Introduction
Colon cancer, the second leading cause of
cancer-related mortality in the USA, has become a common malignancy
in Asia with recent changes in diet (1). Radiotherapy is a standard therapy in
the adjuvant treatment of resected colon and rectum cancers
(2), and its combination with
chemotherapy has been shown to reduce local failure and distant
metastasis further, thereby improving the outcome of treatment
(3,4). One potential chemotherapeutic agent
for this, sorafenib (Nexavar, BAY43-9006), is an oral multikinase
inhibitor that blocks tumor cell proliferation and angiogenesis,
and induces tumor cell apoptosis by inhibiting serine/threonine
kinases (c-RAF and mutant and wild-type BRAF) as well as the
receptor tyrosine kinases vascular endothelial growth factor
receptor 2 and 3 (VEGFR2 and VEGFR3), platelet-derived growth
factor receptor β, FLT3 and c-KIT (5). Sorafenib is currently used in clinics
to treat patients with advanced renal cell carcinoma,
hepatocellular carcinoma and thyroid cancer (6–10).
Moreover, preliminary data from a series of studies in which
sorafenib was used in combination with a variety of anticancer
agents for various solid tumors has been published (11).
In the present study, we investigated the mechanism
by which sorafenib enhances radiation-induced antitumor and
anti-angiogenesis effects in colorectal cancer cells. Collectively,
our results showed that sorafenib can be successfully combined with
a radiation regimen to potentiate its antitumor and
anti-angiogenesis activities. Our study provides a scientific
rationale to evaluate this combination strategy in clinical
trials.
Materials and methods
Antibodies and chemicals
Anti-cyclin B, anti-cyclin A, anti-β-actin and
anti-matrix metalloproteinase-9 (MMP-9) were purchased from Santa
Cruz Biotechnology. Anti-cleaved poly(ADP-ribose) polymerase-1
(PARP1) antibody, anti-cleaved caspase 3, anti-pDNA-dependent
protein kinase (DNA-PK; S2059), anti-PERK and anti-pAkt were
purchased from Cell Signaling Technology (Danvers, MA, USA), and
anti-phosphorylated H2AX (γH2AX) was obtained from Millipore
(Billerica, MA, USA). Sorafenib was purchased from Selleckchem. For
in vitro experiments, sorafenib was dissolved in dimethyl
sulfoxide to make a 16 mmol/l stock solution and was stored at
4°C.
Cell culture
The human colorectal cancer cell lines HT29, HCT116
and SW480 were grown in RPMI-1640 medium supplemented with 10%
fetal bovine serum (FBS), glutamine, HEPES and antibiotics at 37°C
in a 5% CO2 humidified incubator. Human umbilical vein
endothelial cells (HUVECs) were maintained in endothelial cell
basal medium (EGM-2; Cambrex) containing EGM-2 SingleQuot growth
supplements (Cambrex) and maintained for no more than 8 culture
passages.
Irradiation
Cells were plated in 60-mm diameter dishes and
incubated at 37°C under humidified conditions and 5% CO2
until they reached 70–80% confluence. Cells were irradiated with a
137Cs γ-ray source (Atomic Energy of Canada, Ltd.,
Ontario, Canada) at a dose rate of 3.81 Gy/min.
Colony-forming assay
Sorafenib was added to cells 1 h after radiation
exposure to a final concentration of 16 μmol/l, and the cells were
then incubated for 72 h. After 14–20 days, colonies were stained
with 0.4% crystal violet (Sigma, St. Louis, MO, USA). The plating
efficiency (PE) represents the percentage of seeded cells that grew
into colonies under the specific culture conditions of a given cell
line. The survival fraction, expressed as a function of
irradiation, was calculated as follows: Survival fraction =
colonies counted/(cells seeded × PE/100). PEs of HT29, HCT116 and
SW480 were 0.52±0.18, 0.50±0.15, and 0.48±0.10, respectively. To
evaluate the radiosensitizing effects of sorafenib, the ratio for
radiation alone and radiation plus sorafenib was calculated as the
dose (Gy) for radiation alone divided by the dose for radiation
plus sorafenib at a surviving fraction of 50% 50% of cells.
Flow cytometry
Cells were cultured, harvested at the indicated
times, stained with propidium iodide (PI; 1 μg/ml, Sigma),
according to the manufacturer’s protocol, and then analyzed using a
FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
A minimum of 10,000 cells was counted for each sample, and data
analysis was performed with the use of CellQuest software (BD
Biosciences).
Detection of apoptotic cells by Annexin V
staining
After the exposure to radiation, sorafenib was added
to the cells, which were then incubated for a further 48 h. Cells
were washed with ice-cold phosphate-buffered saline (PBS),
trypsinized and re-suspended in 1X binding buffer [10 mm HEPES/NaOH
(pH 7.4), 140 mm NaCl and 2.5 mm CaCl2] at
1×106 cells/ml. Aliquots (100 μl) of cell solution were
mixed with 5 μl Annexin V FITC (Pharmingen) and 10 μl PI stock
solution (50 μg/ml in PBS) by gentle vortexing, followed by 15 min
of incubation at room temperature in the dark. Buffer (400 μl, 1X)
was added to each sample and analyzed on a FACScan flow cytometer
(Becton Dickinson). A minimum of 10,000 cells was counted for each
sample, and data analysis was performed using CellQuest software
(BD Biosciences).
Fluorescent measurement of intracellular
reactive oxygen species
The fluorescent probe 2′,7′-dichlorofluorescin
diacetate (DCFH-DA) was used for the assessment of intracellular
reactive oxygen species (ROS). For fluorocytometrical analysis,
cells were plated in 60-mm diameter dishes (1×105
cells/dish) and loaded for 30 min at room temperature with 10 μM
DCFH-DA in 5 ml PBS. Unincorporated DCFH-DA was removed with 2
washes in PBS. DCFH-DA-loaded cells were treated with sorafenib or
radiation alone, or in combination. Fluorescence was measured using
a flow cytometer (Becton Dickinson).
Immunohistochemistry
Immunohistochemistry was performed to determine the
nuclear distribution of γ-H2AX in individual cells. Cells were
grown on chambered slides 1 day prior to irradiation or sorafenib
treatment. After radiation exposure, sorafenib was added to the
cells, and the cells were treated for various lengths of time. All
treatments were performed while cells remained attached to the
slides, followed by fixing with 4% paraformaldehyde and
permeabilization with 0.2% Triton X-100 in PBS. Detection was
performed after blocking the slides in 10% FBS/1% bovine serum
albumin for 1 h at a 1:1,000 dilution of FITC-labeled mouse
monoclonal antibody against γ-H2AX (Millipore) in the
background-reducing antibody diluent (DAKO plus S3022;
Millipore).
Western blotting
Following irradiation, sorafenib was added to the
colon cancer cells, which were then incubated for 1 or 24 h. The
cells were then lysed with RIPA buffer. Proteins were separated by
sodium-polyacrylamide gel electrophoresis and transferred to
nitrocellulose membranes. The membranes were blocked with 1% (v/v)
non-fat dried milk in Tris-buffered saline with 0.05% Tween-20 and
incubated with the required antibodies. Primary antibodies were
used at a 1:1,000 dilution and secondary antibodies at a 1:5,000
dilution. Immunoreactive protein bands were visualized by enhanced
chemiluminescence (Amersham Biosciences) and scanned.
Tumor cell motility assay
The cell motility assay was conducted in 6-well
plates. A fine scratch in the form of a groove was made using a
sterile pipette tip in a layer of cells at ~90% confluency. Cells
were then treated with sorafenib, radiation or a combination of
both. The migration of cells was monitored using a Nikon Eclipse Ti
microscope with a DS-Fi1 camera.
Invasion assay
The invasive ability was measured in vitro
using Transwell chambers, according to the manufacturer’s protocol.
Briefly, cells were seeded onto the membrane of the upper chamber
of the Transwell at a concentration of 4×105 cells/ml in
150 μl of RPMI medium and were left untreated or treated with the
indicated doses of sorafenib, radiation, or a combination of both
for 24 h. The medium in the upper chamber was serum-free, while the
medium in the lower chamber contained 10% FBS as a source of
chemo-attractants. Cells that passed through the Matrigel-coated
membrane were stained with Cell Stain Solution containing crystal
violet supplied in the Transwell invasion assay (Chemicon,
Millipore) and images were captured after 24 h of incubation.
Matrigel in vitro endothelial tube
formation assay
Endothelial cell tube formation was assessed using
Matrigel-coated chamber slides as previously described (12). The results of each assay were
photographed (Nikon Eclipse Ti microscope with DS-Fi1 camera) at
×40 magnification. Tube formation was quantified by counting the
number of connected cells in randomly selected fields at ×400
magnification with a microscope, and dividing that number by the
total number of cells in the same field.
Statistical analysis
All data were plotted as the mean ± standard error
of the mean (SEM). Results of colony forming assays were analyzed
using a paired t-test with SPSS 18.0 software (SPSS, Chicago, IL,
USA). All other data were analyzed by parametric repeated measure
one-way analysis of variance (ANOVA), followed by Tukey’s honestly
significant difference test (SPSS 18.0). Statistical significance
was set at P<0.05.
Results
Cooperative effect of radiation and
sorafenib on colon cancer cell proliferation
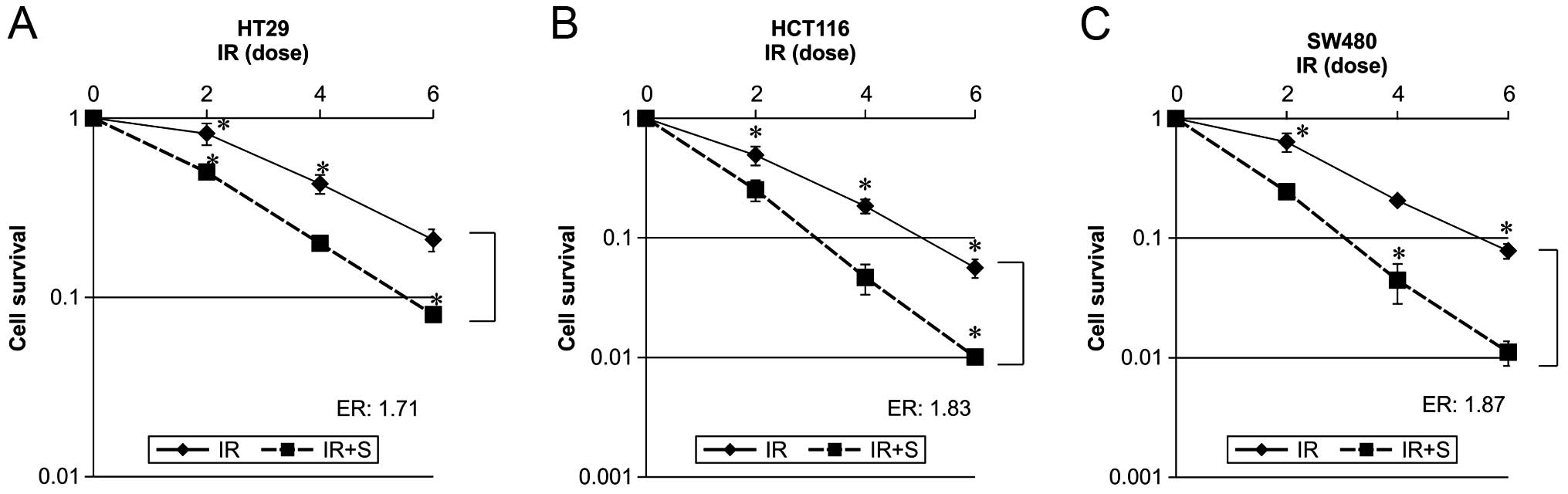
In order to evaluate the effects of sorafenib on
radiation-induced cytotoxicity, a clonogenic survival assay was
performed using HT29, HCT116 and SW480 cells. We used a sorafenib
concentration resulting in a 25% growth inhibition after a 48-h
exposure in each experiment (data not shown), and added this drug
post-radiation as this was previously shown to be more efficacious
than pre-radiation treatment (13).
Dose-response curves of the 3 colon cancer cell lines irradiated in
the presence or absence of sorafenib are shown in Fig. 1. Combined radiation and sorafenib
treatment was more effective than radiation alone in all cases. The
effect of sorafenib on radiation-mediated cell killing was
expressed as an enhancement ratio (D50).
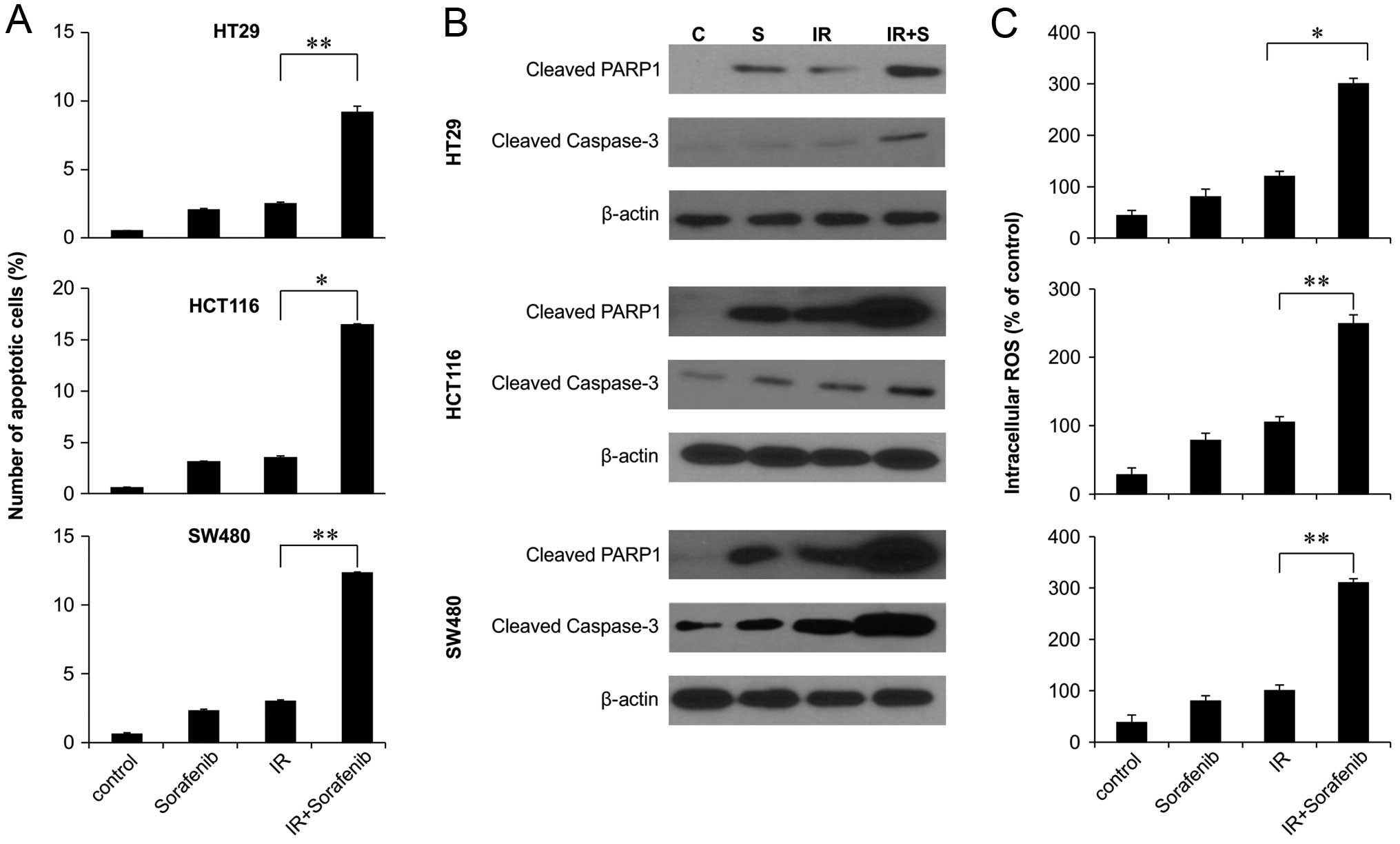
Sorafenib enhances radiation-induced
apoptosis
To investigate whether sorafenib and radiation
induce apoptosis, we assessed early apoptosis by Annexin V and PI
staining. In colon cancer cell lines, combined 48-h sorafenib
treatment and radiation exposure significantly increased the
proportion of cells in early apoptosis in all 3 colon cancer cell
lines (Fig. 2A). We next
investigated whether sorafenib-enhanced radiation cytotoxicity
resulted from the increased activation of caspase, resulting in
enhanced apoptotic cell death. We observed increased activation of
caspase-3 and increased PARP cleavage in response to combined
radiation and sorafenib treatment compared to sorafenib alone
(Fig. 2B). We also investigated the
relationship between ROS production and enhancement of
radiation-induced apoptosis by sorafenib. The production of ROS was
synergistically induced by the combined treatment of sorafenib and
radiation in colon cancer cell lines (Fig. 2C), indicating that ROS generated by
the combined treatment increases intracellular caspase signaling
and, thus, apoptosis.
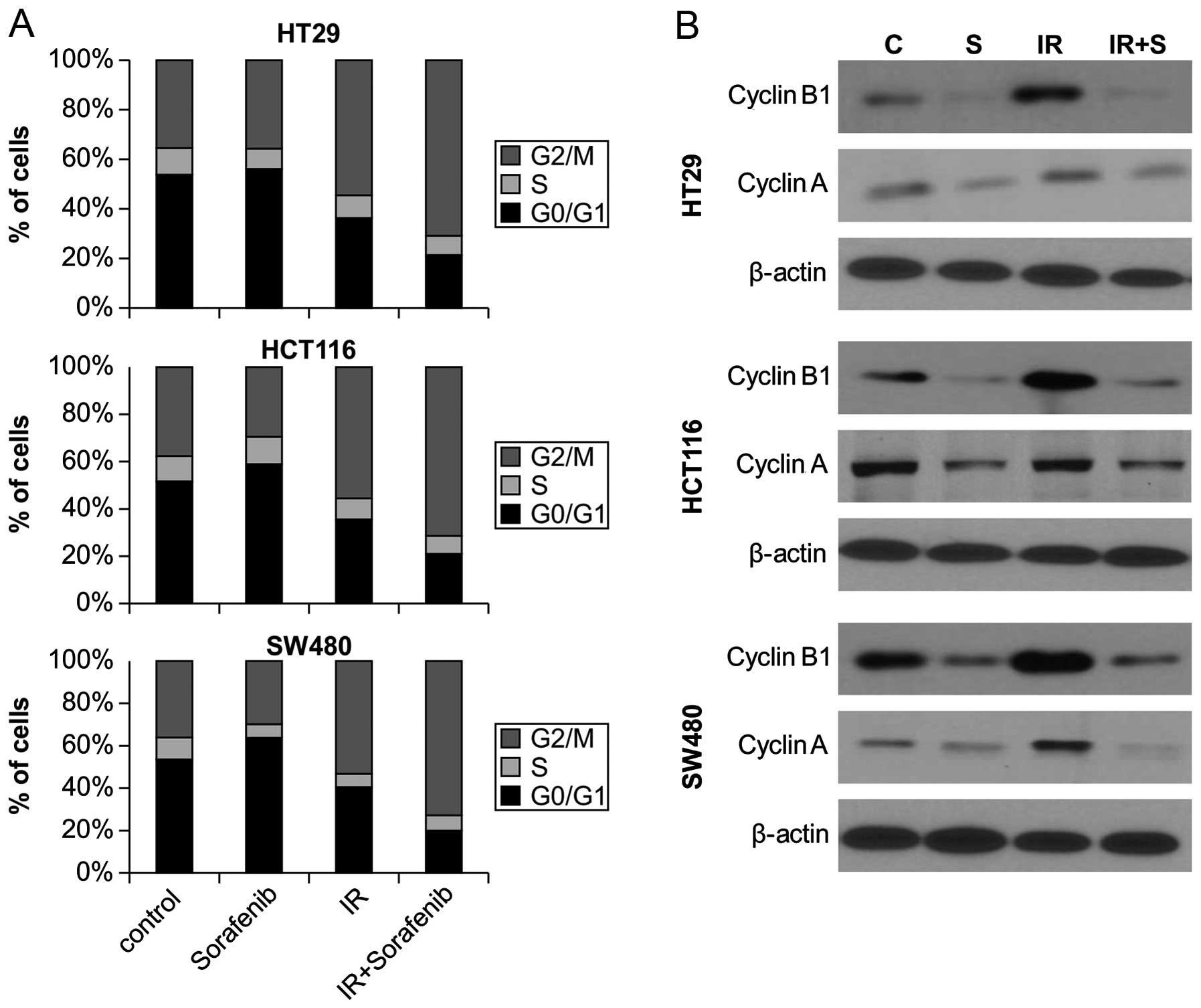
Effects of sorafenib and radiation on the
cell cycle
We next investigated whether combined
sorafenib/radiation-induced cytotoxicity resulted from differences
in cell cycle regulation by analyzing cell cycle progression
through flow cytometry. Sorafenib treatment combined with radiation
significantly increased the proportion of cells in G2 to M phase
for all 3 cell lines, compared to radiation alone (Fig. 3A). We also examined the expression
of cell cycle regulators after combined sorafenib and radiation
treatment. Western blotting revealed that radiation alone resulted
in a significant accumulation of cyclin A and cyclin B, a key cell
cycle regulator involved in the G2/M transition, whereas sorafenib
alone and combined treatment reduced expression of cyclin A and
cyclin B levels (Fig. 3B).
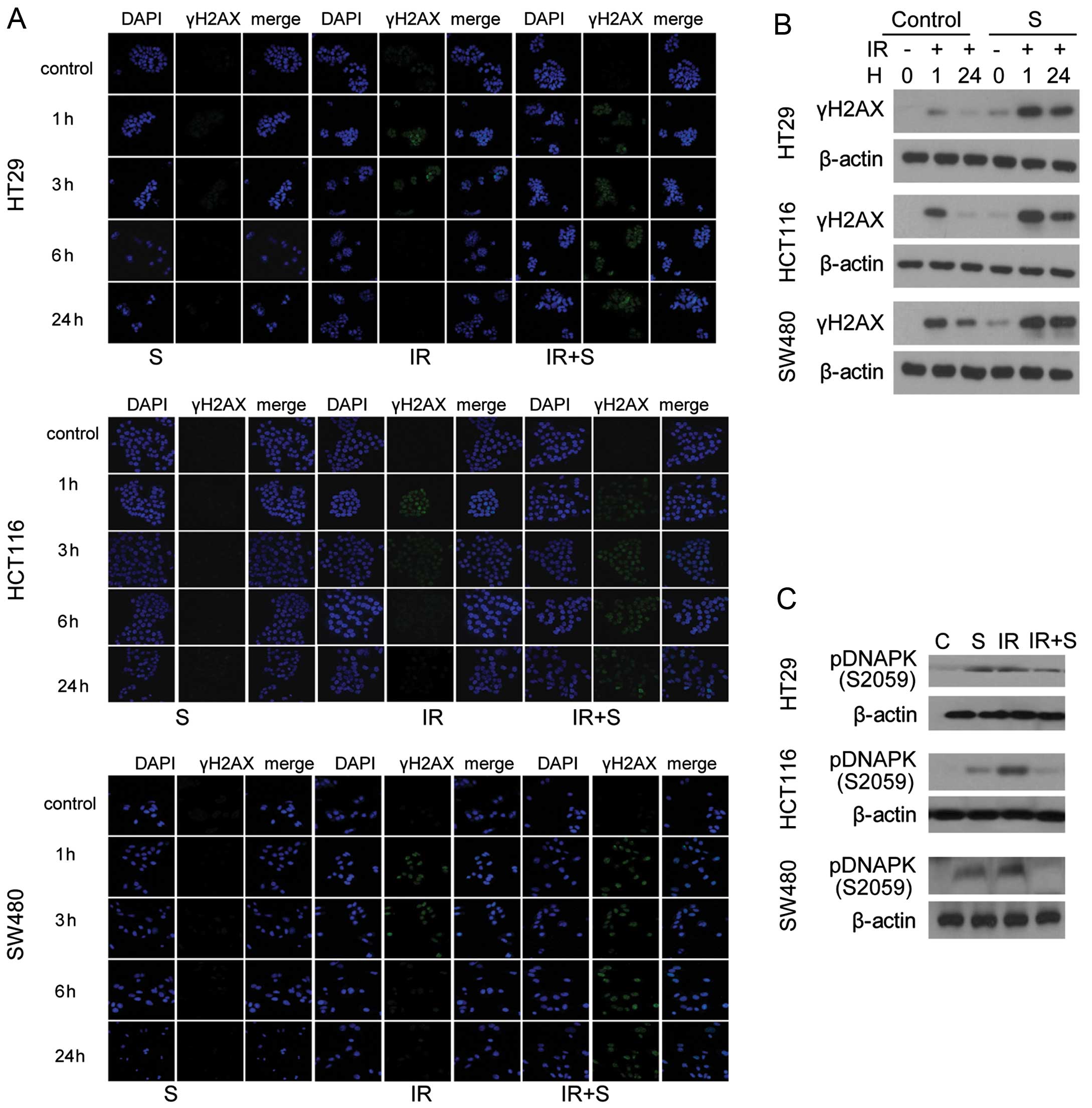
Influence of sorafenib on
radiation-induced DNA damage and DNA repair activity
To analyze the effect of sorafenib on double-strand
break (DSB) repair, the level of γH2AX, a marker for DSB, was
examined by immunofluorescence and western blotting 0, 1 and 24 h
after treatment. Prolonged expression of γH2AX was observed after
24-h radiation exposure in the presence of sorafenib (Fig. 4A). All 3 colon cancer cell lines
treated with combined sorafenib and irradiation exhibited damaged
DNA foci, which appeared 1 h after treatment and were still present
24 h after exposure (Fig 4A). In
addition, cell lysates were immunoblotted with anti-γH2AX antibody
(Fig. 4B), which confirmed the
results of the immunofluorescence assay, and the levels of
phosphorylated DNA-PK were slightly reduced after combined
treatment (Fig. 4C).
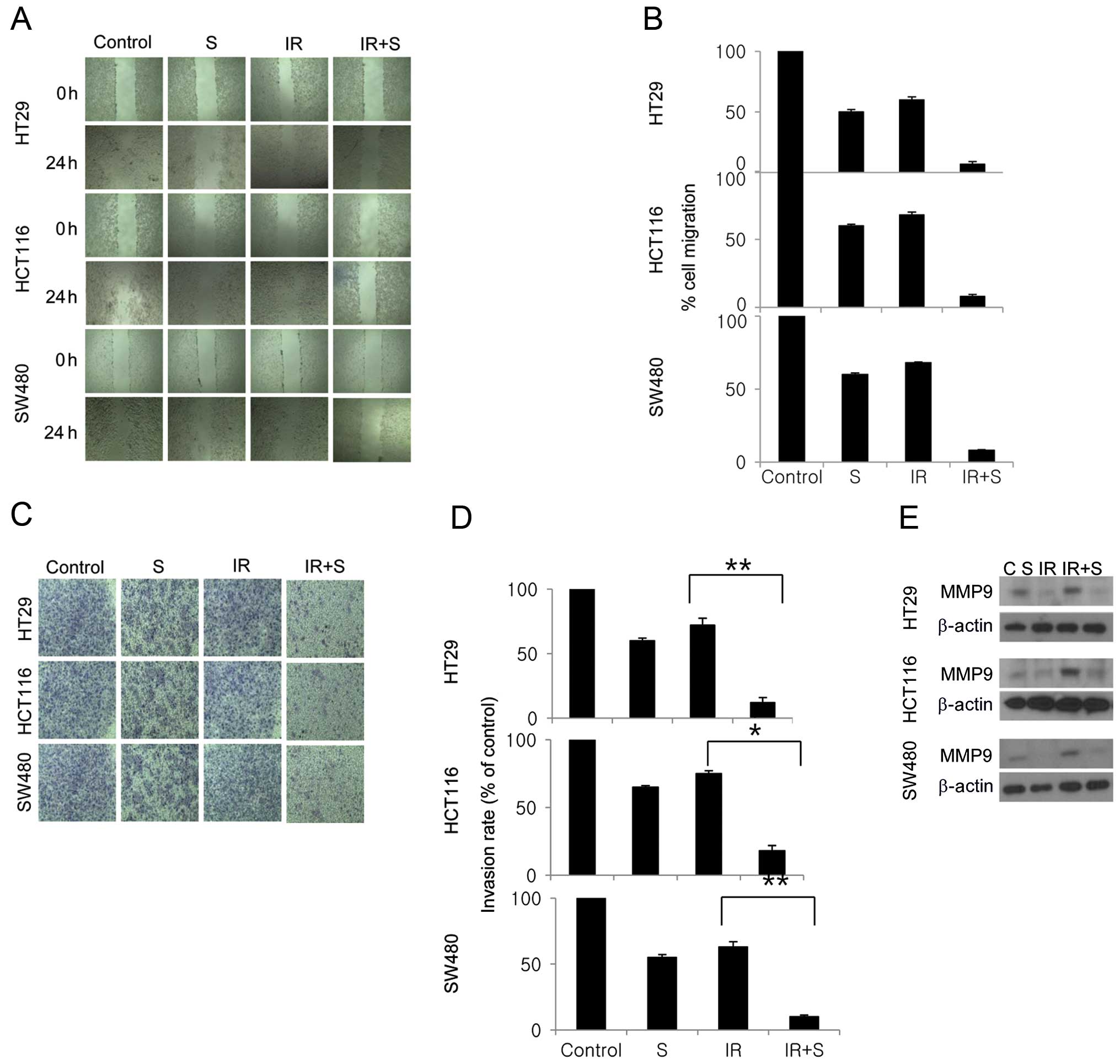
Combination treatment significantly
inhibits tumor cell motility and tumor cell invasion
We next evaluated the effects of sorafenib and
radiation on the invasive and migratory capacities of colon cancer
cells using Matrigel and scratch assays. In the latter, combined
sorafenib and radiation treatment significantly inhibited cell
migration compared with sorafenib or radiation alone (Fig. 5A and B). In the Matrigel invasion
assay, combined sorafenib and radiation treatment was highly
effective in inhibiting the invasive behavior of all 3 colon cancer
cell lines, by 10, 15 and 8% (Fig. 5C
and D). We also evaluated the extent to which MMP-9 expression
is altered in colon cancer cells treated with sorafenib and
radiation. Notably, sorafenib suppressed the upregulation of MMP-9
caused by irradiation (Fig.
5E).
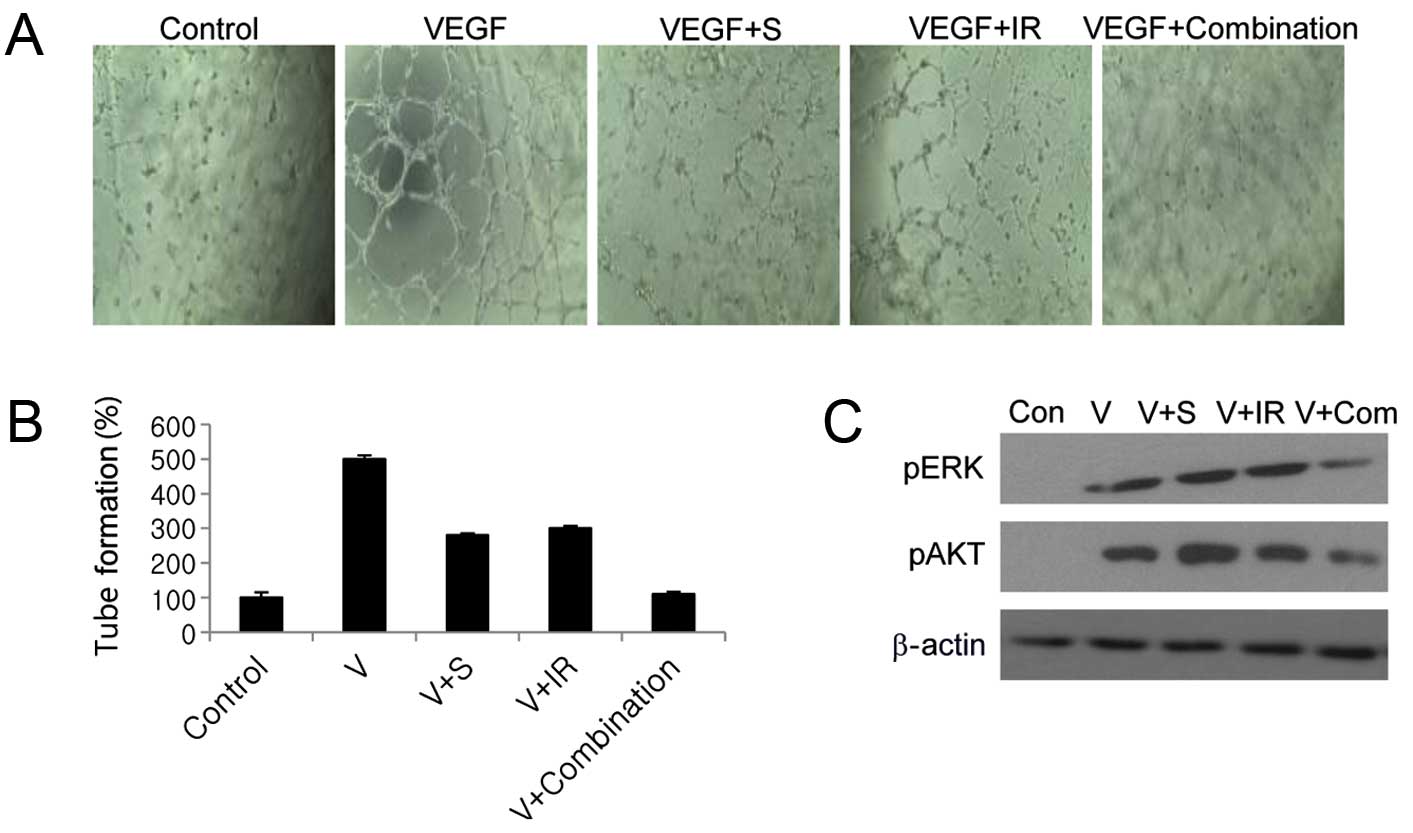
Combination treatment significantly
inhibits tumor angiogenesis
We next examined whether combined sorafenib and
radiation treatment blocks angiogenesis by inhibiting VEGF.
Combined sorafenib and radiation completely inhibited VEGF-mediated
endothelial tube formation in HUVECs, whereas either treatment
alone inhibited tube formation by only 33 and 36%, respectively
(Fig. 6A and B). VEGF predominantly
mediates angiogenesis via the PI3K/Akt and MAPK signaling cascades
(5), and combination treatment
significantly inhibited Akt and ERK1/2 activation compared with
sorafenib or radiation alone, as shown by western blotting
(Fig. 6C).
Discussion
The purpose of the present study was to investigate
the mechanism of radiosensitization by sorafenib on colorectal
cancer cells. Several clinical trials have been initiated to
evaluate the use of sorafenib in combination with a variety of
anticancer agents to treat a range of tumor types. The most
promising evidence of antitumor activity was observed when
sorafenib was combined with interferon-α in renal cell carcinoma,
dacarbazine in melanoma, doxorubicin in hepatocellular carcinoma
and gemcitabine in ovarian cancer (14). Moreover, the combination of
sorafenib and another targeted agent, bevacizumab, also showed
promising antitumor activity in patients with ovarian cancer
(15), and other clinical studies
have shown that the combination of sorafenib and radiation may
provide clinical benefits in other types of cancer (16–18).
However, the mechanism by which radiation-enhancement occurred
appeared to be somewhat more complex than predicted in previous
studies. In this study, we provided a scientific rationale for the
clinical application of sorafenib as a radiosensitizer in
colorectal cancer. On the basis of our results, we suggest that
sorafenib significantly enhances the therapeutic efficiency of
radiation by inhibiting tumor cell survival, cell cycle regulation,
DNA repair activity, tumor cell invasiveness, and angiogenesis in
human colon adenocarcinoma cell lines. Sorafenib combined with
radiation significantly reduced the clonogenic survival and
enhanced the radiosensitivity of colon cancer cells by promoting
apoptosis through increased ROS levels. Previous studies found that
post-radiation sorafenib treatment resulted in greater
radiosensitization than pre-radiation treatment (13), therefore we employed this schedule
in our study. When sorafenib was administered after irradiation,
the cells failed to complete mitosis owing to a block in the G2 to
M phase transition, which was possibly mediated by the
downregulation of cyclin B1.
Radiotherapy is one of the major therapeutic
strategies for cancer treatment and results in DNA damage,
including DSBs, which in turn initiates a variety of signaling
events in cancer cells (19). DSBs
lead to the phosphorylation of H2AX, and we and others have used
this as a marker for the cellular response to radiation-induced DNA
damage (20–22). Our results showed that the combined
treatment of sorafenib and radiation delayed the clearance of
γH2AX, suggesting that sorafenib prevents DNA repair and hence
increases radiosensitivity. Homologous recombination and
non-homologous end-joining (NHEJ) are two major pathways for the
repair of DNA DSBs. NHEJ, which does not require the presence of a
homologous template, is the predominant repair pathway for DSBs
produced by ionizing radiation, and DNA-PK plays a central role in
regulating the NHEJ of DSBs in the course of radiation therapy
(23). DNA-PK is a trimeric protein
consisting of a heterodimer formed between ku70 and ku880, which is
recruited to the break first, and activates the catalytic subunit,
DNA-PKcs (24). DNA-PKcs is a
serine/threonine protein kinase belonging to the phosphoinositide
3-kinase-like family of protein kinase (PIKK) (20). Numerous studies have shown that it
undergoes a series of phosphorylations in response to DSBs at the
clusters of ABCDE (6 sites between Thr2609 and Thr2647) and PQR (5
sites between residues 2023 and 2056) (25). In order to investigate how sorafenib
suppressed the repair of radiation-induced DSBs, we studied the
effects of sorafenib on activated DNA-PK. We found that the level
of activated DNA-PK was slightly reduced by combined sorafenib and
radiation treatment, in a time-dependent manner.
In addition to its action as a direct
radiosensitizer, sorafenib may also reduce tumor cell invasion by
blocking MMP-9 production through the inhibition of the Raf-MAPK
pathway, which was previously shown to induce the production of
matrix metalloproteinases (26).
MMP-9 is known to enhance the invasion of tumor cells through the
controlled degradation of the extra-cellular matrix in a range of
tumor types (27,28). Our findings suggest that sorafenib
also blocks MMP-9 in colon cancer cells, an effect that is enhanced
by concomitant radiation. In addition to MMP-9, VEGF, which
mediates angiogenesis via the activation of the PI3K/Akt and MAPK
signaling cascade, has also been identified as an important target
of sorafenib (5). We observed that,
although phosphorylated VEGF2 was elevated as a result of
irradiation alone, it was significantly downregulated by both
combined sorafenib and radiation treatment. ERK is a key downstream
component of the RAF/MEK/ERK signaling pathway and aberrant
signaling through the ERK pathway could promote cell
immortalization, proliferation and resistance to radiation
(29). Western blot analysis
demonstrated that radiation could result in the phosphorylation and
hence activation of ERK, but that this was also suppressed by
post-irradiation treatment with sorafenib. This was supported by
our additional observation that VEGF treatment of endothelial cells
significantly enhanced tube formation, and this too was blocked by
sorafenib combined with radiation.
In summary, this study demonstrated that sorafenib
can radiosensitize colon cancer cells through the inhibition of
tumor cell survival, cell cycle regulation, DNA repair activity,
tumor cell invasiveness and angiogenesis. These findings provide
molecular evidence for the use of chemoradiation to treat colon
cancer, and in vivo modeling should be used to further
assess its suitability for clinical applications. The interaction
between sorafenib and radiation in normal colorectal cells for
normal tissue toxicity and complication requires further study.
Furthermore, as the sensitizing effect of sorafenib in photon beam
treatment is well characterized, it is important to compare its
sensitizing effect and underlying mechanism for carbon beams in
high LET, as well as other types of radiation in clinical
applications to enhance the efficacy and safety of these forms of
radiotherapy.
Acknowledgements
This study was supported by the National Research
Foundation of Korea (NRF) grant funded by the Korea government
(MSIP) (2014029534).
References
|
1
|
Jemal A, Siegel R, Ward E, et al: Cancer
statistics, 2008. CA Cancer J Clin. 58:71–96. 2008. View Article : Google Scholar
|
|
2
|
Saltz LB and Minsky B: Adjuvant therapy of
cancers of the colon and rectum. Surg Clin North Am. 82:1035–1058.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Krook JE, Moertel CG, Gunderson LL, et al:
Effective surgical adjuvant therapy for high-risk rectal carcinoma.
N Engl J Med. 324:709–715. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Seiwert TY, Salama JK and Vokes EE: The
concurrent chemoradiation paradigm - general principles. Nat Clin
Pract Oncol. 4:86–100. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wilhelm SM, Carter C, Tang L, et al: BAY
43-9006 exhibits broad spectrum oral antitumor activity and targets
the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in
tumor progression and angiogenesis. Cancer Res. 64:7099–7109. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wilhelm SM, Adnane L, Newell P, Villanueva
A, Llovet JM and Lynch M: Preclinical overview of sorafenib, a
multikinase inhibitor that targets both Raf and VEGF and PDGF
receptor tyrosine kinase signaling. Mol Cancer Ther. 7:3129–3140.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Iyer R, Fetterly G, Lugade A and Thanavala
Y: Sorafenib: a clinical and pharmacologic review. Expert Opin
Pharmacother. 11:1943–1955. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Siegel AB, Olsen SK, Magun A and Brown RS
Jr: Sorafenib: where do we go from here? Hepatology. 52:360–369.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Escudier BET, Stadler WM, Szczylik C,
Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA,
Rolland F, Demkow T, Hutson TE, Gore M, Freeman S, Schwartz B, Shan
M, Simantov R and Bukowski RM; TARGET Study Group. Sorafenib in
advanced clear-cell renal-cell carcinoma. New Engl J Med.
356:125–134. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gupta-Abramson V, Troxel AB, Nellore A, et
al: Phase II trial of sorafenib in advanced thyroid cancer. J Clin
Oncol. 26:4714–4719. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takimoto CH and Awada A: Safety and
anti-tumor activity of sorafenib (Nexavar) in combination with
other anti-cancer agents: a review of clinical trials. Cancer
Chemother Pharmacol. 61:535–548. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kumar P, Benedict R, Urzua F, Fischbach C,
Mooney D and Polverini P: Combination treatment significantly
enhances the efficacy of antitumor therapy by preferentially
targeting angiogenesis. Lab Invest. 85:756–767. 2005. View Article : Google Scholar
|
|
13
|
Plastaras JP, Kim SH, Liu YY, et al: Cell
cycle-dependent and schedule-dependent antitumor effects of
sorafenib combined with radiation. Cancer Res. 67:9443–9454. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dal Lago L, D’Hondt V and Awada A:
Selected combination therapy with sorafenib: a review of clinical
data and perspectives in advanced solid tumors. Oncologist.
13:845–858. 2008.PubMed/NCBI
|
|
15
|
Azad N, Annunziata C, Barrett T, et al:
Dual targeting of vascular endothelial growth factor (VEGF) with
sorafenib and bevacizumab: clinical and translational results. J
Clin Oncol (Meeting abstracts). 25:35422007.
|
|
16
|
Kasibhatla M, Steinberg P, Meyer J,
Ernstoff MS and George DJ: Radiation therapy and sorafenib:
clinical data and rationale for the combination in metastatic renal
cell carcinoma. Clin Genitourin Cancer. 5:291–294. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Horgan AM, Dawson LA, Swaminath A and Knox
JJ: Sorafenib and radiation therapy for the treatment of advanced
hepatocellular carcinoma. J Gastrointest Cancer. 43:344–348. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jeong YK, Kim MS, Lee JY, et al: Sorafenib
acts synergistically in combination with radiotherapy without
causing intestinal damage in colorectal cancer. Tumori. 99:176–182.
2013.PubMed/NCBI
|
|
19
|
Jackson SP: Sensing and repairing DNA
double-strand breaks. Carcinogenesis. 23:687–696. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rogakou EP, Pilch DR, Orr AH, Ivanova VS
and Bonner WM: DNA double-stranded breaks induce histone H2AX
phosphorylation on serine 139. J Biol Chem. 273:5858–5868. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bonner WM, Redon CE, Dickey JS, et al:
GammaH2AX and cancer. Nat Rev Cancer. 8:957–967. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bourton EC, Plowman PN, Smith D, Arlett CF
and Parris CN: Prolonged expression of the γ-H2AX DNA repair
biomarker correlates with excess acute and chronic toxicity from
radiotherapy treatment. Int J Cancer. 129:2928–2934. 2011.
|
|
23
|
Collis SJ, DeWeese TL, Jeggo PA and Parker
AR: The life and death of DNA-PK. Oncogene. 24:949–961. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Smith GC and Jackson SP: The DNA-dependent
protein kinase. Genes Dev. 13:916–934. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Douglas P, Sapkota G, Morrice N, et al:
Identification of in vitro and in vivo phosphorylation sites in the
catalytic subunit of the DNA-dependent protein kinase. Biochem J.
368:243–251. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pignochino Y, Grignani G, Cavalloni G, et
al: Sorafenib blocks tumour growth, angiogenesis and metastatic
potential in preclinical models of osteosarcoma through a mechanism
potentially involving the inhibition of ERK1/2, MCL-1 and ezrin
pathways. Mol Cancer. 8:1182009. View Article : Google Scholar
|
|
27
|
Basset P, Okada A, Chenard MP, et al:
Matrix metalloproteinases as stromal effectors of human carcinoma
progression: therapeutic implications. Matrix Biol. 15:535–541.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Johnsen M, Lund LR, Rømer J, Almholt K and
Danø K: Cancer invasion and tissue remodeling: common themes in
proteolytic matrix degradation. Curr Opin Cell Biol. 10:667–671.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|