Introduction
Triple-negative breast cancer (TNBC) is the most
invasive subtype and accounts for 20% of all molecular subtypes of
breast cancer (1). Owing to its
lack of estrogen receptor (ER), progesterone receptor (PR) and
human epidermal growth factor receptor 2 (HER2/neu), neither
endocrine nor anti-HER2 molecular targeting treatment yield
promising results, and standard chemotherapy is the backbone of
systemic treatment. Over the past decade, preclinical findings have
suggested various protein targets and pathways as possible TNBC
treatments, such as growth factor receptors, proteins involved in
cellular DNA repair capacities and epigenetic regulation (2). Nevertheless, these novel molecular
targeting treatments have achieved little clinical progression and
there are no effective therapeutic targets to date available
against TNBC (1,3).
EGFR overexpression in breast cancer is correlated
with large tumor size, more stem-cell like properties and poor
prognosis (4). Its overexpression
is present in more than 50% of TNBC cases, which is more frequent
than in other subtypes (5).
Gefitinib, approved for lung cancers, is a tyrosine kinase
inhibitor (TKI) that targets the adenosine triphosphate binding
site in the cytoplasmic domain of EGFR (6). Unfortunately, gefitinib has shown
little efficacy in most clinical studies of breast cancer (7–9). The
complex interplay downstream pathways of EGFR with other cellular
components lead to their continued activation and insensitivity
toward EGFR inhibitors (10). In
this regard, many studies have reported that both the Raf/MEK/MAPK
and phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathways
together promote the resistance of cancer cells to gefitinib
(11,12).
p53 is critical to the induction of cell cycle
arrest, DNA repair, cellular apoptosis and senescence in response
to a wide array of stimuli (13).
Its mutation has been related to the poor prognosis and drug
resistance of cancer cells (14,15).
The p53 mutation is common in TNBC (16,17),
and the mutant p53 endows tumor cells with invasive and metastatic
abilities (15,18). Huang et al (19) reported that p53 regulates the
sensitivity to EGFR inhibitors and induces apoptosis by modulating
EGFR downstream signaling in lung cancer cells. Recombinant human
p53 adenovirus (Ad-p53), a replication incompetent human type 5
adenovirus whose E1 region is replaced by an expression cassette
containing the human wild-type p53 cDNA (20), was shown to restore p53 activity in
p53-deficient hepatocytes, therefore inducing G2/M
arrest and apoptosis. However, its effect was not apparent as a
single agent treatment in breast cancer. In the present study,
Ad-p53 was used in combination with gefitinib to treat a TNBC cell
line in vitro and in vivo. A significant sensitivity
toward gefitinib was observed after p53 activity was restored.
Materials and methods
Reagents
MDA-MB-468 cells were purchased from the Cell Bank
of Shanghai Institute of Cell Biology, Chinese Academy of Sciences.
Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum
(FBS) were purchased from Gibco (Grand Island, NY, USA). Gefitinib
was obtained from Tocris Bioscience Company (Bristol, UK), minimum
purity >98%, and dissolved in 100% dimethyl sulfoxide (DMSO;
Fisher Scientific, Pittsburgh, PA, USA). A 100-mM stock solution
was prepared and stored at −20°C; gefitinib (Iressa®)
tablets were kindly provided by AstraZeneca (Macclesfield,
Cheshire, UK). Ad-p53 (Gendicine, Shenzhen, China),
1×1012 virus particles (VP), was stored at −20°C;
Annexin V-FITC apoptosis kit was purchased from BD Biosciences
Pharmingen (San Diego, CA, USA). p53, caspase-9, Akt,
phosphorylation of protein kinase B (p-Akt) (S473), extracellular
signal-regulated kinase (ERK) and phosphorylated ERK (p-ERK) (Y204)
were purchased from ImmunoWay Biotechnology (Grand Island, NY,
USA). EGFR, GAPDH and cleaved caspase-3 were purchased from Cell
Signaling Technology (Beverly, MA, USA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
and the BCA protein assay kit were purchased from Beyotime
Institute of Biotechnology (Jiangsu, China).
Cell culture
The human breast cancer cell line MDA-MB-468 was
seeded in DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100
mg/ml streptomycin and 2 mM glutamine. Cells were grown in a
humidified atmosphere of 5% CO2 at 37°C.
MTT assay
The MDA-MB-468 cell line was plated in 96-well
plates in triplicate with 3×103 cells/well. Next, the
cells were treated with Ad-p53 at a multiplicity of infection (MOI)
of 100 for 24 h, while the vehicle-treated control cells were
incubated with 0.5% of DMSO. Next, fixed-ratio concentrations (0,
1.25, 2.5, 5, 10 and 20 μM) of gefitinib were administered. After
48 h, 20 μl of 5 mg/ml MTT solution was added to each well for 4 h.
MTT was carefully aspirated and replaced with 200 μl DMSO/well.
Absorbance was measured on a Bio-Rad 680 microplate reader
(Bio-Rad, Hercules, CA, USA) at 570 nm, and the results were
reported relative to a reference wavelength of 630 nm. The
inhibitory rate of growth was calculated according to the following
equation: Growth inhibition rate = (1 - the mean OD of the
samples/the mean OD of the controls) × 100%.
Clonogenic survival assay
MDA-MB-468 cells were plated in 6-well plates (300
cells/well) and treated with 3 μl of gefitinib and/or an MOI of 100
of Ad-p53 for 48 h. Then, the cells were washed with
phosphate-buffered saline (PBS) and replaced with fresh medium.
After 14 days, the cells were fixed with 70% ethanol and stained
with crystal violet. The number of colonies (>50 cells) was
counted under a microscope. Survival was expressed relative to the
untreated controls.
Measurement of apoptotic cells
The cells were treated with either gefitinib (3 μM)
or Ad-p53 (MOI of 100) alone or in combination for 48 h, and then
washed twice with cold PBS. An indirect immunofluorescence assay
was performed using the Annexin V-FITC apoptosis kit according to
the manufacturer’s instructions. The samples were assessed by flow
cytometry (FACSCalibur; BD Biosciences) using CellQuest software
within 1 h.
Analysis of the cell cycle
distribution
MDA-MB-468 cells (1×106) were treated
with gefitinib (3 μM) and/or Ad-p53 (MOI of 100) for 48 h and then
fixed in 70% of ethanol. After being washed twice with PBS, the
cells were stained with propidium iodide (PI) for 30 min. Flow
cytometric analysis was performed on the FACSCalibur (BD, Bedford,
MA, USA). ModiFIT software (Topsham, ME, USA) was used to analyze
the cell cycle distribution.
Western blot analysis
Cells were treated with gefitinib (3 μM) and/or
Ad-p53 (MOI of 100), and then lysed in RIPA buffer (50 mM Tris pH
7.4, 0.15 M NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1%
SDS) with 1 mM PMSF after 48 h. Supernatants were recovered, and
the total protein concentration was detected using the BCA protein
assay kit. The proteins were applied to sodium dodecyl
polyacrylamide gel electrophoresis, and electrophoretically
transferred onto a PVDF membrane (Amersham Biosciences,
Buckinghamshire, UK). Appropriate antibodies were applied to
determine the membrane. The band density was normalized to
GAPDH.
Effect of gefitinib and Ad-p53 on the
growth of MDA-MB-468 xenografts
Sixteen healthy BALB/C female nude mice (4 weeks
old) were obtained from the Beijing HFK Bioscience Company. All
animals were housed and treated according to the guidelines
outlined by the Institutional Animal Care and Use Committee of
Shandong Cancer Hospital and maintained under a germ-free
controlled environment. The mice were grown until 6 weeks before
being subcutaneously injected in the right axillary with MDA-MB-468
cells (1×107). After the tumor xenograft had grown to 1
cm in diameter, the mice were randomized into four treatment
groups: i) vehicle; ii) Ad-p53; iii) gefitinib; and iv) the
combination. Ad-p53 (1×1010 VP) was dissolved in 100 μl
physiological saline. All were injected peri-/intratumorally.
Gefitinib (100 mg/kg) was administered via oral gavage. The
combined treatment was the same as the single agent treatment.
Physiological saline was administered to the vehicle-treated group.
The greatest longitudinal diameter (a) and the greatest transverse
diameter (b) were measured on day 0, 3, 6, 9, 12 and 14. Tumor
volume (TV) was calculated by the following formula: TV =
1/2ab2. Tumor inhibition rate (TIR) = (average TV of the
vehicle-treated group - average TV of the experimental
group)/average TV of the vehicle-treated group × 100%.
Statistical analysis
Statistical analysis was carried out using SPSS 17.0
statistical software. Significant differences between two groups
(Table I) were conducted by t-test.
The analysis of variance (>2) was analyzed by one-way analysis
of variance (ANOVA) to determine statistical significance. Tukey’s
multiple comparison was applied to compare two subsequent samples.
All statistical tests were two-sided. P<0.05 was considered to
indicate a statistically significant result.
 | Table IIC50 value of MDA-MB-468
cells for gefitinib with or without Ad-p53. |
Table I
IC50 value of MDA-MB-468
cells for gefitinib with or without Ad-p53.
| Group | IC50
gefitinib (μM) |
|---|
| Ad-p53 | 4.3±0.4a |
|
Vehicle-treated | 8.5±0.3 |
Results
Ad-p53 enhances the cytotoxic effect of
gefitinib on MDA-MB-468 cells
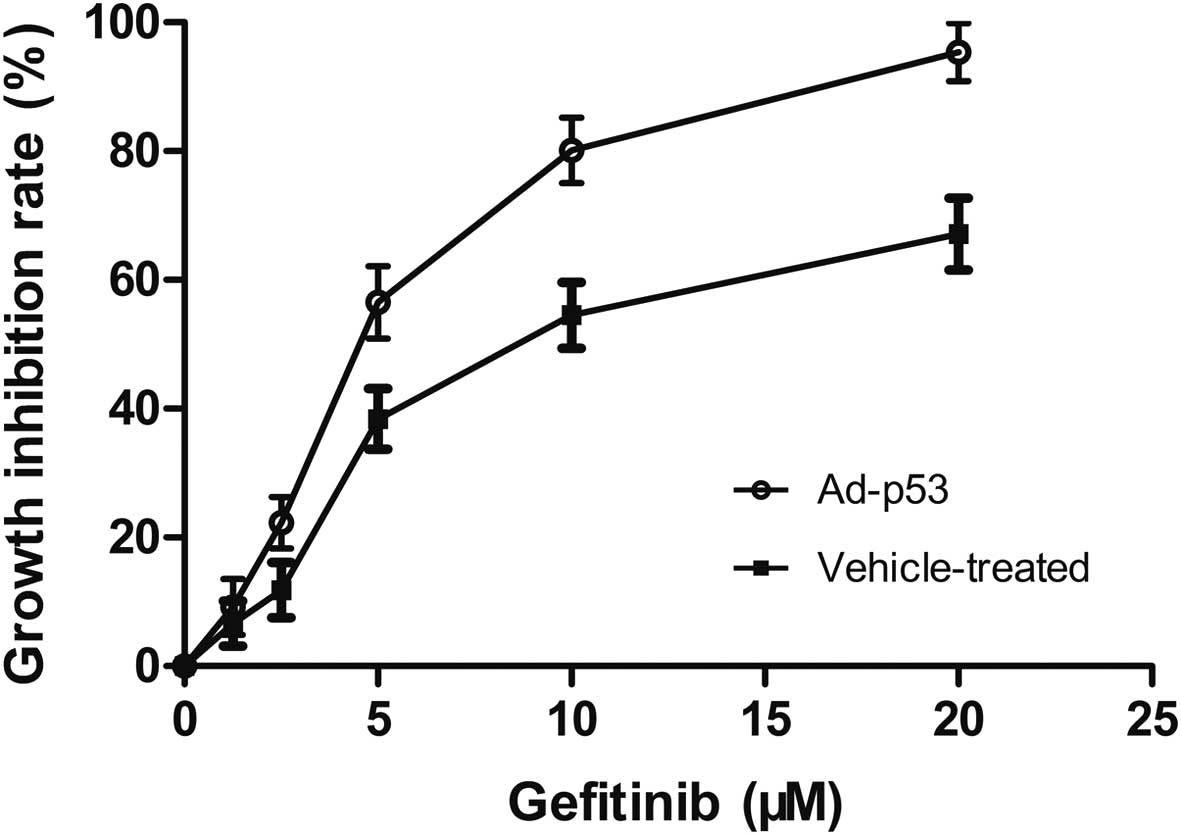
Gefitinib alone or combined with Ad-p53 inhibited
the proliferation of MDA-MB-468 cells in a dose-dependent manner
(Fig. 1). Combined treatment of
Ad-p53 with gefitinib synergistically inhibited the proliferation
of the MDA-MB-468 cells with a lower IC50 value of 4.3
μM; in comparison, cells that were not pretreated with Ad-p53 were
relatively resistant to gefitinib with a higher IC50
value of 8.5 μM (Table I). When
extra wild-type p53 gene was integrated into the MDA-MB-468 cells,
the cancer cells became more sensitive to gefitinib.
Combination of Ad-p53 and gefitinib
inhibits clonogenic cell survival
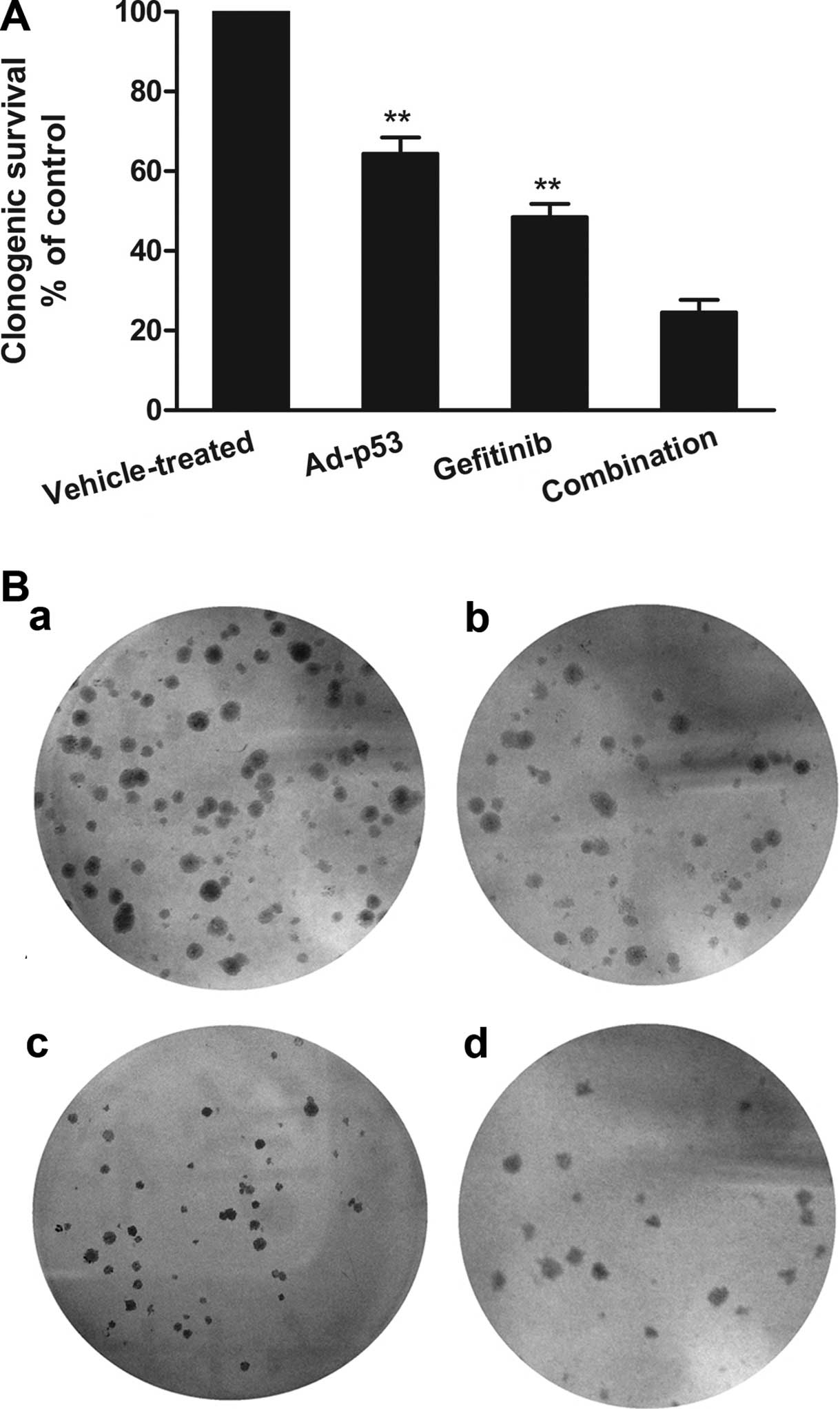
A clonogenic assay was performed to further
investigate the separate and combined effects of Ad-p53 and
gefitinib on cell proliferation. Survival was expressed relative to
the vehicle-treated cells. Ad-p53 or gefitinib could only slightly
weaken colony formation. An approximate 64.4 and 48.5% clonogenic
survival rate was detected when cells were treated with Ad-p53 and
gefitinib, respectively. Nevertheless, when Ad-p53 and gefitinib
were administered in combination, colony formation was
significantly decreased with a lower clonogenic survival rate of
24.5% (Fig. 2).
p53 is required for gefitinib-induced
cellular apoptosis and cell cycle arrest
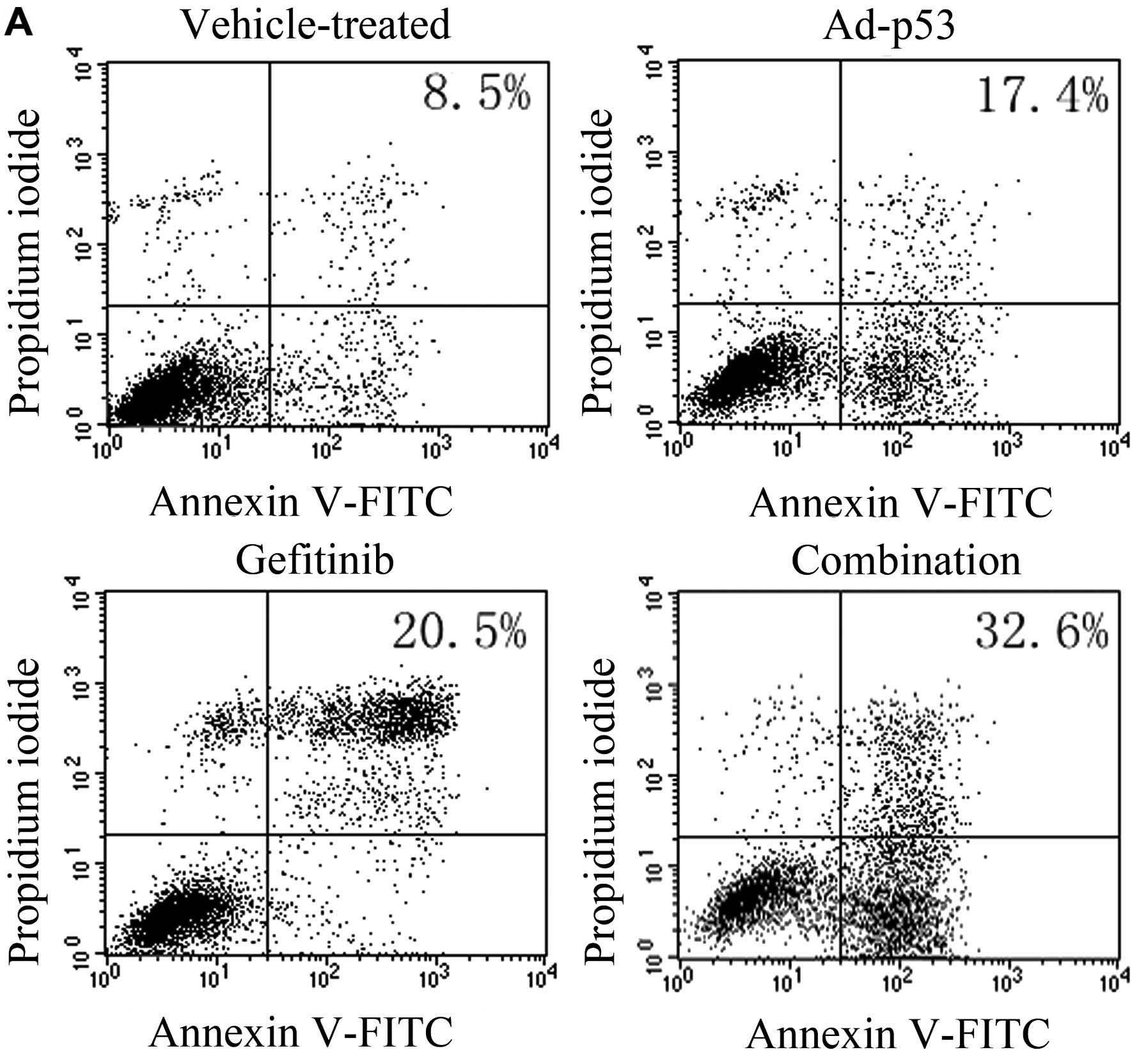
MDA-MB-468 cells were treated with either drug alone
or in combination with Ad-p53 for 48 h. According to flow
cytometry, the apoptosis rate in the Ad-p53, gefitinib, combination
and vehicle-treated group was 17.4, 20.5, 32.6 and 8.5%,
respectively. Treatment with Ad-p53 or gefitinib alone slightly
induced cellular apoptosis (Fig.
3). The combined treatment increased cellular apoptosis to
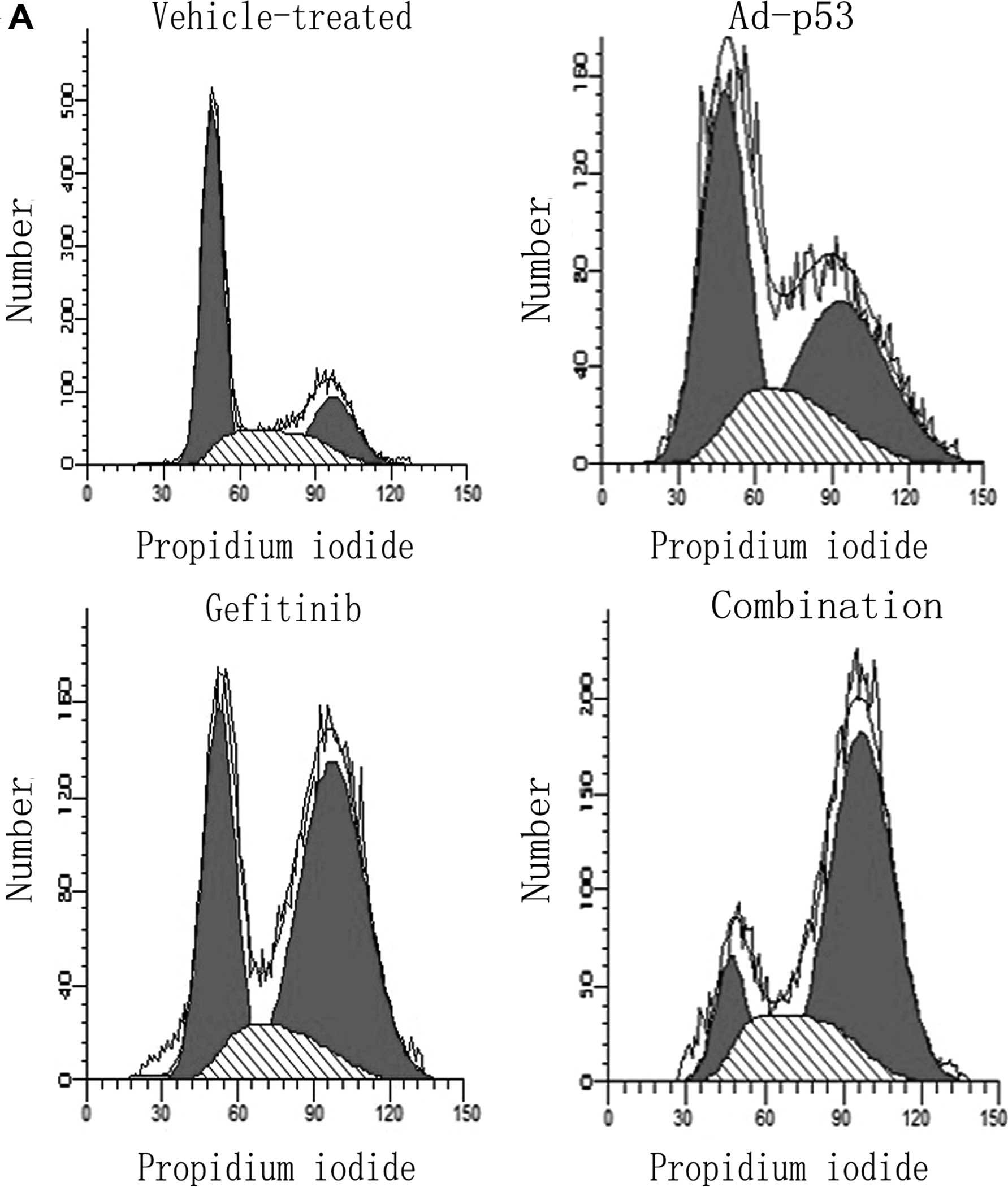
4-fold when compared with the vehicle-treated control (Fig. 3). As shown in Fig. 4, gefitinib induced G2/M
phase arrest from 21.9 to 45.4% compared to the vehicle-treated
groups; G2/M phase increased to no more than 31.5% after
exposure to Ad-p53. In comparison, when the combined treatment was
performed, G2/M arrest was enhanced evidently from 21.9
to 65.3%.
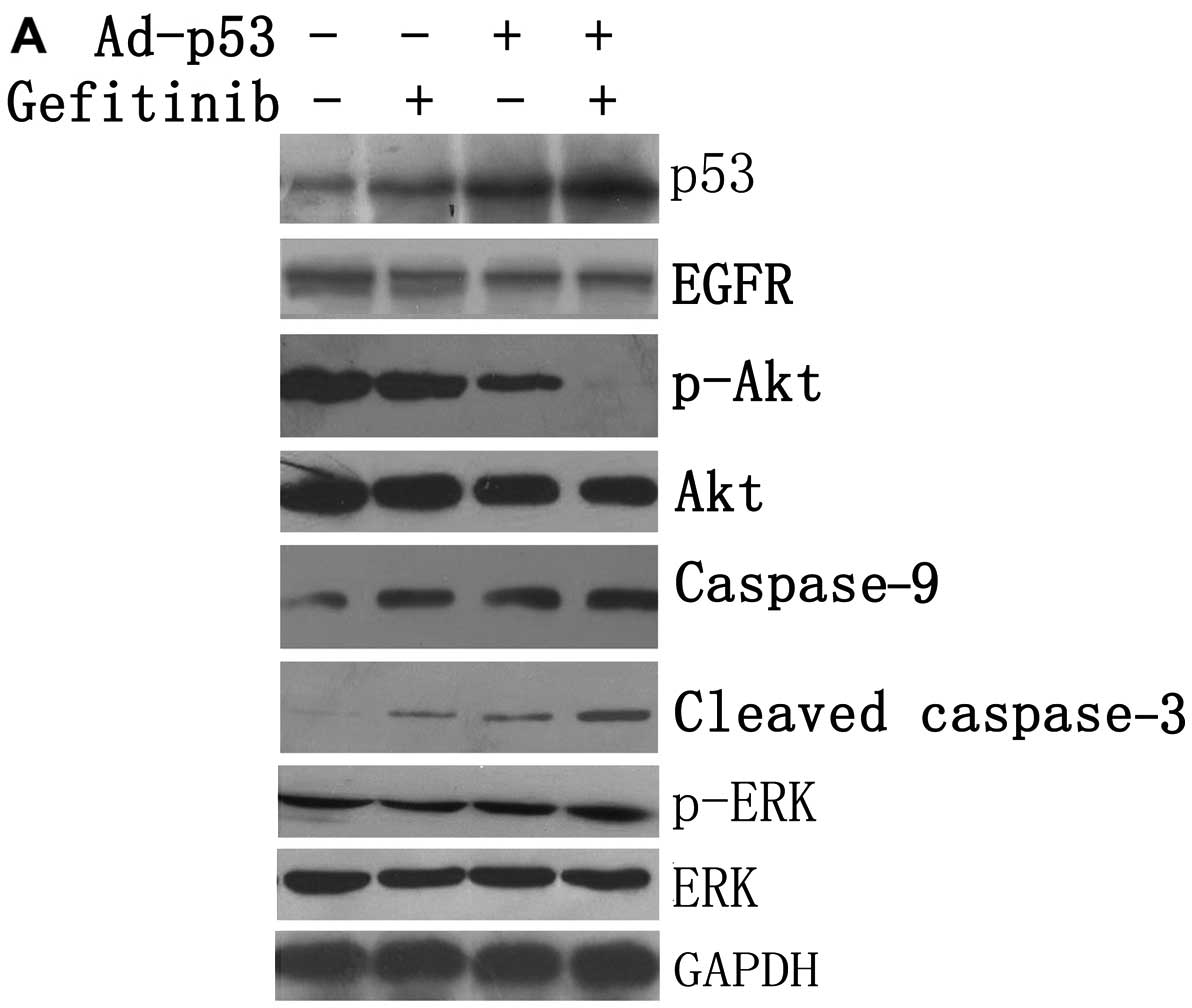
Effect of treatment with Ad-p53 and/or
gefitinib on intracellular signaling
Ad-p53 or gefitinib, as a single agent, produced a
slight reduction in the levels of p-Akt (S473) in the MDA-MB-468
cells (Fig. 5). Surprisingly,
combined treatment synergistically produced a significant reduction
in p-Akt (S473) (Fig. 5). The
expression levels of caspase-9 and cleaved caspase-3 were low in
the vehicle-treated cells. In contrast, when Ad-p53 or gefitinib
was used as a single agent, the levels of caspase-9 and cleaved
caspase-3 increased (Fig. 5).
Nevertheless, when they were administered in combination, the
levels were markedly increased. However, none of the treatments led
to an obvious change in ERK and p-ERK. These results suggest that
Ad-p53 may enhance the sensitivity of MDA-MB-468 cells to gefitinib
by blocking the PI3K/Akt pathway rather than the Raf/MEK/ERK
pathway.
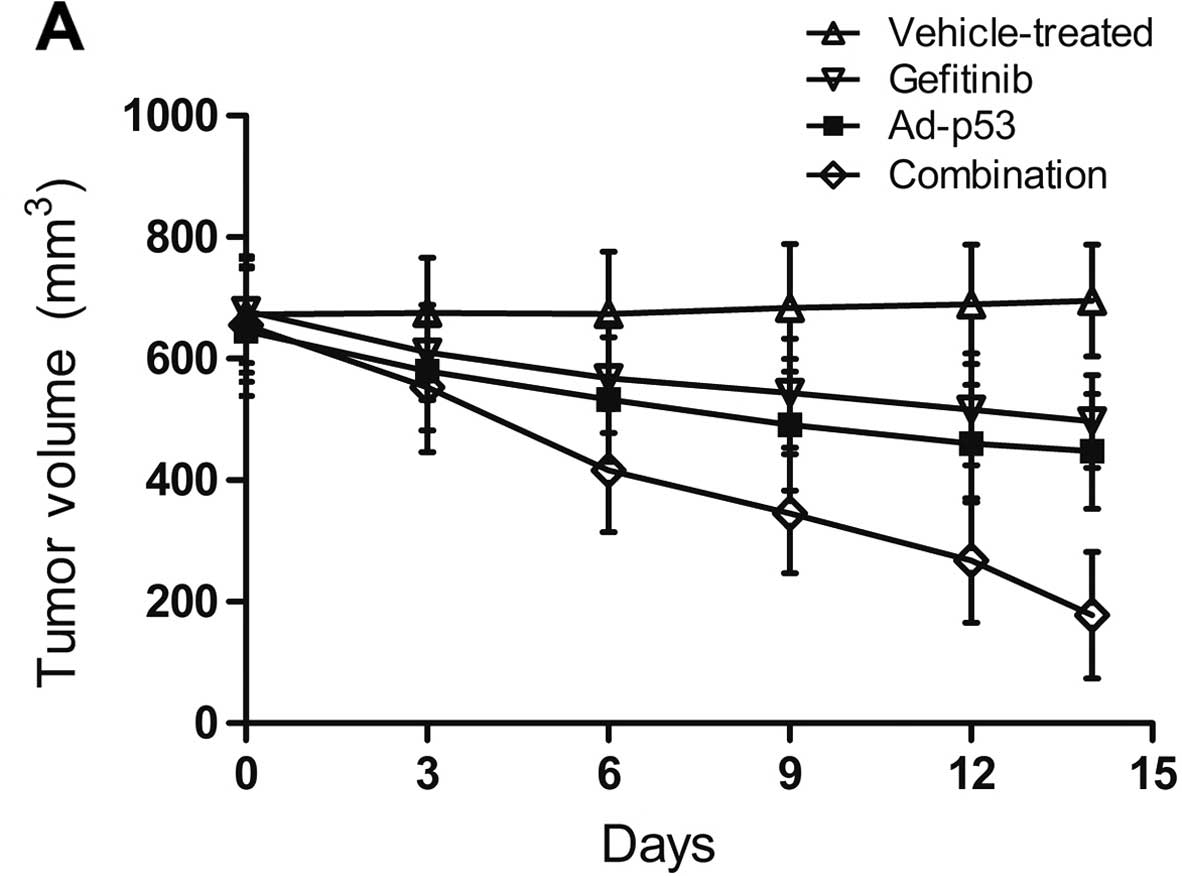
Efficacy of the treatments on the
MDA-MB-468 xenografts in vivo
Ad-p53 and gefitinib were administered to the
MDA-MB-468 xenografts and tumor volume was measured periodically.
Ad-p53 or gefitinib alone caused slight tumor volume shrinkage
(Fig. 6). The tumor inhibition rate
(TIR) in the Ad-p53, gefitinib and combination group was 35.7, 28.7
and 74.4%, respectively. The xenografts were significantly reduced
in size when Ad-p53 and gefitinib were administered simultaneously
(Fig. 6).
Discussion
p53 is a tumor suppressor gene known to be the most
commonly altered gene in human cancer (21). Due to the aggressiveness of cancer
cells with p53 mutation, many studies have been devoted to
eradicating the mutant p53 through a variety of methods such as
isolating the interaction between p53 and p63 with peptide aptamers
(PA) (22), restoring the function
of p53 (23), and disrupting the
interaction between EGFR and colony stimulating factor 1 receptor
(CSF-1R) (24). All these
approaches may remedy the defect of p53 dysfunction. The p53
mutation is kept at a low level in breast cancer, yet mutant p53 is
more prevalent in TNBC (25).
Perhaps this is the reason why TNBCs progress rapidly and have a
poor prognosis. MDA-MB-468 cells harboring EGFR overexpression, as
a gefitinib-resistant model, have been demonstrated in different
studies (26,27).
Small-molecule tyrosine kinase inhibitors (TKIs)
against EGFR have been evaluated in breast cancer. Baselga et
al (9) reported that gefitinib
demonstrated minimal single-agent activity when treating metastatic
breast cancer. Resistance to EGFR inhibitors present a huge
obstacle to breast cancer patients (3). In the present study, in order for us
to explore whether p53 increases the sensitivity of an EGFR
inhibitor, Ad-p53 and EGFR TKI gefitinib were used to treat a TNBC
cell line. Notably, the sensitivity of MDA-MB-468 cells to
gefitinib was significantly increased when they were pretreated
with Ad-p53. The cell proliferation assay indicated that when cells
were treated with gefitinib, the IC50 value of
Ad-p53-infected cells was almost half as much as the
vehicle-treated cells (Fig. 1,
Table I). Furthermore, the results
of the in vitro experiments, such as the clonogenic and
apoptosis assays and cell cycle distribution, revealed that p53
enhanced the sensitivity to gefitinib by inhibiting colony
formation (Fig. 2), by regulating
cellular apoptosis (Fig. 3) and by
inducing cell cycle arrest (Fig.
4).
MDA-MB-468 cells pretreated with Ad-p53 showed
enhanced sensitivity to gefitinib with downregulation of p-Akt,
according to western blotting results, while ERK and p-ERK
exhibited little or no change. Both the PI3K/Akt and Raf/MEK/ERK
pathways are downstream of EGFR activation. The former can resist
apoptosis, while the latter is involved mostly in anti-apoptosis as
well as cell proliferation (28,29).
MDA-MB-468 cells possess an elevated level of p-Akt, and their
persistent activation of p-Akt is relevant to their resistance to
EGFR inhibitors (12). Previous
studies suggest that p53 may participate in the modulation of the
PI3K/Akt and Raf/MEK/ERK pathways in cancer cells (30–32).
Our data, however, indicate that Ad-p53 may interfere with the
PI3K/Akt pathway rather than the Raf/MEK/ERK pathway, leading to an
increase in the sensitivity to gefitinib. Moreover, caspase-9 is a
crucial component of the apoptosis pathway, and activated caspase-9
initiates the caspase cascade by driving the activity of downstream
caspases such as caspase-3, -6 and -7. In the present study,
caspase-9 and cleaved caspase-3 increased synergistically when
MDA-MB-468 cells were exposed to both Ad-p53 and gefitinib in
comparison to the single agent treatment, suggesting that
caspase-mediated apoptosis was triggered in this TNBC treatment.
Similarly, Chang et al (33)
reported that gefitinib induced apoptosis via a p53-dependent
pathway in a lung cancer cell model, which was accompanied by the
upregulation of pro-apoptotic molecules (such as Fas and PUMA) and
the downregulation of anti-apoptotic molecules (such as XIAP and
survivin). Therefore, the synergistic effect of the combined
treatment could be attributed to the effect of gefitinib in
triggering caspase-dependent apoptosis via inhibiting the PI3k/Akt
pathway potentiated by Ad-p53. Recently, Yu et al (34) reported that caspase-dependent
apoptosis and inactivation of the PI3K/Akt pathway were the main
apoptotic mechanisms of human gastric carcinoma AGS cells. Further
studies of expanded TNBC cells should be conducted in order to
obtain more detailed mechanisms related to the dysfunction of the
PI3K/Akt pathway and caspase cascade activation.
Ad-p53 is effective for treating numerous
malignancies, including colon, glioma, lung, ovarian and head and
neck tumors (35–39). In the present study, the in
vivo experiment was designed to mimic a clinical situation in
order to document whether Ad-p53 and gefitinib together
synergistically inhibit the growth of transplanted breast tumors in
nude mice. According to our results, the combination of Ad-p53 and
gefitinib significantly alleviated the bulk of the tumor burden in
the nude mice. These results may pave the way for the clinical
treatment of patients who are resistant to EGFR TKIs. Moreover,
when these two agents are used together, they not only compensate
for the shortcomings of one another, but also maximize their
benefit. For example, their effective combination could potentially
reduce EGFR inhibitor-related side-effects, while exhibiting better
antitumor abilities, thus enhancing the quality of life of these
patients. To further judge the clinical applicability of both
agents, clinical trials should be conducted in follow-up
studies.
In conclusion, finding an effective therapeutic
regimen to cure TNBCs has become an urgent need; TNBCs are
diagnosed in nearly 20% of breast cancer patients, most of whom are
young and have a high rate of recurrence. In the present study, we
demonstrated the feasibility of combining Ad-p53 and the EGFR
inhibitor gefitinib to treat TNBC cells, which are relatively
resistant to gefitinib. Wild-type p53 has a good application
perspective for sensitizing EGFR inhibitors both in vitro
and in vivo. This may stimulate researchers to restore
wild-type p53 in order to enhance the effectiveness of EGFR
targeted therapies.
Acknowledgements
This study was supported by grants from the Natural
Science Foundation of Shandong Province (ZR2012HL34).
References
|
1
|
Podo F, Buydens LM, Degani H, et al:
Triple-negative breast cancer: present challenges and new
perspectives. Mol Oncol. 4:209–229. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Herold CI and Anders CK: New targets for
triple-negative breast cancer. Oncology. 27:846–854.
2013.PubMed/NCBI
|
|
3
|
Alvarez RH, Valero V and Hortobagyi GN:
Emerging targeted therapies for breast cancer. J Clin Oncol.
28:3366–3379. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Viale G, Rotmensz N, Maisonneuve P, et al:
Invasive ductal carcinoma of the breast with the ‘triple-negative’
phenotype: prognostic implications of EGFR immunoreactivity. Breast
Cancer Res Treat. 116:317–328. 2009. View Article : Google Scholar
|
|
5
|
Dent R, Trudeau M, Pritchard KI, et al:
Triple-negative breast cancer: clinical features and patterns of
recurrence. Clin Cancer Res. 13:4429–4434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Herbst RS, Giaccone G, Schiller JH, et al:
Gefitinib in combination with paclitaxel and carboplatin in
advanced non-small-cell lung cancer: a phase III trial - INTACT 2.
J Clin Oncol. 22:785–794. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
von Minckwitz G, Jonat W, Fasching P, et
al: A multicentre phase II study on gefitinib in taxane- and
anthracycline-pretreated metastatic breast cancer. Breast Cancer
Res Treat. 89:165–172. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baselga J and Arteaga CL: Critical update
and emerging trends in epidermal growth factor receptor targeting
in cancer. J Clin Oncol. 23:2445–2459. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Baselga J, Albanell J, Ruiz A, et al:
Phase II and tumor pharmacodynamic study of gefitinib in patients
with advanced breast cancer. J Clin Oncol. 23:5323–5333. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Janmaat ML and Giaccone G: Small-molecule
epidermal growth factor receptor tyrosine kinase inhibitors.
Oncologist. 8:576–586. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Normanno N, De Luca A, Maiello MR, et al:
The MEK/MAPK pathway is involved in the resistance of breast cancer
cells to the EGFR tyrosine kinase inhibitor gefitinib. J Cell
Physiol. 207:420–427. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ferrer-Soler L, Vazquez-Martin A, Brunet
J, Menendez JA, De Llorens R and Colomer R: An update of the
mechanisms of resistance to EGFR-tyrosine kinase inhibitors in
breast cancer: Gefitinib (Iressa™)-induced changes in the
expression and nucleo-cytoplasmic trafficking of HER-ligands
(Review). Int J Mol Med. 20:3–10. 2007.PubMed/NCBI
|
|
13
|
Vousden KH and Lu X: Live or let die: the
cell’s response to p53. Nat Rev Cancer. 2:594–604. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Petitjean A, Mathe E, Kato S, Ishioka C,
Tavtigian SV, Hainaut P and Olivier M: Impact of mutant p53
functional properties on TP53 mutation patterns and tumor
phenotype: lessons from recent developments in the IARC TP53
database. Hum Mutat. 28:622–629. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brosh R and Rotter V: When mutants gain
new powers: news from the mutant p53 field. Nat Rev Cancer.
9:701–713. 2009.PubMed/NCBI
|
|
16
|
Grob TJ, Heilenkötter U, Geist S, et al:
Rare oncogenic mutations of predictive markers for targeted therapy
in triple-negative breast cancer. Breast Cancer Res Treat.
134:561–567. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cancer Genome Atlas Network. Comprehensive
molecular portraits of human breast tumours. Nature. 490:61–70.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ding L, Ellis MJ, Li S, et al: Genome
remodelling in a basal-like breast cancer metastasis and xenograft.
Nature. 464:999–1005. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang S, Benavente S, Armstrong EA, Li C,
Wheeler DL and Harari PM: p53 modulates acquired resistance to EGFR
inhibitors and radiation. Cancer Res. 71:7071–7079. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brand K, Klocke R, Possling A, Paul D and
Strauss M: Induction of apoptosis and G2/M arrest by infection with
replication-deficient adenovirus at high multiplicity of infection.
Gene Ther. 6:1054–1063. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guida E, Bisso A, Fenollar-Ferrer C, et
al: Peptide aptamers targeting mutant p53 induce apoptosis in tumor
cells. Cancer Res. 68:6550–6558. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Selivanova G and Wiman KG: Reactivation of
mutant p53: molecular mechanisms and therapeutic potential.
Oncogene. 26:2243–2254. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Coniglio SJ, Eugenin E, Dobrenis K,
Stanley ER, West BL, Symons MH and Segall JE: Microglial
stimulation of glioblastoma invasion involves epidermal growth
factor receptor (EGFR) and colony stimulating factor 1 receptor
(CSF-1R) signaling. Mol Med. 18:519–527. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Curtis C, Shah SP, Chin SF, et al: The
genomic and transcriptomic architecture of 2,000 breast tumours
reveals novel subgroups. Nature. 486:346–352. 2012.PubMed/NCBI
|
|
26
|
Bianco R, Shin I, Ritter CA, et al: Loss
of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells
counteracts the antitumor action of EGFR tyrosine kinase
inhibitors. Oncogene. 22:2812–2822. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yi YW, Hong W, Kang HJ, et al: Inhibition
of the PI3K/AKT pathway potentiates cytotoxicity of EGFR kinase
inhibitors in triple-negative breast cancer. J Cell Mol Med.
17:648–656. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Elrod HA, Lin YD, Yue P, Wang X, Lonial S,
Khuri FR and Sun SY: The alkylphospholipid perifosine induces
apoptosis of human lung cancer cells requiring inhibition of Akt
and activation of the extrinsic apoptotic pathway. Mol Cancer Ther.
6:2029–2038. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
McCubrey JA, Steelman LS, Chappell WH, et
al: Roles of the Raf/MEK/ERK pathway in cell growth, malignant
transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar
|
|
30
|
Zwang Y, Sas-Chen A, Drier Y, et al: Two
phases of mitogenic signaling unveil roles for p53 and EGR1 in
elimination of inconsistent growth signals. Mol Cell. 42:524–535.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bouali S, Chrétien AS, Ramacci C, et al:
P53 and PTEN expression contribute to the inhibition of EGFR
downstream signaling pathway by cetuximab. Cancer Gene Ther.
16:498–507. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kojima K, Konopleva M, Samudio IJ, et al:
Mitogen-activated protein kinase kinase inhibition enhances nuclear
proapoptotic function of p53 in acute myelogenous leukemia cells.
Cancer Res. 67:3210–3219. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chang GC, Yu CT, Tsai CH, et al: An
epidermal growth factor inhibitor, Gefitinib, induces apoptosis
through a p53-dependent upregulation of pro-apoptotic molecules and
downregulation of anti-apoptotic molecules in human lung
adenocarcinoma A549 cells. Eur J Pharmacol. 600:37–44. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu HY, Kim SO, Jin CY, Kim GY, Kim WJ, Yoo
YH and Choi YH: β-lapachone-induced apoptosis of human gastric
carcinoma AGS cells is caspase-dependent and regulated by the
PI3K/Akt pathway. Biomol Ther. 22:184–192. 2014. View Article : Google Scholar
|
|
35
|
Spitz FR, Nguyen D, Skibber JM, Meyn RE,
Cristiano RJ and Roth JA: Adenoviral-mediated wild-type p53 gene
expression sensitizes colorectal cancer cells to ionizing
radiation. Clin Cancer Res. 2:1665–1671. 1996.PubMed/NCBI
|
|
36
|
Lang FF, Bruner JM, Fuller GN, et al:
Phase I trial of adenovirus-mediated p53 gene therapy for recurrent
glioma: biological and clinical results. J Clin Oncol.
21:2508–2518. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schuler M, Herrmann R, De Greve JL, et al:
Adenovirus-mediated wild-type p53 gene transfer in patients
receiving chemotherapy for advanced non-small-cell lung cancer:
results of a multicenter phase II study. J Clin Oncol.
19:1750–1758. 2001.PubMed/NCBI
|
|
38
|
Buller RE, Runnebaum IB, Karlan BY, et al:
A phase I/II trial of rAd/p53 (SCH 58500) gene replacement in
recurrent ovarian cancer. Cancer Gene Ther. 9:553–566. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pan JJ, Zhang SW, Chen CB, et al: Effect
of recombinant adenovirus- p53 combined with radiotherapy on
long-term prognosis of advanced nasopharyngeal carcinoma. J Clin
Oncol. 27:799–804. 2009. View Article : Google Scholar
|