Introduction
Lung cancer is the leading cause of death from
cancer globally (1). Non-small cell
lung cancer (NSCLC) accounts for more than 80% of all lung cancer
cases. Few NSCLC patients are diagnosed at an early stage and
patients with advanced disease are treated with platinum-based
combination chemotherapy; however, the objective response rate is
very low (2). Resent advances in
understanding the molecular basis of lung cancer has led to
practical implementation of epidermal growth factor receptor
(EGFR)-targeted treatment. The EGFR tyrosine kinase inhibitor (TKI)
gefitinib was approved for the treatment of NSCLC in Japan in
January 2002, and activating somatic mutations in EGFR,
conferring sensitivity to EGFR TKIs were discovered in 2004
(3). Since then, EGFR TKIs, such as
gefitinib and the equally effective erlotinib, have become the
first-line treatment option for NSCLC patients in which the tumor
harbors activating EGFR mutations, based on the results of a
number of phase III trials (4–9).
Therefore, in modern clinical settings, EGFR mutation
testing has become essential for offering the most suitable therapy
for a patient with advanced NSCLC.
The historical standard for EGFR mutation
testing has been direct sequencing of DNA extracted from samples of
resected tumor or from biopsies. This method is advantageous as it
can be applied to discover ‘new’ mutations; to date, nearly 30
mutations in exons 18–21 have been detected in lung cancer
specimens (3,10–14).
However, direct sequencing has several limitations. The method
requires complex steps and a few days to obtain a result. More
importantly, the sensitivity of this method is low; mutant DNA must
comprise ~20% of all the DNA in a sample in order to be reliably
detected (15). Therefore, when the
diagnosis is based on cytology samples that contain a very low
percentage of tumor cells, direct sequencing is not applicable.
More recently, based on findings that the most
common EGFR mutations are a 15-bp in-frame deletion in exon
19 (delE746-A750) and a point mutation in exon 21 (L858R), which
together account for ~90% of cases with EGFR mutations
(16), more focused and
mutation-specific approaches have been developed. These methods,
PCR-Invader (17,18), peptide nucleic acid-locked nucleic
acid (PNA-LNA) PCR clamp (19),
cycleave PCR (20), and Scorpion
Amplification Refractory Mutation System (ARMS) (21) are PCR-based methods that can detect
known EGFR mutations with higher sensitivity and a shorter
turnaround time than direct sequencing. Therefore, these methods
are now frequently used in modern clinical laboratory practice.
However, these methods still have several
limitations. These methods adopt relatively complex PCR
technologies with pre-designed fluorogenic probes, are packaged by
manufacturers, and are often available through outside reference
laboratories at relatively expensive rates. The turnaround time for
receiving results is 3–5 days, which can sometimes create a
bottleneck for immediately starting TKI therapy in patients.
Moreover, the cost of the testing renders repeated examination
impossible; yet, this may sometimes be required for patients in
whom the disease recurs after prior TKI therapy. Therefore, more
rapid and less expensive EGFR mutation testing is
required.
Here, we developed a new, simple, PCR-based method
for the detection of the two most common EGFR mutations.
This assay involves a pair of mutation-specific primers used in
combination with a newly developed PCR machine that is equipped
with a novel thermo-control mechanism that makes ultrarapid PCR
cycling possible. In the present study, we evaluated this approach
for EGFR mutation detection in tumor tissue gathered during
resection and showed the feasibility of using this approach in a
cytology sample collected by bronchoscopic examination.
Materials and methods
Cell lines and DNA samples
All lung cancer cell lines used in the present study
originated from adenocarcinoma. The 11–18 cell line was obtained
from the Cell Resource Center for Biomedical Research (Tohoku
University, Sendai, Japan). The Ma1 cell line was provided by Dr
Hirashima (Osaka Prefectural Habikino Hospital, Osaka, Japan). The
A549 cell line was purchased from the American Type Culture
Collection (Rockville, MD, USA). The EGFR mutation status of
these cell lines was examined in our previous study (22). Cells were maintained in DMEM (Wako,
Osaka, Japan) supplemented with 10% fetal bovine serum (Life
Technologies, Carlsbad, CA, USA), 50 U/ml penicillin, and 50 U/ml
streptomycin (both from Wako). Genomic DNA was prepared using a
Wizard® Genomic DNA Purification kit (A1120; Promega,
Madison, WI, USA) according to the manufacturer’s instructions.
Clinical samples
Ethical approval was obtained from the Tottori
University Hospital and fully informed written consent was obtained
from all patients involved prior to the surgery or tissue
collection.
Tumor tissues were obtained from surgical specimens
of resected tumors, from 143 lung cancer patients treated at
Tottori University Hospital; these samples were embedded in
Tissue-Tek OCT Compound (Sakura Finetechnical, Tokyo, Japan), and
were immediately frozen at −80°C. Macrodissection of the
OCT-embedded tissue samples was performed to enrich the final
proportion of the tumor DNA, and DNA was extracted using the
Wizard® Genomic DNA Purification kit. For samples with
discordant results between direct sequencing and mutation-specific
PCR, a PCR-Invader method was performed by BML, Inc. (Tokyo, Japan)
as a reference test.
Direct sequence analysis
For direct sequence analysis exon 19 and 21 of
EGFR, the following PCR primers were used: EGFR exon
19F, 5′-GCAATATCAGCCTTAGGTGCGGCTC-3′ and EGFR exon 19R,
5′-CATAGAAAGTGAACATTTAGGAT GTG-3′; and EGFR exon 21F,
5′-CTAACGTTCGCCAGCC ATAAGTCC-3′ and EGFR exon 21R,
5′-GCTGCGAGCTCA CCCAGAATGTCTGG-3′. The PCR conditions were as
follows: 1 cycle at 94°C for 9 min, followed by 40 cycles each
consisting of 94°C for 1 min, 57°C for 1 min, and 72°C for 2 min,
and a final cycle at 72°C for 5 min. The PCR products were purified
with a MultiScreen-PCR filter plate (Millipore, Tokyo, Japan) and
then sequenced using a BigDye Terminator v3.1 cycle sequencing kit
and an ABI PRISM 3130xl genetic analyzer (Applied Biosystems,
Foster City, CA, USA).
Design of mutation-specific PCR primer
sets
We designed a deletion-specific primer for the
delE746-A750 mutation within exon 19 and a point mutation-specific
primer for the L858R mutation within exon 21 of EGFR.
Sequences of the primer sets were as follows: PCR forward primer
for delE746-A750, 5′-CACAATTGCCAGTTAACGTCTTC-3′ (19DF) and PCR
reverse primer for delE746-A750, 5′-TGTTGGCTTTCGGAG ATGTTTTG-3′
(19DR3); PCR forward primer for L858R, 5′-TCCCATGATGATCTGTCCCT-3′
(21F2f) and PCR reverse primer for L858R, 5′-CACCCAGCAGTTTGGTCC-3′
(21ARMS3).
Mutation-specific PCR using a
conventional thermal cycler
For conventional PCR amplification using the
mutation-specific primer sets, PCR conditions were as follows: the
reaction mixtures contained 2 μl of 10X PCR buffer, 0.5 μl of
dNTPs, 1 μl of each allelic-specific primer (10 μM), 0.2 μl of
AmpliTaq® Gold DNA polymerase (Applied Biosystems), 1 μl
of template DNA, and 14.3 μl of ddH2O in a total volume
of 20 μl. Thermal cycling conditions on a PCR Thermal Cycler Dice
(Takara, Shiga, Japan) were as follows for the delE746-A750
mutation: 1 cycle at 94°C for 9 min, followed by 35 cycles each
consisting of 94°C for 1 min, 59°C for 1 min, and 72°C for 2 min,
and a final cycle at 72°C for 5 min. Similarly, for the L858R
mutation, conditions involved 1 cycle at 94°C for 9 min, followed
by 35 cycles at 94°C for 1 min, 64°C for 1 min, and 72°C for 2 min,
and a final cycle at 72°C for 5 min. The PCR products were then
electrophoresed on agarose gels and stained with ethidium
bromide.
Mutation-specific PCR using an ultrarapid
PCR machine
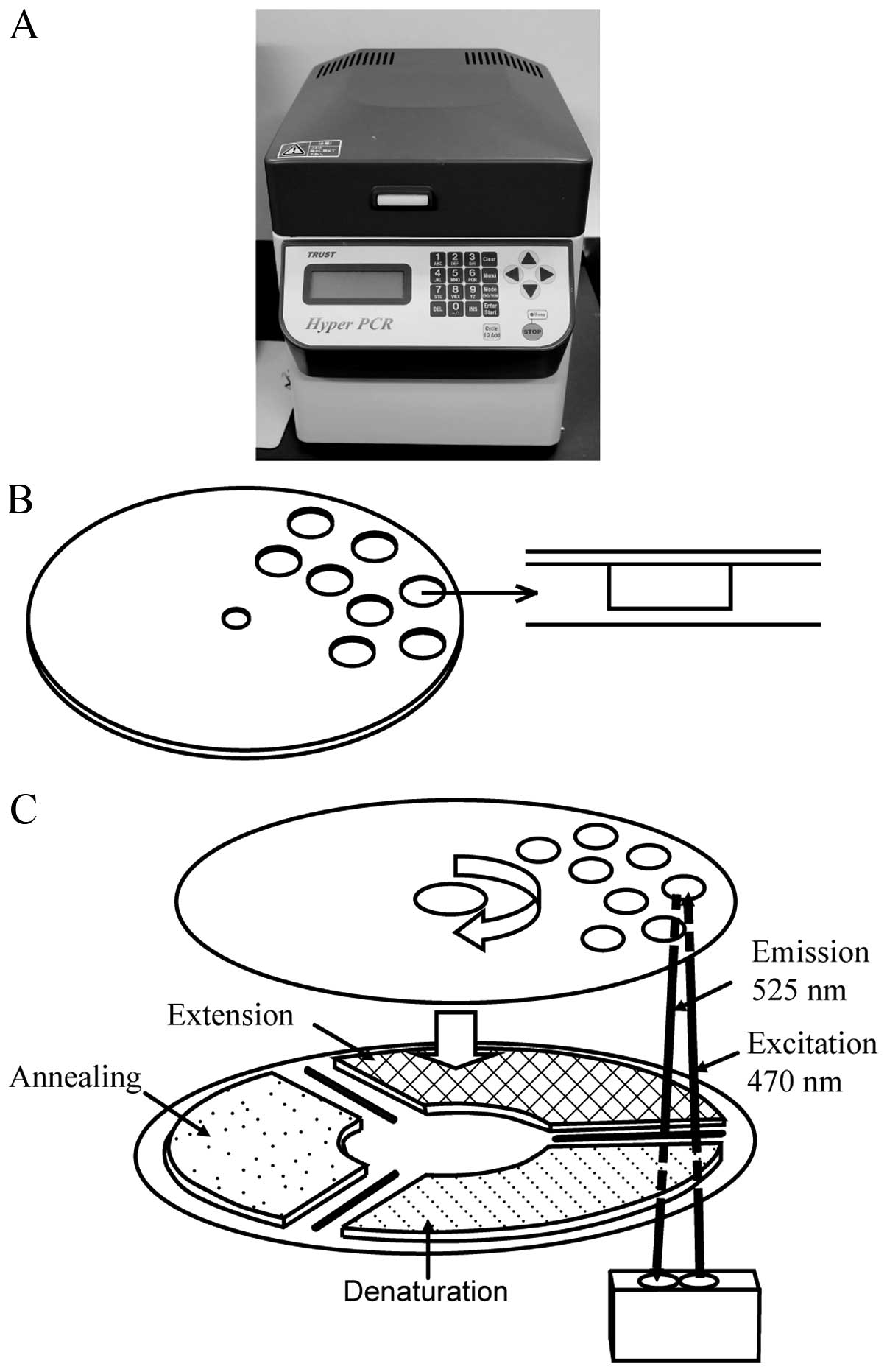
For ultrarapid PCR-based EGFR mutation
detection, we utilized a newly developed high-speed real-time PCR
machine, termed the ‘Hyper-PCR’ UR104MK III (Trust Medical, Hyogo,
Japan), which was jointly developed by ourselves and Trust Medical
(23). The UR104MK III employs a
novel temperature control technology. In this system, the PCR
mixture is enclosed in a small vessel on a thin, flexible plastic
disk and sealed with adhesive film, and the disk is rotated rapidly
onto three separated heat elements. By controlling the speed of
rotation and the temperature of the three heat elements, rapid PCR
can be accomplished. Real-time monitoring of the fluorescent dsDNA
dye produced during PCR progression, and the ability to perform
melting curve analysis of the PCR product are also incorporated
into this machine (Fig. 1). The
typical time for amplification and detection when using this
apparatus is <10 min.
The optimized reaction mixtures for use with this
machine contained 1.6 μl of 10X Fast Buffer I, 1.3 μl of a 2.5 mM
dNTP mixture, 0.4 μl of each allele-specific primer (10 μM), 0.2 μl
of SpeedSTAR HS DNA Polymerase (5 U/μl) (Takara), 1 μl of template
DNA, 1.6 μl of 1:2,000 SYBR®-Green I nucleic acid gel
stain (Cambrex Biosciences, Rockland, ME, USA), and 9.5 μl of
ddH2O in a total volume of 16 μl. Furthermore, dimethyl
sulfoxide (DMSO) Hybri-Max® (Sigma, St. Louis, MO, USA)
was added to a final concentration of 5%. Thermal cycling
conditions for ultrarapid PCR were as follows for the delE746-A750
mutation: 1 cycle at 94°C for 1 min, followed by 35 cycles each
including 98°C for 1.30 sec, 55°C for 5.00 sec, and 72°C for 3.00
sec. Similarly, for the L858R mutation, conditions entailed 1 cycle
at 94°C for 1 min, followed by 30 cycles each consisting of 98°C
for 1.30 sec, 68°C for 8.00 sec, and a further 68°C for 8.00 sec.
Total PCR cycling time for the delE746-A750 and the L858R mutation
detection was within 6 and 9 min, respectively. Following PCR
cycling, melting curve analysis was performed within 4 min.
For interpretation of the ultrarapid PCR results,
criteria used in other studies of qualitative real-time PCR
analysis were applied (24). In
brief, to be considered as a positive result, a fluorescence signal
generated during ultrarapid PCR should display an exponential
amplification above the threshold level and the obvious crossing
point (Cp) (25), with a single
peak upon melting curve analysis, giving a unique melting
temperature (Tm) value. A signal was considered as negative when no
Cp value was obtained within the amplification cycles.
Results
Establishment of EGFR mutation-specific
ultrarapid PCR
We first established a specific PCR to detect
mutations within exons 19 and 21 of EGFR, which are
representative mutations underlying the responsiveness of NSCLC to
EGFR inhibitors (3). The genomic
sequence of EGFR was retrieved from the NCBI database
(NM_005228). For the in-frame deletion within exon 19, which
removes nucleotides 2235–2249, causing a deletion of amino acids
746 through 750 (delE746-A750), we designed a deletion-specific
primer, 19DR3 (Fig. 2A). This
primer was designed to anneal only to the genomic sequence
harboring the nucleotide 2235–2249 deletion, by connecting the
flanking sequences on either side of the deletion. By shortening
the 3′-end of the primer that corresponded to the upstream genomic
sequence, we could improve the specificity of the primer.
For the exon 21 amino acid substitution, in which G
is substituted for T at nucleotide 2573, causing an amino acid
substitution of L to R (L858R), a point mutation-specific primer,
21ARMS3, was designed (Fig. 2B).
Here, we employed the ARMS technique, and designed the primers to
be refractory to PCR amplification of non-matching target sequences
(26,27), by also including an additional
mismatch in the candidate point mutation-specific primers
(ARMS1-10) at positions −2 or −3 from the 3′-end of the primers
(data not shown). Among these candidate primers, we chose the
primer (ARMS3) that allowed discrimination without decreasing the
sensitivity and specificity of amplification in a series of
experiments in which these candidate primers were tested under the
same temperature and time conditions in ultrarapid PCR.
The forward primers for each mutation-specific
primer were designed to match the stable area of each EGFR
(19F and 21F). The concentration of the PCR primers and magnesium,
the annealing temperature and other cycling parameters, and the
type of DNA polymerase used were determined by exploration, and the
conditions described here are the final optimized conditions.
Specificity of the mutation-specific
primers
To evaluate the specificity of the mutation-specific
primers, ultrarapid PCR using the mutation-specific primers was
performed on lung cancer cell lines, and the concordance of these
results with those of conventional PCR was evaluated. EGFR
genotyping of the lung cancer cell lines Ma1, 11–18 and A549 had
been performed by sequence analysis in our previous study, and were
revealed as delE746-A750, L858R and wild-type, respectively
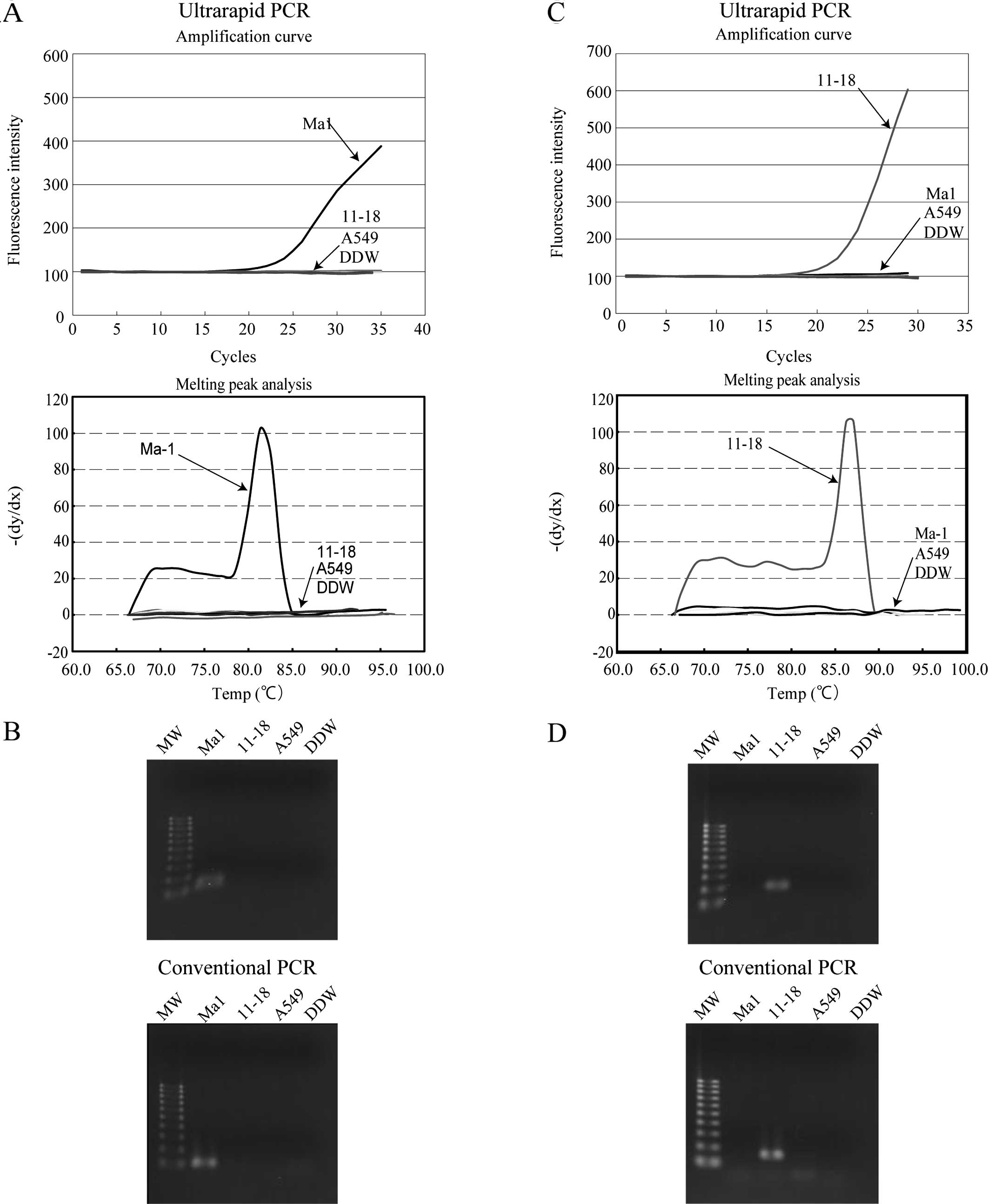
(22). As shown in Fig. 3A, using primer sets, 19DF and 19DR3,
a significant increase in fluorescence intensity was observed upon
ultrarapid PCR only in Ma1 cells that harbor delE746-A750, and
melting curve analysis revealed a clear peak at the expected Tm
(81.3°C). The PCR product was visualized by agarose gel
electrophoresis and the expected product of 113 bp was detected. To
validate the data, conventional PCR was performed using the same
primer sets, and a product of the same size was detected only in
Ma1 cells (Fig. 3B).
A similar experiment was performed using the L858R
point mutation-specific primers (21F2f and 21ARMS3) in ultrarapid
PCR. As shown in Fig. 3C, a
significant increase in fluorescence intensity and a clear melting
curve peak at the expected Tm (87.1°C) was observed only in 11–18
cells that harbor the exon 21 L858R point mutation, and the size of
the amplicon was confirmed as 166 bp by agarose gel
electrophoresis. The same result was obtained by conventional PCR
using this primer set (Fig. 3D).
These data revealed that the combination of mutation-specific
primers and ultrarapid PCR yielded satisfactory discrimination of
the mutant alleles.
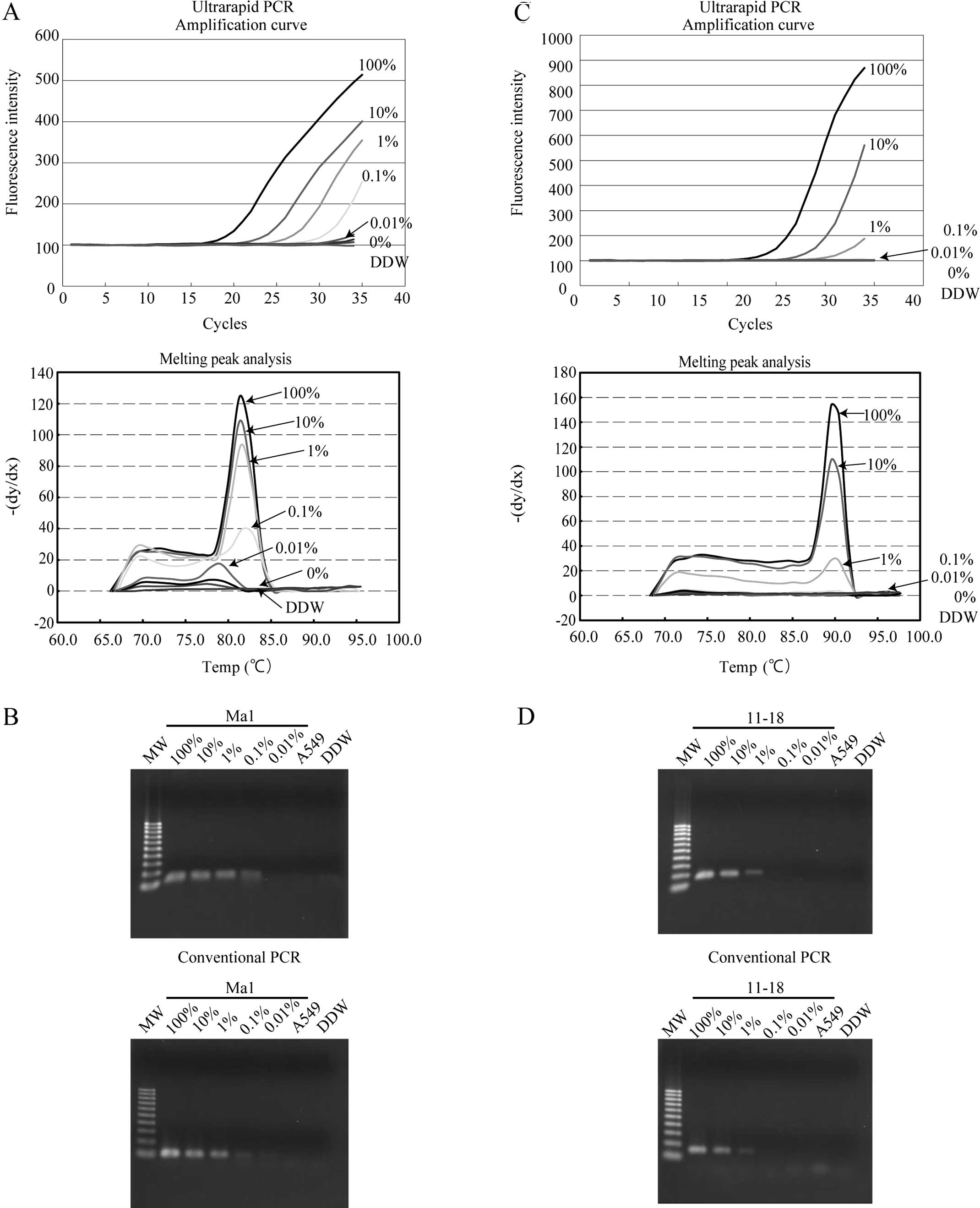
Sensitivity of mutation-specific PCR
To evaluate the sensitivity of the assay, a serial
dilution of lung cancer cell lines carrying EGFR mutations
(Ma1 or 11–18) into wild-type cell lines (A549) was analyzed by
ultrarapid PCR using the mutation-specific primers. Using the
delE746-A750 primers in ultrarapid PCR, a mixture containing 0.1%
Ma1 cells could be detected as positive from the amplification
curve and melting curve analysis, and this result was confirmed by
gel electrophoresis (Fig. 4A). The
same result was also obtained by conventional PCR (Fig. 4B). Using the L858R primer sets, a
cell mixture containing 1% 11–18 cells was detected as positive by
ultrarapid PCR, and the result was confirmed by gel electrophoresis
of the PCR product (Fig. 4C). The
same detection limit was also observed with this primer set with
conventional PCR (Fig. 4D). These
data revealed that ultrarapid PCR combined with delE746-A750 and
L858R mutation-specific primers allowed detection of 0.1 or 1%
mutation-carrying lung cancer cells among wild-type cells,
respectively.
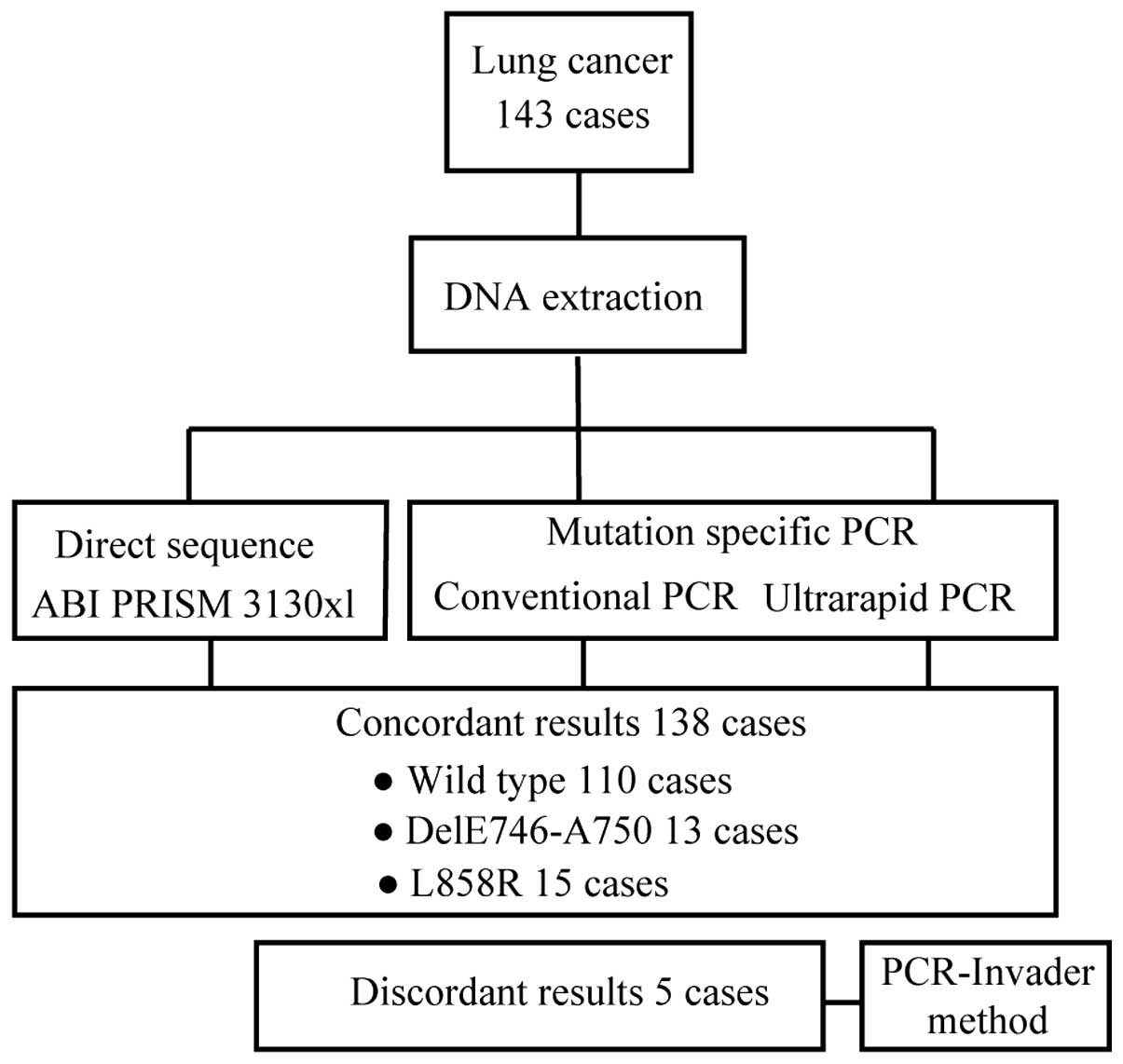
Clinical sample testing
We evaluated the concordance of the results obtained
by direct sequencing and using mutation-specific primers in
ultrarapid PCR or conventional PCR in 143 lung cancer tumors.
Overall, the results of 138 of 143 samples were concordant among
direct sequencing, ultrarapid and conventional PCR (Fig. 5). The remaining five samples that
demonstrated inconsistent results are shown in Table I. In these samples, direct
sequencing could not detect any mutations, whereas the results with
ultrarapid PCR and conventional PCR were concordant: two
delE746-A750 and three L858R mutations were identified. To evaluate
these discrepant samples, we used PCR-Invader analysis, which
confirmed the PCR-based results. Therefore, we concluded that the
indicated mutations were indeed present in these samples, yet could
not be detected by direct sequencing, possibly due to the lower
sensitivity of detection of direct sequencing methodology.
 | Table ISamples showing discordant results
between sequencing and PCR detection methods. |
Table I
Samples showing discordant results
between sequencing and PCR detection methods.
| Case | Direct
sequence | Conventional
PCR | Ultrarapid PCR | PCR-Invader
method |
|---|
| 1 | Wild-type | DelE746-A750 | DelE746-A750 | DelE746-A750 |
| 2 | Wild-type | DelE746-A750 | DelE746-A750 | DelE746-A750 |
| 3 | Wild-type | L858R | L858R | L858R |
| 4 | Wild-type | L858R | L858R | L858R |
| 5 | Wild-type | L858R | L858R | L858R |
When compared with the concluded EGFR
mutation status, the sensitivity and specificity of direct
sequencing was 84.8 and 100% respectively, whereas those of
ultrarapid PCR were 100 and 100%, respectively (Table II). These data revealed that
ultrarapid PCR has superior sensitivity and specificity in clinical
samples to direct sequencing, and parallels that of the PCR-Invader
method, which is one of the commonly used PCR-based methodologies
in current clinical practice.
| Table IISensitivity and specificity of direct
sequencing and mutation-specific ultrarapid PCR. |
Table II
Sensitivity and specificity of direct
sequencing and mutation-specific ultrarapid PCR.
| Direct
sequence | Ultrarapid PCR |
|---|
| Sensitivity | 84.8% (28/33) | 100% (33/33) |
| Specificity | 100% (110/110) | 100% (110/110) |
Ultrarapid detection of EGFR mutation in
a patient with adenocarcinoma
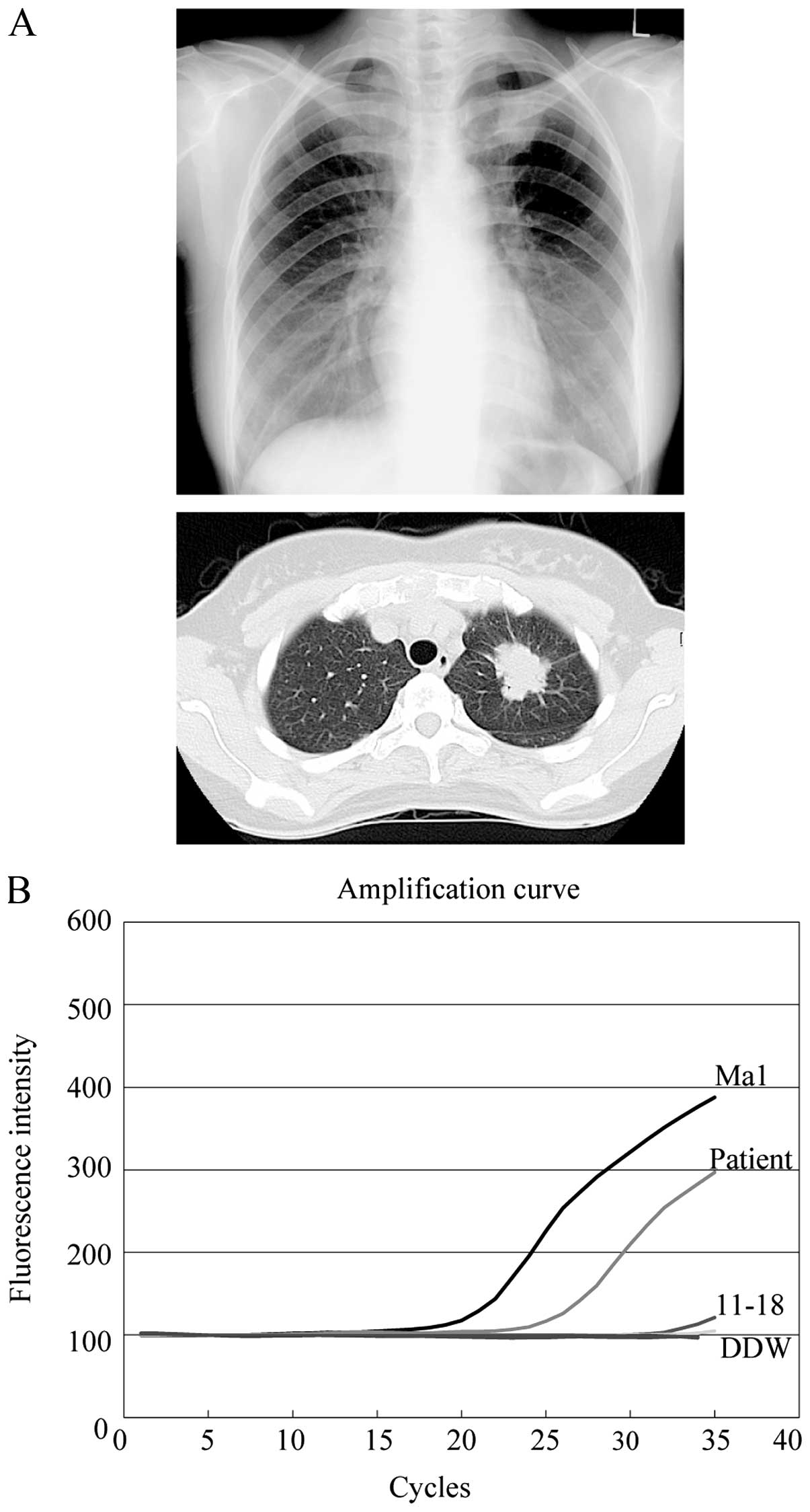
A 39-year-old woman with no history of smoking was
referred to our hospital due to the presence of an abnormal shadow
in her left upper lung field that was noticed during her regular
medical checkup (Fig. 6A, upper
panel). A computed tomography (CT) scan of the chest of this
patient revealed a spiculated nodular shadow with a 35-mm diameter
in the superior division of the left upper lobe (Fig. 6A, lower panel).
F-2-deoxy-2-fluoro-D-glucose (FDG)-positron emission tomography
revealed multiple lesions with high uptake in her liver and
vertebrae. Based on the suspicion of adenocarcinoma with multiple
metastases, we preformed flexible bronchoscopic examination with
washing, brushing and forceps biopsy. In addition to the
cytological and histological examination of the lavage and biopsy
specimens, we applied ultrarapid PCR analysis to the lavage sample.
The lavage was centrifuged and DNA was extracted within 40 min
after sample collection. Ultrarapid PCR analysis subsequently
revealed the presence of the delE746-A750 EGFR mutation in
her lavage sample, within 6 min (Fig.
6B). After waiting for confirmation of positive results by
cytology and histology, which were obtained 3–4 days after
bronchoscopic examination, the patient was diagnosed as having
adenocarcinoma with an EGFR mutation (cT2aN0M1b, stage IV).
She commenced treatment with 150 mg erlotinib/day immediately, and
her lung CT scan at 6 weeks after the initiation of treatment
revealed a marked improvement.
Discussion
In the present study, we newly developed an
ultrarapid PCR approach for detecting EGFR mutations. This
method showed excellent specificity and sensitivity for clinical
samples, which is superior to direct sequencing and is comparable
to other PCR-based methodologies that are frequently used in modern
laboratory practice. In addition, to our knowledge, this method has
superior rapidity among the methodologies reported for EGFR
mutation detection. Therefore, introduction of this technology to
clinical practice may open new opportunities for diagnosis and
therapy of lung cancer patients.
In the present study, we used direct sequencing as a
method against which to compare our newly developed ultrarapid
mutation-specific PCR, since it is the historical standard and many
studies reporting novel methodology have used it for comparison
(28). However, even when using
macrodissection to enrich tumor DNA in a sample, the sensitivity of
direct sequencing was 84.8%, whereas that of ultrarapid PCR was
100%. In addition, it is noteworthy that the mutation analysis
results of the five sequencing-discordant samples were consistent
between ultrarapid PCR and PCR-Invader, one of the current
laboratory-standard PCR-based assays. In a recent study, these
frequently used PCR-based assays, i.e., PCR-Invader, PNA-LNA PCR
clamp, Scorpion ARMS and cycleave PCR, could detect mutations in at
least 1% of mutant/wild-type allele-admixture samples, and showed
equal sensitivity and specificity in clinical specimens (tissue and
cytology samples) (29). Based on
the detection limit in the admixture analysis and the results of
surgical resected tissues in our study, the ultrarapid PCR we
present here appears to be comparable to current sensitive
laboratory-standard PCR-based methodologies for detecting
EGFR mutations.
Moreover, compared to the other current PCR-based
methods, ultrarapid PCR has several advantages. The first
significant advantage is the rapid turnaround time for
amplification and detection (<10 min in the experiments reported
here). This machine employed a novel thermo-control mechanism, with
a thermal ramp rate of up to 20°C/sec; this is the shortest ramp
rate among the published ramp rate for thermal cyclers (30). By combining this machine with our
newly designed mutation-specific primers, choosing an adequate
polymerase, optimizing the composition of the reaction mixture, and
adjusting the thermal conditions, we have here developed the
fastest real-time detection system for EGFR mutation
screening to date.
The second advantage of our approach is its
simplicity and cost-effectiveness. We used the double-strand
DNA-binding dye, SYBR-Green I, for real-time detection of the PCR
product, whereas other systems use fluorogenic probes. Since
EGFR mutation detection is a fundamentally qualitative
detection, we believe the simplicity, flexibility, and
cost-effectiveness of detection using SYBR-Green I may be
sufficient for the purpose. Besides their relative expense, the use
of fluorogenic probes complicates modification and optimization of
real-time PCR, and primers/probes need to be designed according to
specific rules, due to the simultaneous annealing of the primers
and probes in real-time PCR (31).
Our system was designed to detect the two major
EGFR mutations for which the majority of clinical evidence
supports the use of EGFR TKIs; it is not able to detect all
EGFR mutations, similar to other PCR-based systems. However,
clinical data supporting the use of EGFR TKIs for less common
mutations are emerging, and adaptation of the detection system to
these more rare mutations will be required in the near feature. Our
system may present a convenient way to do this, since it would only
require the design of new primers and adjustment of the PCR
conditions.
Our ultrarapid mutation-specific PCR will open new
potential applications for the clinical management of lung cancer
patients. The representative case we presented in the present study
illustrates three important characteristics of this test. First,
this test can be applied to cytology samples obtained in routine
clinical practice, such as bronchoscopic lavage. Second, since the
test result can be obtained within <50 min after sample
collection, treatment with TKIs can be started very rapidly. Third,
since this system is simple and less expensive, this test may
enable clinicians to request repeat tests for EGFR mutation
for a given patient. Repeated testing for EGFR mutations is
in demand, particularly for recurrent or metastatic lesions, to
allow selection of optimal treatment. Eventually, this test can be
used as a point-of-care approach, which has not been available for
EGFR testing to date.
In conclusion, we developed a simple, rapid, and
less expensive EGFR mutation detection system. This system
has comparable sensitivity and specificity to that of recently
developed laboratory-standardized PCR-based methods, and, unlike
these methods, offers high speed performance. Given the simplicity
of the methodology, this system may help to usher in a new phase in
EGFR mutation testing for the diagnosis and treatment of
lung cancer patients.
References
|
1
|
Ferlay J, Parkin DM and Steliarova-Foucher
E: Estimates of cancer incidence and mortality in Europe in 2008.
Eur J Cancer. 46:765–781. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Azzoli CG, Baker S Jr, Temin S, et al:
American Society of Clinical Oncology Clinical Practice Guideline
update on chemotherapy for stage IV non-small-cell lung cancer. J
Clin Oncol. 27:6251–6266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lynch TJ, Bell DW, Sordella R, et al:
Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maemondo M, Inoue A, Kobayashi K, et al:
Gefitinib or chemotherapy for non-small-cell lung cancer with
mutated EGFR. N Engl J Med. 362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mitsudomi T, Morita S, Yatabe Y, et al:
Gefitinib versus cisplatin plus docetaxel in patients with
non-small-cell lung cancer harbouring mutations of the epidermal
growth factor receptor (WJTOG3405): an open label, randomised phase
3 trial. Lancet Oncol. 11:121–128. 2010. View Article : Google Scholar
|
|
6
|
Mok TS, Wu YL, Thongprasert S, et al:
Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N
Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rosell R, Carcereny E, Gervais R, et al:
Erlotinib versus standard chemotherapy as first-line treatment for
European patients with advanced EGFR mutation-positive
non-small-cell lung cancer (EURTAC): a multicentre, open-label,
randomised phase 3 trial. Lancet Oncol. 13:239–246. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou C, Wu YL, Chen G, et al: Erlotinib
versus chemotherapy as first-line treatment for patients with
advanced EGFR mutation-positive non-small-cell lung cancer
(OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase
3 study. Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Han JY, Park K, Kim SW, et al:
First-SIGNAL: first-line single-agent iressa versus gemcitabine and
cisplatin trial in never-smokers with adenocarcinoma of the lung. J
Clin Oncol. 30:1122–1128. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Paez JG, Janne PA, Lee JC, et al: EGFR
mutations in lung cancer: correlation with clinical response to
gefitinib therapy. Science. 304:1497–1500. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pao W, Miller V, Zakowski M, et al: EGF
receptor gene mutations are common in lung cancers from ‘never
smokers’ and are associated with sensitivity of tumors to gefitinib
and erlotinib. Proc Natl Acad Sci USA. 101:13306–13311. 2004.
View Article : Google Scholar
|
|
12
|
Shigematsu H, Lin L, Takahashi T, et al:
Clinical and biological features associated with epidermal growth
factor receptor gene mutations in lung cancers. J Natl Cancer Inst.
97:339–346. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Han SW, Kim TY, Hwang PG, et al:
Predictive and prognostic impact of epidermal growth factor
receptor mutation in non-small-cell lung cancer patients treated
with gefitinib. J Clin Oncol. 23:2493–2501. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kosaka T, Yatabe Y, Endoh H, Kuwano H,
Takahashi T and Mitsudomi T: Mutations of the epidermal growth
factor receptor gene in lung cancer: biological and clinical
implications. Cancer Res. 64:8919–8923. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pao W and Ladanyi M: Epidermal growth
factor receptor mutation testing in lung cancer: searching for the
ideal method. Clin Cancer Res. 13:4954–4955. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mitsudomi T and Yatabe Y: Mutations of the
epidermal growth factor receptor gene and related genes as
determinants of epidermal growth factor receptor tyrosine kinase
inhibitors sensitivity in lung cancer. Cancer Sci. 98:1817–1824.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hall JG, Eis PS, Law SM, et al: Sensitive
detection of DNA polymorphisms by the serial invasive signal
amplification reaction. Proc Natl Acad Sci USA. 97:8272–8277. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Naoki K, Soejima K, Okamoto H, et al: The
PCR-invader method (structure-specific 5′ nuclease-based method), a
sensitive method for detecting EGFR gene mutations in lung cancer
specimens; comparison with direct sequencing. Int J Clin Oncol.
16:335–344. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nagai Y, Miyazawa H, Huqun, et al: Genetic
heterogeneity of the epidermal growth factor receptor in non-small
cell lung cancer cell lines revealed by a rapid and sensitive
detection system, the peptide nucleic acid-locked nucleic acid PCR
clamp. Cancer Res. 65:7276–7282. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yatabe Y, Hida T, Horio Y, Kosaka T,
Takahashi T and Mitsudomi T: A rapid, sensitive assay to detect
EGFR mutation in small biopsy specimens from lung cancer. J Mol
Diagn. 8:335–341. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kimura H, Kasahara K, Kawaishi M, et al:
Detection of epidermal growth factor receptor mutations in serum as
a predictor of the response to gefitinib in patients with
non-small-cell lung cancer. Clin Cancer Res. 12:3915–3921. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Takata M, Chikumi H, Miyake N, et al: Lack
of AKT activation in lung cancer cells with EGFR mutation is a
novel marker of cetuximab sensitivity. Cancer Biol Ther.
13:369–378. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fujimoto T, Konagaya M, Enomoto M, et al:
Novel high-speed real-time PCR method (Hyper-PCR): results from its
application to adenovirus diagnosis. Jpn J Infect Dis. 63:31–35.
2010.PubMed/NCBI
|
|
24
|
Barbau-Piednoir E, Botteldoorn N, Yde M,
Mahillon J and Roosens NH: Development and validation of
qualitative SYBR®Green real-time PCR for detection and
discrimination of Listeria spp. and Listeria monocytogenes. Appl
Microbiol Biotechnol. 97:4021–4037. 2013. View Article : Google Scholar :
|
|
25
|
Bustin SA: Absolute quantification of mRNA
using real-time reverse transcription polymerase chain reaction
assays. J Mol Endocrinol. 25:169–193. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Newton CR, Graham A, Heptinstall LE, et
al: Analysis of any point mutation in DNA. The amplification
refractory mutation system (ARMS). Nucleic Acids Res. 17:2503–2516.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Newton CR, Kalsheker N, Graham A, et al:
Diagnosis of α1-antitrypsin deficiency by enzymatic
amplification of human genomic DNA and direct sequencing of
polymerase chain reaction products. Nucleic Acids Res.
16:8233–8243. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ellison G, Zhu G, Moulis A, Dearden S,
Speake G and McCormack R: EGFR mutation testing in lung cancer: a
review of available methods and their use for analysis of tumour
tissue and cytology samples. J Clin Pathol. 66:79–89. 2013.
View Article : Google Scholar :
|
|
29
|
Goto K, Satouchi M, Ishii G, et al: An
evaluation study of EGFR mutation tests utilized for non-small-cell
lung cancer in the diagnostic setting. Ann Oncol. 23:2914–2919.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim YH, Yang I, Bae YS and Park SR:
Performance evaluation of thermal cyclers for PCR in a rapid
cycling condition. Biotechniques. 44:495–496. 498500passim. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nitsche A: Oligonucleotide design for
in-house real-time PCR applications in microbiology. Real-Τime PCR
in Microbiology. Mackay IM: Caister Academic Press; Norfolk: pp.
41–69. 2007
|