such as Bcl2, Bclxl and inhibition of
pro-apoptotic factors, including Bad, has been correlated with the
oncogenic potential of ERK activity (48).
Deregulation of ERK has also been described in HNSCC
and various studies have suggested that ERK inhibition correlates
with reduced proliferation and induced apoptosis in OSCC (49,50).
Wang et al suggested that overexpression of activated Erk1/2
and cyclin D1 might be related to cell cycle regulation and cell
proliferation in OSCC (51).
Bancroft et al (52) showed
the involvement of Erk1/2 in AP-1 and NF-κB induction of VEGF
expression in OSCC cell lines SCC9 and SCC11, and Duvvuri et
al (53) found that
overexpression of Erk1/2, induced by receptor-activated
calcium-dependent chloride channel (TMEM16A), is associated with
enhanced cell proliferation in HNSCC. Recently, Li et al
demonstrated that knockdown of kinase suppressor of Ras 1 and
suppression of the Raf-MEK-ERK pathway reduced proliferation and
induced apoptosis in OSCC cells (54).
In agreement with the aforementioned findings, our
results are in support of the oncogenic role of ERK in oral cancer.
In the present study, possible effects of ERK modulation on OSCC
cell proliferation and viability were assessed. In particular,
Erk1/2 inhibition caused a dose-dependent decrease in the absolute
number of living cells along with downregulation of cyclin D1
levels in both OSCC cell lines tested. These findings corroborate
previous studies that demonstrated negative regulation of cell
growth and cyclin D1 levels following Erk1/2 inhibition by means of
U0126 treatment (44,47,55,56).
In our study, selective siRNA Erk1/2 inhibition
induced similar results to those of Erk1/2 pharmacological
inhibition in a dose-dependent manner in both cell lines, followed
by a corresponding decrease in the absolute number of living cells
and cell viability levels. Using similar siRNA techniques, Bessard
et al underlined the crucial role of the MEK/ERK pathway in
liver cancer cell growth in vitro and in vivo
demonstrating that RNAi-mediated Erk2 knockdown inhibits tumor cell
growth (44). Similarly, Si et
al showed that downregulation of Erk1/2 by siRNA inhibited the
growth and invasion of human osteosarcoma cells, and found that the
knockdown of Erk1/2 made cancer cells more sensitive to cisplatin
treatment (57). Moreover, Dumesic
et al reported that combined siRNA-induced knockdown of
Erk1/2 caused epidermal hypoplasia and hypoproliferation without
disrupting differentiation in human epidermis (58). Finally, Duvvuri et al found
that genetic inactivation of Erk1/2 using siRNA abrogated the
Erk1/2 mediated growth effects of TMEM16A in HNSCC (53).
Furthermore, we demonstrated that pharmacological
induction of Erk1/2 appears to have the opposite effects. Notably,
the increase in absolute cell number is accompanied by a relative
increase in cyclin D1. In agreement, Gao et al suggested
that activation of the c-Raf-MEK-Erk1/2 pathway leads to subsequent
increase in cell cycle proteins (cyclin D1, p27kip1), accompanied
by an increased growth rate and transition of cells from G0/G1 into
the S phase of the cell cycle in both ovarian and non-small cell
lung cancer (47). Similarly, Wang
et al indicated that overexpression of activated Erk1/2 and
cyclin D1 proteins are involved in oral tongue carcinogenesis
(51). Finally, Judd et al
(59) reported that MEK1 activation
enhances CD44 expression and promotes aggressiveness in HNSCC,
while Katada et al (60) and
Zuo et al (61) suggested
that Erk1/2 activation correlates with increased cell migration and
invasion in HNSCC.
A number of findings indicate that STAT3 is
constitutively activated and participates in the regulation of cell
proliferation, differentiation and apoptosis in HNSCC (17,18).
Aberrant STAT3 activation, manifested by increases in STAT3
tyrosine phosphorylation, is considered as a potent promoter of
HNSCC initiation and progression, thereby inhibition of STAT3 has
been identified as a potential therapeutic target for the treatment
of HNSCC (16,18,62).
STAT3 constitutive activation in HNSCC is driven by a number of
upstream signal transduction pathways. Ligand-induced stimulation
of the receptor results in phosphorylation of tyrosine residues
within the receptor, which is the first critical event for their
activation and has been shown to correlate with STAT DNA binding
and transcriptional activity (4,5,7–9).
The oncogenic potential of STAT3 depends mainly on the
phosphorylation status of Tyr705 whereas the role of STAT3 serine
phosphorylation is more controversial (7). There are several lines of evidence
supporting a negative impact of Ser727 residue phosphorylation on
STAT3 activity. Previous studies have demonstrated that Ser727
phosphorylation negatively regulates STAT3 tyrosine
phosphorylation, which is required for dimer formation and the
subsequent nuclear translocation and transcriptional activity
(8). Similarly, Decker and Kovarik
proposed that STAT3 phosphorylation on Ser727 either inhibits
tyrosine phosphorylation or increases tyrosine dephosphorylation
(9). A negative relationship
between STAT3 serine and tyrosine phosphorylation has also been
suggested by Venkatasubbarao et al (10) and Wakahara et al (63), who described that phospho-Ser727
determines the duration of STAT3 activity by enhancing
dephosphorylation of phospho-Tyr705 largely through TC45
phosphatase.
However, other investigators suggested that STAT3
serine phosphorylation is associated with increased nuclear
translocation and serves to maximize transcriptional activity
(6,64). Hazan-Halevy et al reported
that constitutive phosphorylation of STAT3 on Ser727 residues is a
critical event in chronic lymphocytic leukemia (CLL) and may serve
as a potent therapeutic target (64). Miyakoshi et al showed that
FBS treatment of mouse hepatic carcinoma cells induced STAT3
phosphorylation on Ser727 via MAPK activation, followed by STAT3
nuclear translocation and cell proliferation (66). In contrast, IL-6 treatment induced
STAT3 phosphorylation on Tyr705 phosphorylation through JAK
activation without increasing STAT3 nuclear translocation and cell
proliferation. Sakaguchi et al investigated the oncogenic
role of Ser727 STAT3 in melanoma cells and suggested that
constitutive Ser727 phosphorylation, partially mediated by the
B-Raf-MEK-Erk1/2 pathway, has a role in the regulation of cell
survival activity and nuclear translocation of STAT3 in melanocytes
(65). Moreover, Gough et al
proposed that the MEK-ERK pathway is required for activated
Ras-induced phosphorylation of STAT3 on Ser727 and that
mitochondrial STAT3 is one of the critical substrates of the
Ras-MEK-ERK-axis during cellular transformation (35).
One of the major downstream signaling routes of
MAPKs are STAT proteins and MAPKs have been implicated in the
regulation of STAT proteins through crosstalk signaling (9).
Considering the significance of STAT3 and ERK
signaling in several types of cancer, the possibility of a
crosstalk between these two major oncogenic pathways in oral cancer
was explored in the present study. In particular, changes in the
expression and activation status of Erk1/2 MAPK and their effect on
STAT3 tyrosine and/or serine phosphorylation and total STAT3 levels
in OSCC cell lines were investigated. Chemical inhibition (via
U0126) or selective targeting (via siRNA) of Erk1/2 MAPK
downregulated STAT3 serine phosphorylation in both cell lines used;
in addition, a moderate increase in STAT3 tyrosine (Y705)
phosphorylation was observed in oral SCC9 cells. In contrast,
induction of ERK in OSCC cells resulted in upregulation of STAT3
serine phosphorylation and downregulation of STAT3 tyrosine
phosphorylation. Previous studies suggested that a crosstalk exists
between ERK and STAT3, indicating that ERK either induces Ser727
phosphorylation (8,28,35) or
downregulates IL-6-mediated STAT3 signaling (29). In pancreatic cancer cells, ERK
activation has been associated with STAT3 negative regulation
manifested by decreased tyrosine phosphorylation levels (10). Similar results were obtained by
Tkach et al, who found that U0126 suppresses phosphorylation
of STAT3 on Ser727 in breast cancer cells (33). In addition, Wierenga et al,
indicated that U0126 abrogated the EPO-mediated STAT3 Ser727
phosphorylation without an effect on tyrosine phosphorylation in
erythroid cells (67). Nelson et
al found that the combination of nifuroxazide and U0126
inhibits STAT3 tyrosine phosphorylation in multiple myeloma cells
(68). Furthermore, Chen et
al reported that Erk1/2 activation phosphorylates STAT3 on
Ser727 and regulates cell proliferation in human bladder cancer
cells (34). On the other hand,
Sumimoto et al reported that U0126 inhibits ERK
phosphorylation, but not STAT3 phosphorylation on Ser727 or Tyr705
residues in human melanoma cells (69). It can be hypothesized that the
significance of Erk1/2 and STAT3 crosstalk differs according to the
cancer cell types studied.
In summary, our data are supportive of the oncogenic
potential of Erk1/2 in OSCC, which appears to contribute to cell
proliferation. It is possible that pharmacologic inhibition of
Erk1/2 activity or targeting Erk1/2 genes by gene therapy may offer
an alternative strategy for the treatment of patients with OSCC. On
the other hand, the oncogenic STAT3 constitutive signaling in OSCC
cells appears to be negatively regulated by Erk1/2. The
Erk1/2-STAT3 crosstalk appears to involve mainly ERK-induced
upregulation of STAT3 Ser727 phosphorylation while Tyr705
phosphorylation does not exhibit major changes. It is possible that
the role of Erk1/2 in STAT3 modulation varies according to the type
and status of the cells studied, indicating the need to identify
the role of MAPK activation in relation to STAT3 signaling in
specific cell types.
Acknowledgements
This study was co-financed by the European Union
(European Social Fund - ESF) and Greek national funds through the
Operational Program ‘Education and Lifelong Learning’ of the
National Strategic Reference Framework (NSRF) - Research Funding
Program: Heracleitus II. Investing in knowledge society through the
European Social Fund.
References
|
1
|
Curado MP and Hashibe M: Recent changes in
the epidemiology of head and neck cancer. Curr Opin Oncol.
21:194–200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Molinolo AA, Amornphimoltham P, Squarize
CH, et al: Dysregulated molecular networks in head and neck
carcinogenesis. Oral Oncol. 45:324–334. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Choi S and Myers JN: Molecular
pathogenesis of oral squamous cell carcinoma: implications for
therapy. J Dent Res. 87:14–32. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bromberg J and Darnell JE Jr: The role of
STATs in transcriptional control and their impact on cellular
function. Oncogene. 19:2468–2473. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rane SG and Reddy EP: Janus kinases:
components of multiple signaling pathways. Oncogene. 19:5662–5679.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Aggarwal BB, Kunnumakkara AB, Harikumar
KB, et al: Signal transducer and activator of transcription-3,
inflammation, and cancer: how intimate is the relationship? Ann NY
Acad Sci. 1171:59–76. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Reich NC and Liu L: Tracking STAT nuclear
traffic. Nat Rev Immunol. 6:602–612. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chung J, Uchida E, Grammer TC and Blenis
J: STAT3 serine phosphorylation by ERK-dependent and -independent
pathways negatively modulates its tyrosine phosphorylation. Mol
Cell Biol. 17:6508–6516. 1997.PubMed/NCBI
|
|
9
|
Decker T and Kovarik P: Serine
phosphorylation of STATs. Oncogene. 19:2628–2637. 2000. View Article : Google Scholar
|
|
10
|
Venkatasubbarao K, Choudary A and Freeman
JW: Farnesyl transferase inhibitor (R115777)-induced inhibition of
STAT3(Tyr705) phosphorylation in human pancreatic cancer cell lines
require extracellular signal-regulated kinases. Cancer Res.
65:2861–2871. 2005. View Article : Google Scholar
|
|
11
|
Lim CP and Cao X: Serine phosphorylation
and negative regulation of STAT3 by JNK. J Biol Chem.
274:31055–31061. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
O’Shea JJ, Gadina M and Schreiber RD:
Cytokine signaling in 2002: new surprises in the Jak/Stat pathway.
Cell. 109:S121–S131. 2002.PubMed/NCBI
|
|
13
|
Bromberg J: Stat proteins and oncogenesis.
J Clin Invest. 109:1139–1142. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Song JI and Grandis JR: STAT signaling in
head and neck cancer. Oncogene. 19:2489–2495. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Grandis JR, Drenning SD, Zeng Q, et al:
Constitutive activation of Stat3 signaling abrogates apoptosis in
squamous cell carcinogenesis in vivo. Proc Natl Acad Sci USA.
97:4227–4232. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nikitakis NG, Siavash H and Sauk JJ:
Targeting the STAT pathway in head and neck cancer: recent advances
and future prospects. Curr Cancer Drug Targets. 4:637–651. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kijima T, Niwa H, Steinman RA, et al:
STAT3 activation abrogates growth factor dependence and contributes
to head and neck squamous cell carcinoma tumor growth in vivo. Cell
Growth Differ. 13:355–362. 2002.PubMed/NCBI
|
|
18
|
Leeman RJ, Lui VW and Grandis JR: STAT3 as
a therapeutic target in head and neck cancer. Expert Opin Biol
Ther. 6:231–241. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Siavash H, Nikitakis NG and Sauk JJ:
Abrogation of IL-6-mediated JAK signalling by the cyclopentenone
prostaglandin 15d-PGJ(2) in oral squamous carcinoma cells. Br J
Cancer. 91:1074–1080. 2004.PubMed/NCBI
|
|
20
|
Lee TL, Yeh J, Van Waes C and Chen Z:
Epigenetic modification of SOCS-1 differentially regulates STAT3
activation in response to interleukin-6 receptor and epidermal
growth factor receptor signaling through JAK and/or MEK in head and
neck squamous cell carcinomas. Mol Cancer Ther. 5:8–19. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Naher L, Kiyoshima T, Kobayashi I, et al:
STAT3 signal transduction through interleukin-22 in oral squamous
cell carcinoma. Int J Oncol. 41:1577–1586. 2012.PubMed/NCBI
|
|
22
|
Jewett A, Head C and Cacalano NA: Emerging
mechanisms of immunosuppression in oral cancers. J Dent Res.
85:1061–1073. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lai SY and Johnson FM: Defining the role
of the JAK-STAT pathway in head and neck and thoracic malignancies:
implications for future therapeutic approaches. Drug Resist Updat.
13:67–78. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Maggioni D, Gaini R, Nicolini G, et al:
MAPKs activation in head and neck squamous cell carcinomas. Oncol
Rev. 5:223–231. 2011. View Article : Google Scholar
|
|
25
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pearce AK and Humphrey TC: Integrating
stress-response and cell-cycle checkpoint pathways. Trends Cell
Biol. 11:426–433. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dunn C, Wiltshire C, MacLaren A and
Gillespie DA: Molecular mechanism and biological functions of c-Jun
N-terminal kinase signalling via the c-Jun transcription factor.
Cell Signal. 14:585–593. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jain N, Zhang T, Fong SL, et al:
Repression of Stat3 activity by activation of mitogen-activated
protein kinase (MAPK). Oncogene. 17:3157–3167. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sengupta TK, Talbot ES, Scherle PA and
Ivashkiv LB: Rapid inhibition of interleukin-6 signaling and Stat3
activation mediated by mitogen-activated protein kinases. Proc Natl
Acad Sci USA. 95:11107–11112. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Quadros MR, Peruzzi F, Kari C and Rodeck
U: Complex regulation of signal transducers and activators of
transcription 3 activation in normal and malignant keratinocytes.
Cancer Res. 64:3934–3939. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kovarik P, Stoiber D, Eyers PA, et al:
Stress-induced phosphorylation of STAT1 at Ser727 requires p38
mitogen-activated protein kinase whereas IFN-gamma uses a different
signaling pathway. Proc Natl Acad Sci USA. 96:13956–13961. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ahmed ST, Mayer A, Ji JD and Ivashkiv LB:
Inhibition of IL-6 signaling by a p38-dependent pathway occurs in
the absence of new protein synthesis. J Leukoc Biol. 72:154–162.
2002.PubMed/NCBI
|
|
33
|
Tkach M, Rosemblit C, Rivas MA, et al:
p42/p44 MAPK-mediated Stat3Ser727 phosphorylation is required for
progestin-induced full activation of Stat3 and breast cancer
growth. Endocr Relat Cancer. 20:197–212. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen RJ, Ho YS, Guo HR and Wang YJ: Rapid
activation of Stat3 and ERK1/2 by nicotine modulates cell
proliferation in human bladder cancer cells. Toxicol Sci.
104:283–293. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gough D, Koetz L and Levy D: The MEK-ERK
pathway is necessary for serine phosphorylation of mitochondrial
STAT3 and Ras-mediated transformation. PLoS One. 8:e833952013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xue P, Zhao Y, Liu Y, Yuan Q, et al: A
novel compound RY10-4 induces apoptosis and inhibits invasion via
inhibiting STAT3 through ERK-, p38-dependent pathways in human lung
adenocarcinoma A549 cells. Chem Biol Interact. 209:25–34. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee MH, Huang Z, Kim DJ, et al: Direct
targeting of MEK1/2 and RSK2 by silybin induces cell-cycle arrest
and inhibits melanoma cell growth. Cancer Prev Res. 6:455–465.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Aguzzi A, Maggioni D, Nicolini G, et al:
MAP kinase modulation in squamous cell carcinoma of the oral
cavity. Anticancer Res. 29:303–308. 2009.PubMed/NCBI
|
|
39
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen Z, Gibson TB, Robinson F, et al: MAP
kinases. Chem Rev. 101:2449–2476. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lee SH, Lee JW, Soung YH, et al:
Colorectal tumors frequently express phosphorylated
mitogen-activated protein kinase. APMIS. 112:233–238. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mendoza MC, Er EE and Blenis J: The
Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends
Biochem Sci. 36:320–328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Anjum R and Blenis J: The RSK family of
kinases: emerging roles in cellular signalling. Nat Rev Mol Cell
Biol. 9:747–758. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bessard A, Frémin C, Ezan F, et al:
RNAi-mediated ERK2 knockdown inhibits growth of tumor cells in
vitro and in vivo. Oncogene. 27:5315–5325. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Obajimi O and Melera P: Suppression of
ERK1/2 with siRNA restores drug sensitivity in DU145 cells selected
for resistance to AG2034 (abstract 77). Cancer Res. 70(Suppl
1)2010. View Article : Google Scholar
|
|
46
|
Smalley KS: A pivotal role for ERK in the
oncogenic behaviour of malignant melanoma? Int J Cancer.
104:527–532. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gao J, Zhao Y, Lv Y, et al: Mirk/Dyrk1B
mediates G0/G1 to S phase cell cycle progression and cell survival
involving MAPK/ERK signaling in human cancer cells. Cancer Cell
Int. 13:22013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Balmanno K and Cook SJ: Tumour cell
survival signalling by the ERK1/2 pathway. Cell Death Differ.
16:368–377. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lin YC, Wu MH, Wei TT, et al: Degradation
of epidermal growth factor receptor mediates dasatinib-induced
apoptosis in head and neck squamous cell carcinoma cells.
Neoplasia. 14:463–475. 2012.PubMed/NCBI
|
|
50
|
Ji WT, Chen HR, Lin CH, et al: Monocyte
chemotactic protein 1 (MCP-1) modulates pro-survival signaling to
promote progression of head and neck squamous cell carcinoma. PLoS
One. 9:e889522014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang L, Liu T, Nishioka M, et al:
Activation of ERK1/2 and cyclin D1 expression in oral tongue
squamous cell carcinomas: relationship between clinicopathological
appearances and cell proliferation. Oral Oncol. 42:625–631. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bancroft CC, Chen Z, Dong G, et al:
Coexpression of proangiogenic factors IL-8 and VEGF by human head
and neck squamous cell carcinoma involves coactivation by MEK-MAPK
and IKK-NF-kappaB signal pathways. Clin Cancer Res. 7:435–442.
2001.PubMed/NCBI
|
|
53
|
Duvvuri U, Shiwarski DJ, Xiao D, et al:
TMEM16A induces MAPK and contributes directly to tumorigenesis and
cancer progression. Cancer Res. 72:3270–3281. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Li B, Lu L, Zhong M, Tan XX, et al:
Terbinafine inhibits KSR1 and suppresses Raf-MEK-ERK signaling in
oral squamous cell carcinoma cells. Neoplasma. 60:406–412. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lavoie JN, L’Allemain G, Brunet A, et al:
Cyclin D1 expression is regulated positively by the p42/p44MAPK and
negatively by the p38/HOGMAPK pathway. J Biol Chem.
271:20608–20616. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhao Y, Lv M, Lin H, et al: Rho-associated
protein kinase isoforms stimulate proliferation of vascular smooth
muscle cells through ERK and induction of cyclin D1 and PCNA.
Biochem Biophys Res Commun. 432:488–493. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Si H, Peng C, Li J, Wang X, et al:
RNAi-mediated knockdown of ERK1/2 inhibits cell proliferation and
invasion and increases chemosensitivity to cisplatin in human
osteosarcoma U2-OS cells in vitro. Int J Oncol.
40:1291–1297. 2012.PubMed/NCBI
|
|
58
|
Dumesic PA, Scholl FA, Barragan DI and
Khavari PA: Erk1/2 MAP kinases are required for epidermal G2/M
progression. J Cell Biol. 185:409–422. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Judd NP, Winkler AE, Murillo-Sauca O, et
al: ERK1/2 regulation of CD44 modulates oral cancer aggressiveness.
Cancer Res. 72:365–374. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Katada K, Tomonaga T, Satoh M, et al:
Plectin promotes migration and invasion of cancer cells and is a
novel prognostic marker for head and neck squamous cell carcinoma.
J Proteomics. 75:1803–1815. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zuo JH, Zhu W, Li MY, et al: Activation of
EGFR promotes squamous carcinoma SCC10A cell migration and invasion
via inducing EMT-like phenotype change and MMP-9-mediated
degradation of E-cadherin. J Cell Biochem. 112:2508–2517. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Li R, You S, Hu Z, et al: Inhibition of
STAT3 by niclosamide synergizes with erlotinib against head and
neck cancer. PLoS One. 8:e746702013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wakahara R, Kunimoto H, Tanino K, et al:
Phospho-Ser727 of STAT3 regulates STAT3 activity by enhancing
dephosphorylation of phospho-Tyr705 largely through TC45. Genes
Cells. 17:132–145. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hazan-Halevy I, Harris D, Liu Z, Liu J, et
al: STAT3 is constitutively phosphorylated on serine 727 residues,
binds DNA, and activates transcription in CLL cells. Blood.
115:2852–2863. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sakaguchi M, Oka M, Iwasaki T, et al: Role
and regulation of STAT3 phosphorylation at Ser727 in melanocytes
and melanoma cells. J Invest Dermatol. 132:1877–1885. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Miyakoshi M, Yamamoto M, Tanaka H and
Ogawa K: Serine 727 phosphorylation of STAT3: an early change in
mouse hepatocarcinogenesis induced by neonatal treatment with
diethylnitrosamine. Mol Carcinog. 53:67–76. 2014. View Article : Google Scholar
|
|
67
|
Wierenga AT, Vogelzang I, Eggen BJ and
Vellenga E: Erythropoietin-induced serine 727 phosphorylation of
STAT3 in erythroid cells is mediated by a MEK-, ERK-, and
MSK1-dependent pathway. Exp Hematol. 31:398–405. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Nelson EA, Walker SR, Kepich A, et al:
Nifuroxazide inhibits survival of multiple myeloma cells by
directly inhibiting STAT3. Blood. 112:5095–5102. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Sumimoto H, Imabayashi F, Iwata T and
Kawakami Y: The BRAF-MAPK signaling pathway is essential for
cancer-immune evasion in human melanoma cells. J Exp Med.
203:1651–1656. 2006. View Article : Google Scholar : PubMed/NCBI
|