Introduction
Lung cancer is the leading cause of cancer-related
death worldwide and is associated with a 5-year survival rate of
15% (1). Non-small cell lung cancer
(NSCLC) accounts for ~80% of all lung cancers and is associated
with relatively low survival rates. Among patients with advanced
NSCLC and good performance status, even standard first-line
platinum-based chemotherapy is associated with response rates of
less than 40% and a median length of survival of 8–14 months after
diagnosis (2). Malignant
mesothelioma is an aggressive tumor type with a very poor prognosis
and a median length of patient survival of 9–12 months after
diagnosis. At this time, there are few effective chemotherapeutic
options for the treatment of malignant mesothelioma, yet available
treatments include cisplatin, vinorelbin and gemcitabine (3). We believe that new strategies based on
better understanding of tumor biology could maximize the efficacy
of current treatments.
Pemetrexed, a multitargeted antifolate cytotoxic
agent, is used to treat malignant mesothelioma and NSCLC, mainly in
nonsquamous cell carcinomas (4–6).
Pemetrexed targets enzymes involved in the de novo
biosynthesis of purines and pyrimidines, including thymidylate
synthase (TS), dihydrofolate reductase (DHFR) and glycinamide
ribonucleotide formyltransferase (GARFT), thereby inducing an
imbalance in the nucleotide pool and DNA damage (7). Cellular studies indicate that
pemetrexed induces DNA damage, growth arrest and apoptosis by
generating reactive oxygen species (ROS), accumulation of which
contributes to the apoptosis of human melanoma cells treated with
pemetrexed (8). On the basis of
these results, we hypothesized that pemetrexed has the potential to
induce apoptosis in malignant mesothelioma and lung cancer cells
via ROS accumulation.
Sirtuin 1 (SIRT1) is a nicotinamide adenine
dinucleotide (NAD+)-dependent deacetylase that exerts
its biological effects by deacetylating histones and non-histone
proteins, and its substrates include many proto-oncogenes and tumor
suppressors (9). Previous studies
have reported that SIRT1 is overexpressed and activated in certain
types of human cancers, and that SIRT1 overexpression blocks
apoptosis and senescence, while promoting cell proliferation and
angiogenesis (10–12). Inhibition of SIRT1 was found to
induce growth arrest and apoptosis in several types of cancer cells
(13,14).
The aim of the present study was to examine the
effects of pemetrexed in malignant mesothelioma and lung cancer
cells and study the mechanisms by which it produces these effects.
This is the first study to report the involvement of ROS and SIRT1
in pemetrexed-induced apoptosis in malignant mesothelioma and NSCLC
cell lines.
Materials and methods
Materials
RPMI-1640, fetal bovine serum (FBS), and antibiotics
were obtained from Gibco-BRL Co. (Grand Island, NY, USA).
Pemetrexed was purchased from Toronto Research Chemicals, Inc.
(Toronto, Ontario, Canada).
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
(MTT), propidium iodide (PI), dimethyl sulfoxide (DMSO),
N-acetylcysteine (NAC) and resveratrol were purchased from
Sigma-Aldrich (St. Louis, MO, USA). JC-1, a lipophilic fluorescent
dye used to detect mitochondrial membrane depolarization, was
obtained from Molecular Probes Co. (Eugene, OR, USA). Primary
antibodies against the following targets, caspase-3, -8 and -9, and
poly(ADP-ribose) polymerase (PARP) were obtained from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). Antibodies against SIRT1,
voltage-dependent anion channel (VDAC) and glyceraldehyde
3-phosphate dehydrogenase (GAPDH) were purchased from Cell
Signaling Technology (Beverly, MA, USA). Antibodies against
cytochrome c were obtained from Pharmingen (San Diego, CA,
USA). Anti-rabbit IgG-conjugated horseradish peroxidase (HRP)
antibodies and enhanced chemiluminescence (ECL) kits were purchased
from Amersham Pharmacia Biotech (Buckinghamshire, UK).
Cell culture and viability test
The human mesothelioma cell line MSTO-211H was
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA), and the human lung adenocarcinoma cancer cell line A549
was obtained from the Korean Cell Line Bank (KCLB, Seoul, Korea).
These cell lines were grown in RPMI-1640 containing 100 U/ml
penicillin, 0.1 mg/ml streptomycin and 10% FBS. The cells were
incubated in a humidified atmosphere of 5% CO2 in air at
37°C and maintained in log phase growth. Cell viability was
determined by measuring the mitochondrial conversion of MTT to
formazan, which was measured spectrophotometrically. After cells
were treated with the specified study drugs, MTT was added to the
cell suspension for 4 h. After 3 washes with phosphate-buffered
saline (PBS; pH 7.4), the insoluble formazan product was dissolved
in DMSO. The optical density (OD) of each well was measured using a
microplate reader (Titertek Multiskan; Flow Laboratories, North
Ryde, New South Wales, Australia) at 590 nm. The OD resulting from
formazan production in the control cells was considered as
representing 100% cell viability, and all other measurements were
expressed as a percentage of the control cell value.
Annexin V assay for the assessment of
apoptosis
MSTO-211 and A549 cells undergoing early/late
apoptosis were analyzed by Annexin V-fluorescein isothiocyanate
(FITC) and PI staining. Cells in the log phase (2.5×105
cells) were seeded in 35-mm2 dishes. Cells were left
untreated or were incubated with the specified drugs for the
indicated times at 37°C. Both adherent and floating cells were
collected and analyzed by the Annexin V assay according to the
manufacturer’s instructions. Pelleted cells were briefly washed
with PBS and resuspended in Annexin binding buffer. Cells were then
incubated with Annexin V-FITC and PI for 15 min at room
temperature. After incubation, the stained cells were analyzed
using a fluorescence-activated cell sorting (FACS)Calibur system
equipped with CellQuest software (Becton-Dickinson, San Jose, CA,
USA). Cells with no drug treatment were used as controls.
Measurement of the mitochondrial membrane
potential (ΔΨm)
MSTO-211 and A549 cells were harvested at the
indicated treatment times, washed with PBS, and then stained with
10 μg/ml JC-1 at 37°C for 30 min. After a brief wash with
PBS, cells were immediately analyzed using a FACSCalibur system
equipped with CellQuest software. At low concentrations, JC-1
exists mainly in a monomeric form, emitting green fluorescence with
an emission maximum at ~530 nM, whereas at higher concentrations it
forms aggregates known as J-aggregates, which emit orange-red
fluorescence with an emission maximum at ~590 nM.
Measurement of ROS
To measure intracellular ROS, cells were incubated
with 10 μmol/l
5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate
(carboxy-H2DCFDA; Molecular Probes Co.) at 37°C for 30
min. Cells were washed, scraped gently, resuspended in PBS and kept
on ice for immediate analysis by FACSCalibur flow cytometry using
an argon laser (488 nm) for excitation (Becton-Dickinson). Green
fluorescence due to DCF trapped inside the cells was measured and
plotted on a log scale. Data were acquired and analyzed with the
CellQuest program (Becton-Dickinson).
Western blotting
Cells were harvested and lysed using
radioimmunoprecipitation assay buffer (50 mM Tris-Cl, pH 7.4, 1%
NP40, 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1
μg/ml each of aprotinin and leupeptin, and 1 mM
Na3VO4). After centrifugation at 12,000 × g
for 30 min, the supernatant was collected and the protein
concentration was determined by the Bradford method (Bio-Rad
Protein Assay; Bio-Rad, Hercules, CA, USA). Equal amounts of
protein were separated by 12% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis (SDS-PAGE) under reducing conditions and
transferred to nitrocellulose membranes. The membranes were blocked
with 5% skim milk in TBS-T (25 mM Tris, pH 7.6, 138 mM NaCl and
0.05% Tween-20) for 1 h and probed with primary antibodies (at
1:1,000–1:5,000). After a series of washes, the membranes were
further incubated with a secondary antibody (at 1:2,000–1:10,000)
conjugated with HRP. Detection of the immunoreactive signals was
carried out using an ECL detection system.
Preparation of cytosolic and
mitochondrial fractions
Cytosolic and mitochondrial fractions were prepared
as previously described (15) with
modifications. Cells were harvested, washed with ice-cold PBS, and
then incubated with 500 μM buffer A [250 mM sucrose, 20 mM
HEPES (pH 7.5), 10 mM KCl, 1.5 mM MgCl2, 1 mM EGTA, 1 mM
EDTA, 1 mM DTT, 1 mM PMSF and 10 μg/ml each of leupeptin,
aprotinin and pepstatin A] on ice for 30 min. Cells were then
disrupted by 20 passages through a 26-gauge needle and centrifuged
at 750 × g for 10 min. The supernatant was centrifuged at 10,000 ×
g for 25 min. After centrifugation, the cytosolic fraction was
frozen at 70°C. The pellet containing the mitochondria was washed
with ice-cold buffer A and then resuspended with cell lysis buffer.
The resuspended pellet was incubated on ice for 30 min and then
centrifuged at 10,000 × g for 25 min. The supernatant thus
collected represented the mitochondrial fraction of the cells.
Gene silencing
Transcriptional expression of SIRT1 was
specifically suppressed by the introduction of a 21-nucleotide
duplex small interfering RNA (siRNA) targeting the coding sequence
of SIRT1 mRNA (16). Cells
(105 cells/well) were plated in 6-well plates and
transiently transfected with 50 nM/well of SIRT1 siRNA (Cell
Signaling Technology) mixed with the X-tremeGENE siRNA transfection
reagent (Roche Applied Science, Penzberg, Germany) according to the
manufacturer’s instructions. Silencer Negative Control siRNA (Roche
Applied Science) was used as a negative control and introduced into
the cells using the same protocol.
Statistical analysis
Each experiment was performed at least 3 times, and
all values are expressed as the mean ± SD of triplicate samples.
The Student’s t-test was used to determine the statistical
significance of the results. Values of p<0.05 were considered to
indicate statistically significant results.
Results
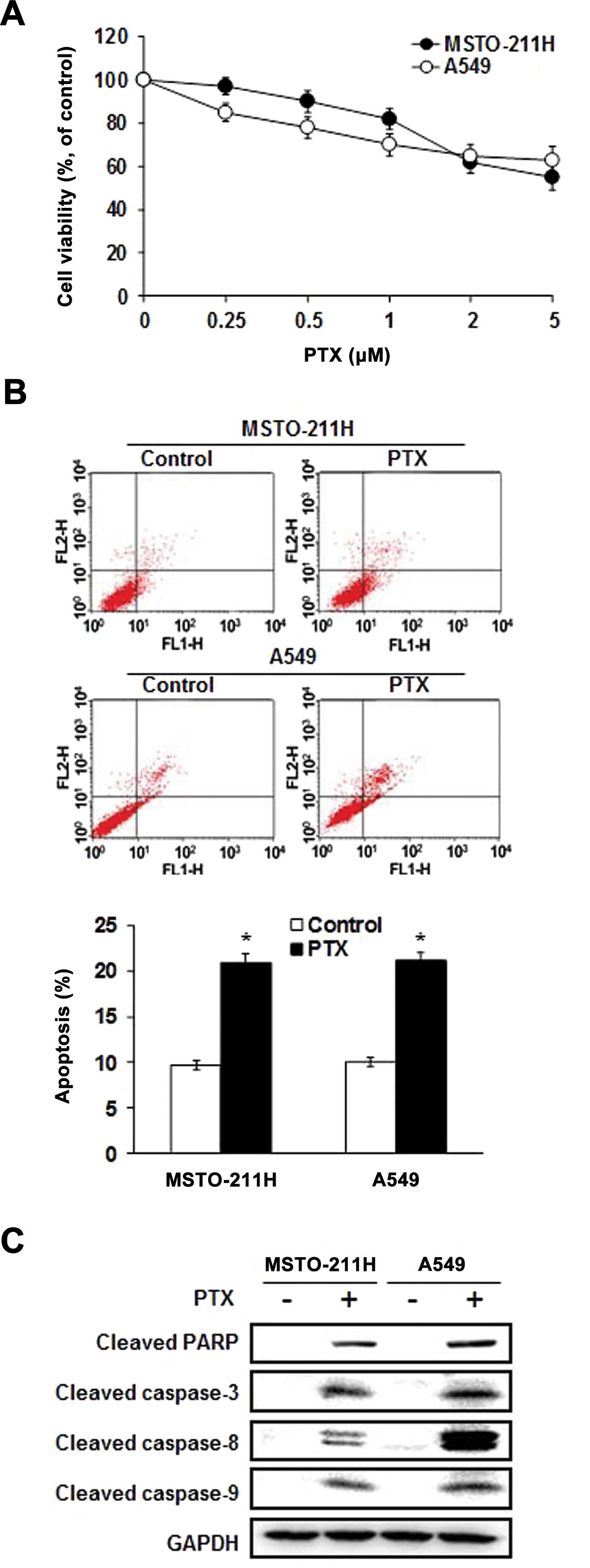
Effect of pemetrexed on the apoptotic
activity of malignant mesothelioma and lung cancer cells
MSTO-211 and A549 cells were treated with different
concentrations of pemetrexed, and the viability was measured by the
MTT assay. As shown in Fig. 1A,
pemetrexed significantly inhibited the growth of MSTO-211 and A549
cells in a dose-dependent manner. To determine whether the observed
growth inhibition was due to enhanced apoptosis, the proportion of
apoptotic cells was measured using Annexin V-PI staining. Annexin V
staining showed that pemetrexed significantly enhanced apoptosis in
the MSTO-211 and A549 cells (Fig.
1B). To further elucidate the mechanism through which
pemetrexed induced apoptosis, cell lysates were evaluated by
immunoblotting (Fig. 1C).
Pemetrexed enhanced the expression of the processed 85-kDa isoform
of PARP, which is known to play a major role in the process of
apoptosis. Moreover, pemetrexed led to a marked increase in the
expression of caspase-3, -8 and -9. These results indicate that
pemetrexed enhanced caspase-dependent apoptosis in the MSTO-211 and
A549 cells.
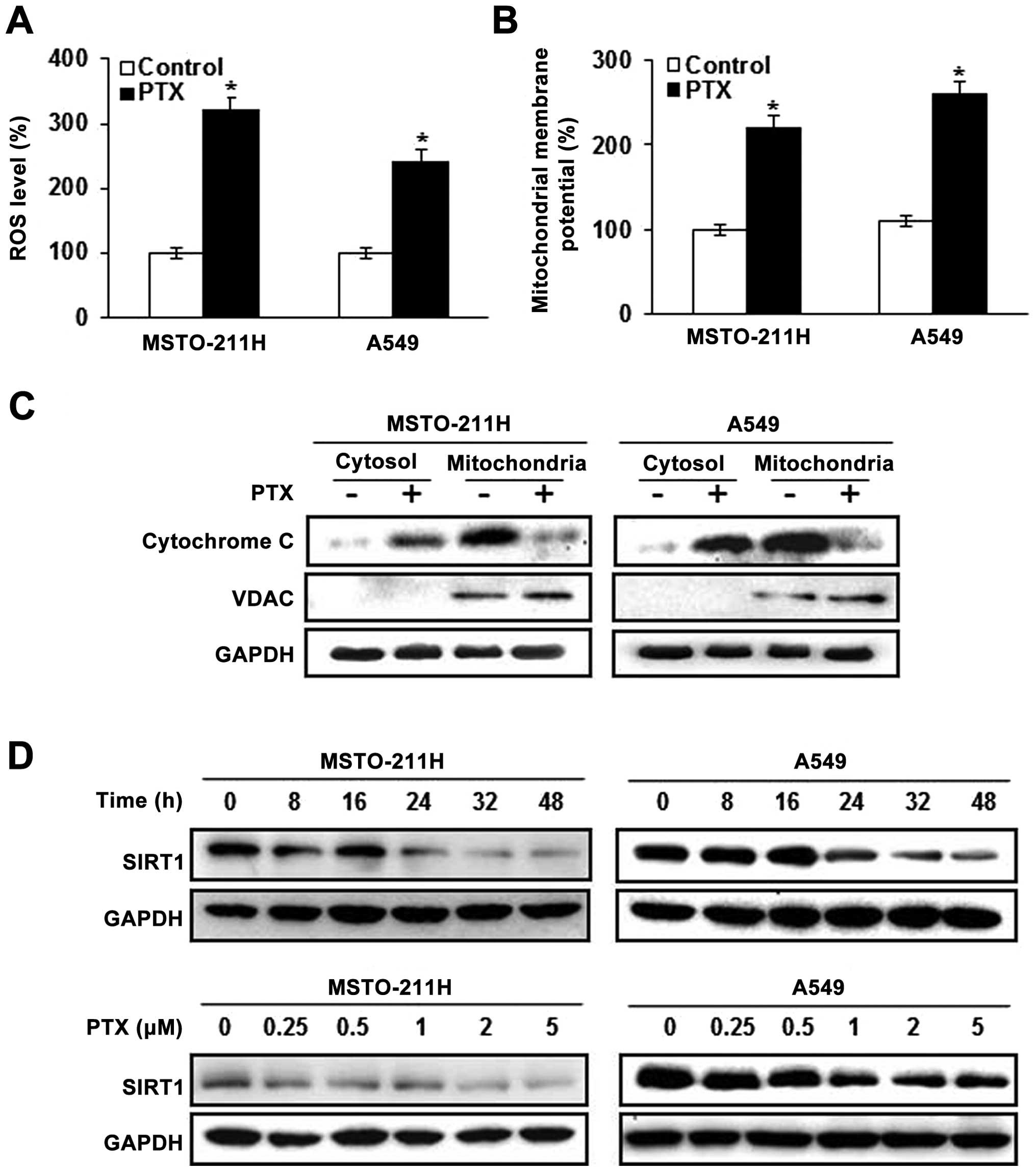
Pemetrexed induces intracellular ROS
production and downregulates SIRT1 in malignant mesothelioma and
lung cancer cells
We examined the role of signal transduction pathways
in modulating pemetrexed-induced apoptosis. Intracellular ROS
generation was assessed by flow cytometry using the total ROS
marker H2DCFDA. Elevated ROS levels in the MSTO-211 and
A549 cells were detectable 24 h after treatment with pemetrexed
(Fig. 2A). Next, markers of
mitochondrial dysfunction were evaluated in the cells treated with
pemetrexed, including mitochondrial membrane potential transition
(MPT) and cytosolic release of cytochrome c. JC-1 has been
used to detect apoptosis induced by mitochondrial depolarization.
As shown in Fig. 2B, JC-1 monomer
levels were increased in the MSTO-211 and A549 cells treated with
pemetrexed. Since the loss of ΔΨm resulted in cytochrome c
release into the cytosol, cytochrome c levels were evaluated
by western blotting in the mitochondrial and cytosolic fractions
(Fig. 2C). Pemetrexed was
associated with increased cytosolic cytochrome c and
decreased mitochondrial cytochrome c. We next examined the
effects of pemetrexed on the expression of SIRT1 in the MSTO-211
and A549 cells. SIRT1 expression was decreased in a time- and
dose-dependent manner after pemetrexed treatment as compared with
the control cells (Fig. 2D).
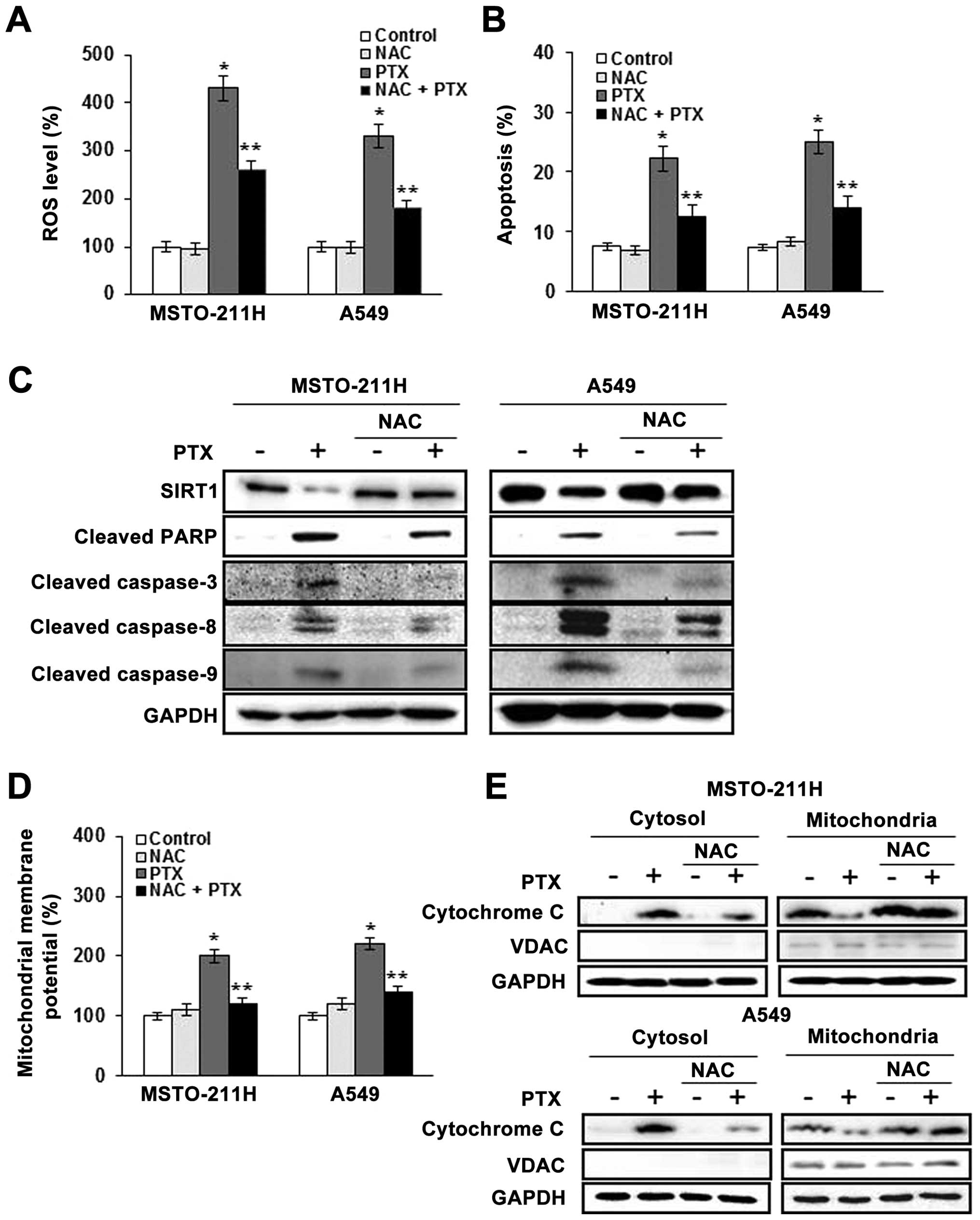
Pretreatment with NAC prevents apoptosis
induced by pemetrexed
We next tested the effect of the free radical
scavenger NAC in pemetrexed-treated MSTO-211 and A549 cells. Cells
were pretreated with 10 mM NAC, followed by the addition of
pemetrexed at 2 μM for 24 h. As shown in Fig. 3A, the enhancement of ROS generation
by pemetrexed was abolished by NAC. To determine whether elevated
ROS mediated the apoptosis induced by pemetrexed, the proportion of
apoptotic cells was determined by Annexin V-PI staining (Fig. 3B). The proportions of Annexin
V-positive MSTO-211 and A549 cells were increased by treatment with
pemetrexed, whereas pretreatment with NAC markedly reduced this
effect. Moreover, western blot analysis of MSTO-211 and A549 cell
lysates (Fig. 3C) showed that
pemetrexed decreased SIRT1 expression, enhanced the cleavage of
PARP, and enhanced the expression of caspase-3, -8 and -9, and
pretreatment with NAC blocked these effects.
We found that JC-1 monomers were increased in the
MSTO-211 and A549 cells treated with pemetrexed, whereas the loss
of ΔΨm was significantly reduced in the cells pretreated with NAC
(Fig. 3D). To provide further
evidence of mitochondrial dysfunction, cytosolic cytochrome
c was measured by western blotting in the mitochondrial and
cytosolic fractions (Fig. 3E).
Cytosolic cytochrome c was increased by pemetrexed, and
pretreatment with NAC reduced this effect. Together, these findings
indicate that ROS generation plays a primary role in apoptosis
induction by pemetrexed through mitochondrial dysfunction and this
effect involves, at least in part, the expression of SIRT1.
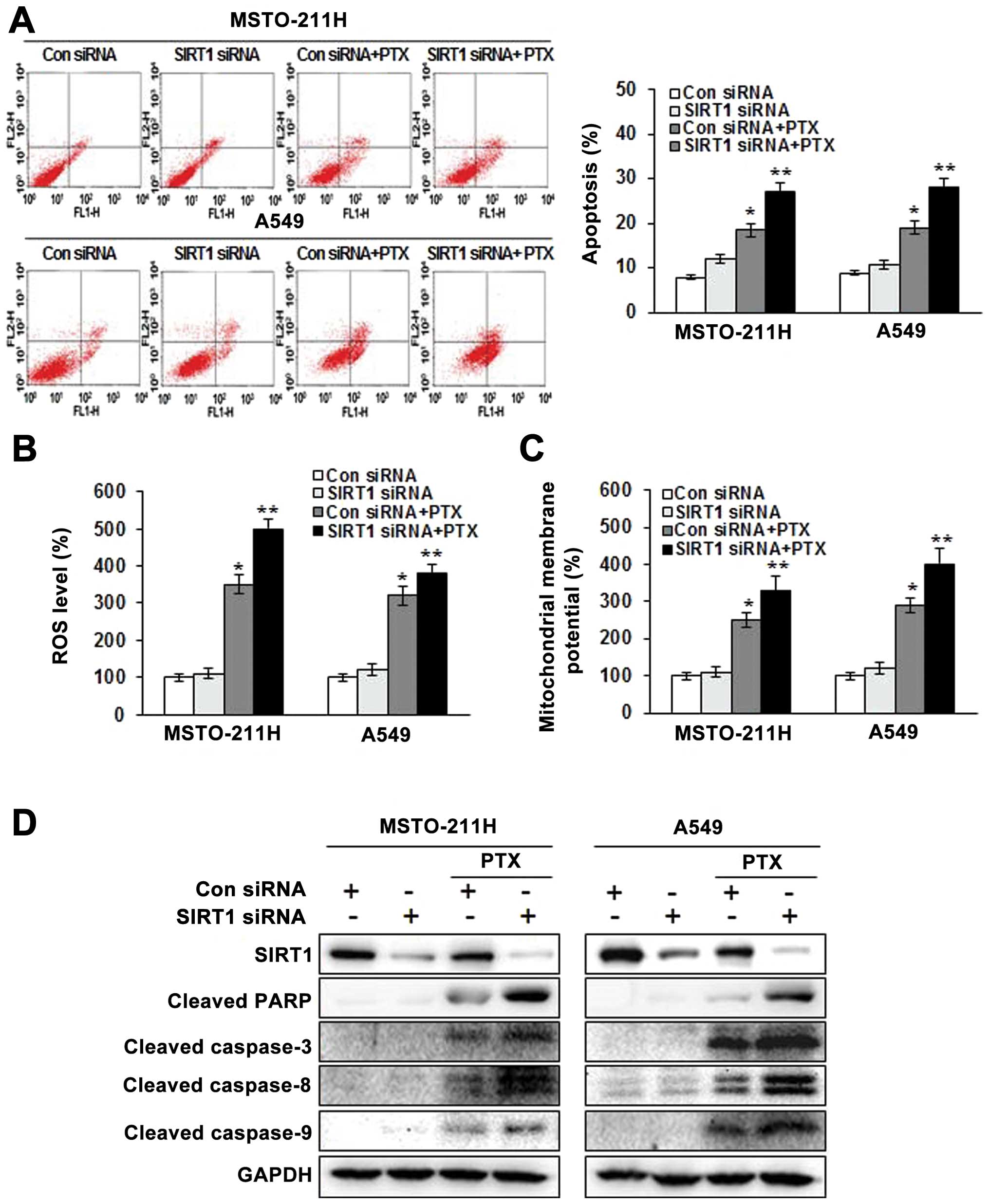
Downregulation of SIRT1 augments
apoptosis induced by pemetrexed
To determine the role of SIRT1 in pemetrexed-induced
apoptosis in MSTO-211 and A549 cells, we knocked down SIRT1 with
siRNA. The introduction of SIRT1 siRNA attenuated SIRT1 protein
expression 48 h after transfection. No reduction in SIRT1 protein
was observed in cells transfected with the scrambled siRNA, which
contained the same number of each nucleotide found in the SIRT1
siRNA. As shown in Fig. 4A, the
number of Annexin V-positive cells was increased in the SIRT1 siRNA
transfectants, and combined treatment with SIRT1 siRNA and
pemetrexed resulted in a greater apoptosis rate than combined
treatment with the control siRNA and pemetrexed.
We next examined whether SIRT1 siRNA affects ROS
generation. ROS generation in the MSTO-211 and A549 cells treated
with SIRT1 siRNA and pemetrexed was increased markedly compared to
that of cells treated with SIRT1 siRNA or pemetrexed alone
(Fig. 4B). JC-1 monomer levels were
consistently enhanced in the MSTO-211 and A549 cells treated with
SIRT1 siRNA and pemetrexed (Fig.
4C). Moreover, the combination of SIRT1 siRNA and pemetrexed
enhanced the expression of the processed 85-kDa isoform of PARP,
caspase-3, -8 and -9 (Fig. 4D).
Together, these data indicate that downregulation of SIRT1 enhanced
apoptosis induced by pemetrexed through mitochondrial dysfunction
that partially involves the generation of ROS.
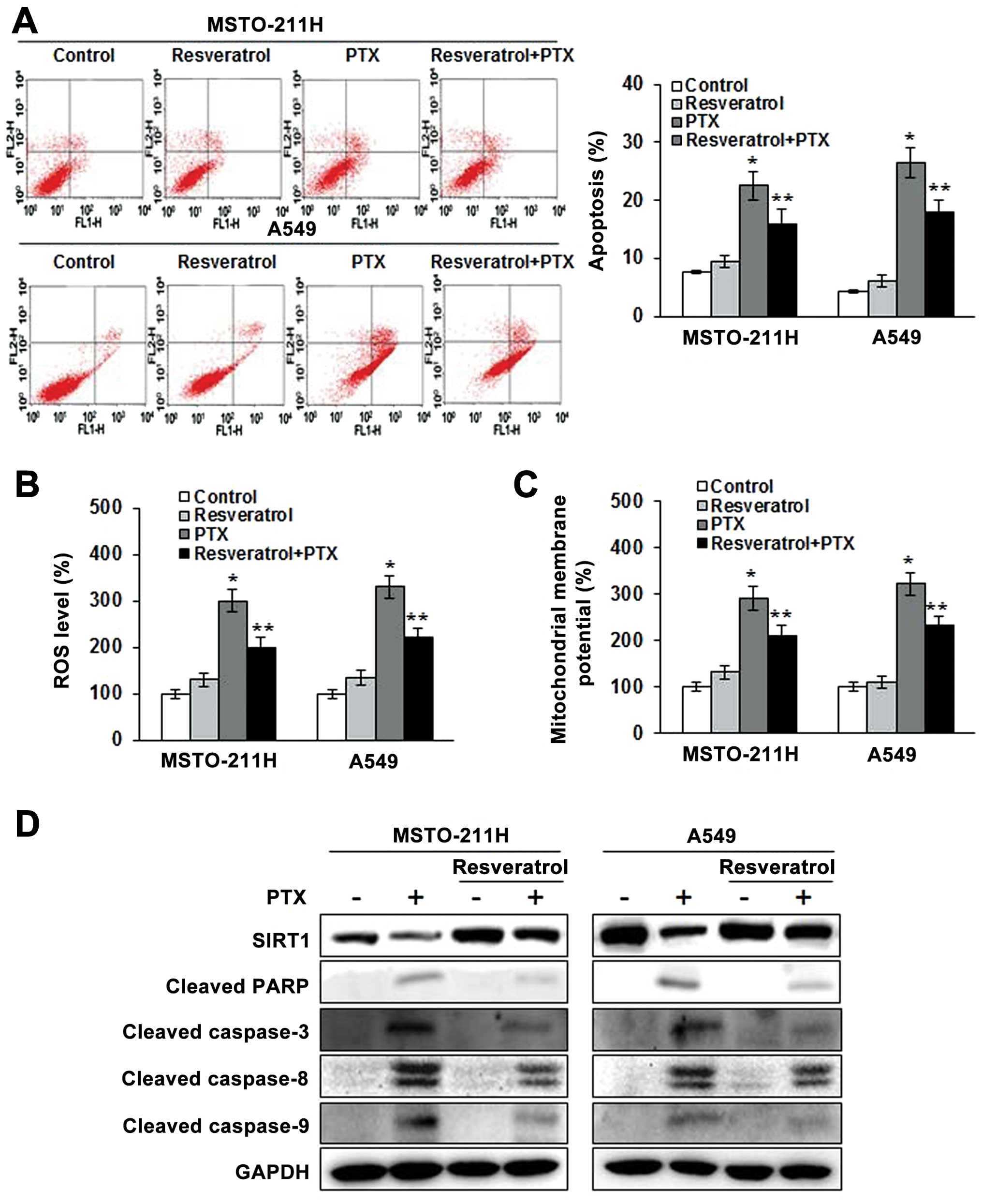
Resveratrol, an activator of SIRT1,
attenuates the apoptosis induced by pemetrexed
We next examined whether resveratrol, an activator
of SIRT1, could confer protection against the apoptosis induced by
pemetrexed. As shown in Fig. 5A,
pemetrexed resulted in 22.5 and 26.8%, respectively, more apoptotic
MSTO-211 and A549 cells than were observed in the untreated cells.
However, resveratrol decreased the number of apoptotic cells after
pemetrexed treatment by only 16.0 and 18.1%, respectively, in the
MSTO-211 and A549 cells. We next examined whether SIRT1
upregulation had a role in ROS generation by pemetrexed. The
results demonstrated that ROS generation in the MSTO-211 and A549
cells treated with resveratrol and pemetrexed was decreased
compared to the ROS generation in cells treated with pemetrexed
alone (Fig. 5B). Consistently, JC-1
monomer levels were decreased in the MSTO-211 and A549 cells
treated with resveratrol and pemetrexed (Fig. 5C) as compared to those treated with
pemetrexed alone. Western blot analyses showed that SIRT1
overexpression decreased the cleavage of PARP and the expression of
caspase-3, -8 and -9, even in the presence of pemetrexed (Fig. 5D). These results suggest that the
reduction of apoptosis by pemetrexed is due, at least in part, to
SIRT1 upregulation.
Discussion
In the present study, we examined the ability of
pemetrexed to induce apoptosis in MSTO-211 malignant mesothelioma
and A549 lung cancer cells, and explored the correlation between
ROS generation and SIRT1 expression. We demonstrated that
pemetrexed induced apoptosis by mitochondrial dysfunction in
malignant mesothelioma cells and A549 NSCLC cells. These effects
were mainly dependent on ROS accumulation and SIRT1
downregulation.
Previous studies have examined the effects of
pemetrexed on various human tumor cells, including malignant
mesothelioma and NSCLC cells. Pemetrexed as a maintenance therapy
after cisplatin-based doublet chemotherapy in NSCLC increases
survival (17). A better
understanding of the mechanisms underlying the antitumor effects of
pemetrexed is needed to optimize therapeutic targeting of the drug.
Pemetrexed has been shown to inhibit cell proliferation and induce
apoptosis in cancer cells (18).
Several studies have demonstrated that pemetrexed-induced apoptosis
is closely related to caspase-dependent and -independent cascades,
S-phase accumulation, deregulated activation of Akt signaling, and
ataxia telangiectasia mutated/p53-dependent and -independent
pathways (19–21). To date, however, the targets and
anticancer mechanisms of this compound remain largely unclear.
Mitochondria play an essential role in cellular
metabolism, ROS production, and regulation of cell proliferation
and death (22,23). A key feature of apoptosis via the
mitochondrial pathway is loss of ΔΨm, followed by the release of
cytochrome c. Cytochrome c is essential to the
formation of a caspase activation platform that is formed when it
activates APAF-1 oligomerization, and the latter then binds to and
activates caspase-9 (24–26). Our results demonstrate that the
release of cytochrome c into the cytosol activates
caspase-9, and subsequently leads to the activation of caspase-3.
Indeed, cleavage of PARP, a downstream target in this pathway,
occurs during pemetrexed-induced apoptosis of malignant
mesothelioma and lung cancer cells.
ROS generated in the mitochondria slow growth and
cause cell cycle arrest and apoptosis (27). Many studies suggest that ROS may act
as important regulators of apoptosis. ROS have been suggested to
play a unique role in apoptosis regulation since they can readily
produce mitochondrial dysfunction without diffusing a long distance
within the cytosol (28–30). A challenge for novel treatment
strategies is the fine-tuning of intracellular ROS signaling for
effective therapeutic gain. Buque et al (8) reported that elevations in
intracellular ROS and p53 are required for pemetrexed-induced
cytotoxicity in melanoma cells. Accordingly, we investigated the
possibility that ROS play a role in pemetrexed-induced apoptosis in
malignant mesothelioma and lung cancer cells. We demonstrated that
pemetrexed increased ROS levels. Moreover, pemetrexed-induced
apoptosis, mitochondrial dysfunction and caspase activation were
greatly reduced by pretreatment with NAC. These results suggest
that, in this model system, ROS generation plays a primary role in
the induction of apoptosis by pemetrexed.
SIRT1 promotes cytoprotection and apoptosis,
depending on the nature and severity of cellular stress. SIRT1
suppresses apoptosis via modulation of a number of cell survival
regulators, including p53, FOXO, HSF1, NF-κB and DNA damage
response proteins (31–33), whereas SIRT1 induces apoptosis
through a number of different routes, including Nrf2, NF-κB and
p53-mediated pathways (34–36). We demonstrated that downregulation
of SIRT1 by the introduction of SIRT1 siRNA into malignant
mesothelioma and NSCLC cells enhanced apoptosis induced by
pemetrexed. Therefore, we hypothesized that the upregulation of
SIRT1 may confer protection against apoptosis induced by
pemetrexed. As expected, the numbers of apoptotic malignant
mesothelioma and NSCLC cells induced by pemetrexed were clearly
decreased by resveratrol, an activator of SIRT1. This observation
indicated that SIRT1 confers resistance against pemetrexed-induced
apoptosis. In the present study, we provide for the first time a
molecular mechanism for the role of SIRT1 by demonstrating that
pemetrexed induces apoptosis by mitochondrial dysfunction.
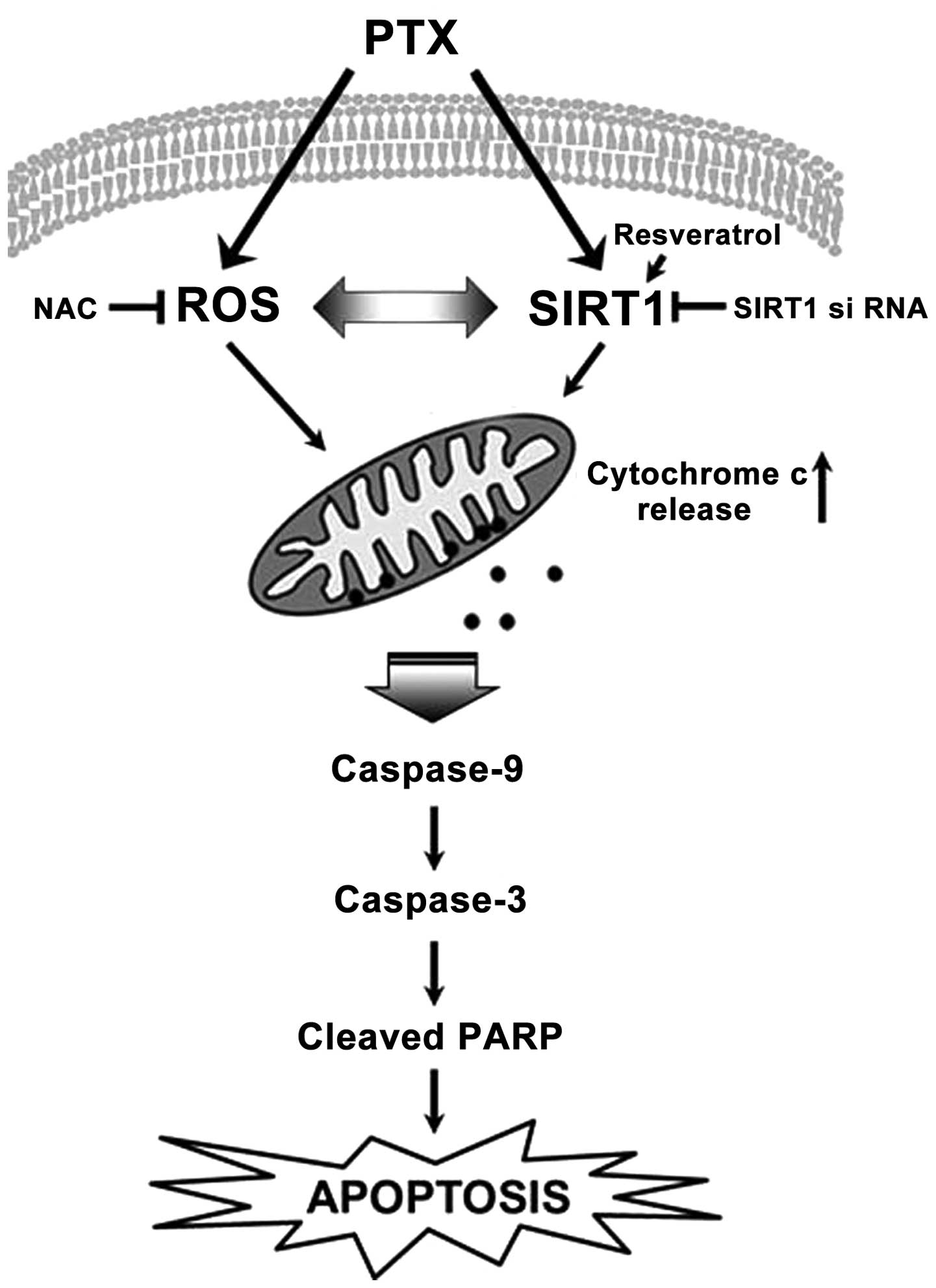
In the present study, we illustrated the molecular
mechanisms that mediate pemetrexed-induced apoptosis (Fig. 6). The present study confirmed that
pemetrexed induced apoptosis in malignant mesothelioma and lung
cancer cells through mitochondrial dysfunction by the regulation of
intracellular ROS generation and SIRT1. This conclusion has both
mechanistic and translational implications for future treatment
strategies for patients with malignant mesothelioma and NSCLC.
Acknowledgments
The present study was supported by a grant from the
Korean Health Technology R&D Project, Ministry of Health and
Welfare, Republic of Korea (A120152).
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hotta K, Matsuo K, Ueoka H, Kiura K,
Tabata M and Tanimoto M: Meta-analysis of randomized clinical
trials comparing cisplatin to carboplatin in patients with advanced
non-small-cell lung cancer. J Clin Oncol. 22:3852–3859. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Belli C, Fennell D, Giovannini M, Gaudino
G and Mutti L: Malignant pleural mesothelioma: Current treatments
and emerging drugs. Expert Opin Emerg Drugs. 14:423–437. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vogelzang NJ, Rusthoven JJ, Symanowski J,
et al: Phase III study of pemetrexed in combination with cisplatin
versus cisplatin alone in patients with malignant pleural
mesothelioma. J Clin Oncol. 21:2636–2644. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Scagliotti GV, Parikh P, von Pawel J, et
al: Phase III study comparing cisplatin plus gemcitabine with
cisplatin plus pemetrexed in chemotherapy-naive patients with
advanced-stage non-small-cell lung cancer. J Clin Oncol.
26:3543–3551. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hong J, Kyung SY, Lee SP, et al:
Pemetrexed versus gefitinib versus erlotinib in previously treated
patients with non-small cell lung cancer. Korean J Intern Med.
25:294–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chattopadhyay S, Moran RG and Goldman ID:
Pemetrexed: Biochemical and cellular pharmacology, mechanisms, and
clinical applications. Mol Cancer Ther. 6:404–417. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Buqué A, Muhialdin JS, Muñoz A, Calvo B,
Carrera S, Aresti U, Sancho A, Rubio I and López-Vivanco G:
Molecular mechanism implicated in pemetrexed-induced apoptosis in
human melanoma cells. Mol Cancer. 11:252012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smith BC, Hallows WC and Denu JM:
Mechanisms and molecular probes of sirtuins. Chem Biol.
15:1002–1013. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huffman DM, Grizzle WE, Bamman MM, Kim JS,
Eltoum IA, Elgavish A and Nagy TR: SIRT1 is significantly elevated
in mouse and human prostate cancer. Cancer Res. 67:6612–6618. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim JE, Chen J and Lou Z: DBC1 is a
negative regulator of SIRT1. Nature. 451:583–586. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen WY, Wang DH, Yen RC, Luo J, Gu W and
Baylin SB: Tumor suppressor HIC1 directly regulates SIRT1 to
modulate p53-dependent DNA-damage responses. Cell. 123:437–448.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Campisi J: Suppressing cancer: The
importance of being senescent. Science. 309:886–887. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ota H, Tokunaga E, Chang K, Hikasa M,
Iijima K, Eto M, Kozaki K, Akishita M, Ouchi Y and Kaneki M: Sirt1
inhibitor, Sirtinol, induces senescence-like growth arrest with
attenuated Ras-MAPK signaling in human cancer cells. Oncogene.
25:176–185. 2006.
|
|
15
|
Wolf CM and Eastman A: The temporal
relationship between protein phosphatase, mitochondrial cytochrome
c release, and caspase activation in apoptosis. Exp Cell Res.
247:505–513. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Elbashir SM, Harborth J, Lendeckel W,
Yalcin A, Weber K and Tuschl T: Duplexes of 21-nucleotide RNAs
mediate RNA interference in cultured mammalian cells. Nature.
411:494–498. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ciuleanu T, Brodowicz T, Zielinski C, et
al: Maintenance pemetrexed plus best supportive care versus placebo
plus best supportive care for non-small-cell lung cancer: A
randomised, double-blind, phase 3 study. Lancet. 374:1432–1440.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ramirez JM, Ocio EM, San Miguel JF and
Pandiella A: Pemetrexed acts as an antimyeloma agent by provoking
cell cycle blockade and apoptosis. Leukemia. 21:797–804.
2007.PubMed/NCBI
|
|
19
|
Tonkinson JL, Worzalla JF, Teng CH and
Mendelsohn LG: Cell cycle modulation by a multitargeted antifolate,
LY231514, increases the cytotoxicity and antitumor activity of
gemcitabine in HT29 colon carcinoma. Cancer Res. 59:3671–3676.
1999.PubMed/NCBI
|
|
20
|
Chen KC, Yang TY, Wu CC, Cheng CC, Hsu SL,
Hung HW, Chen JW and Chang GC: Pemetrexed induces S-phase arrest
and apoptosis via a deregulated activation of Akt signaling
pathway. PLoS One. 9:e978882014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang TY, Chang GC, Chen KC, Hung HW, Hsu
KH, Wu CH, Sheu GT and Hsu SL: Pemetrexed induces both intrinsic
and extrinsic apoptosis through ataxia telangiectasia
mutated/p53-dependent and -independent signaling pathways. Mol
Carcinog. 52:183–194. 2013. View
Article : Google Scholar
|
|
22
|
Copeland WC, Wachsman JT, Johnson FM and
Penta JS: Mitochondrial DNA alterations in cancer. Cancer Invest.
20:557–569. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim HR, Yang SH and Jeong ET: Combination
treatment with arsenic trioxide and sulindac induces apoptosis of
NCI-H157 human lung carcinoma cells via ROS generation with
mitochondrial dysfunction. Tuberc Respir Dis. 59:30–38. 2005.
|
|
24
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li LY, Luo X and Wang X: Endonuclease G is
an apoptotic DNase when released from mitochondria. Nature.
412:95–99. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tournier C, Hess P, Yang DD, Xu J, Turner
TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA and Davis RJ:
Requirement of JNK for stress-induced activation of the cytochrome
c-mediated death pathway. Science. 288:870–874. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Burdon RH: Control of cell proliferation
by reactive oxygen species. Biochem Soc Trans. 24:1028–1032.
1996.PubMed/NCBI
|
|
28
|
Balaban RS, Nemoto S and Finkel T:
Mitochondria, oxidants, and aging. Cell. 120:483–495. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ozben T: Oxidative stress and apoptosis:
Impact on cancer therapy. J Pharm Sci. 96:2181–2196. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tan S, Sagara Y, Liu Y, Maher P and
Schubert D: The regulation of reactive oxygen species production
during programmed cell death. J Cell Biol. 141:1423–1432. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim EJ, Kho JH, Kang MR and Um SJ: Active
regulator of SIRT1 cooperates with SIRT1 and facilitates
suppression of p53 activity. Mol Cell. 28:277–290. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brunet A, Sweeney LB, Sturgill JF, et al:
Stress-dependent regulation of FOXO transcription factors by the
SIRT1 deacetylase. Science. 303:2011–2015. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Westerheide SD, Anckar J, Stevens SM Jr,
Sistonen L and Morimoto RI: Stress-inducible regulation of heat
shock factor 1 by the deacetylase SIRT1. Science. 323:1063–1066.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Raynes R, Brunquell J and Westerheide SD:
Stress inducibility of SIRT1 and its role in cytoprotection and
cancer. Genes Cancer. 4:172–182. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kawai Y, Garduño L, Theodore M, Yang J and
Arinze IJ: Acetylation-deacetylation of the transcription factor
Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its
transcriptional activity and nucleocytoplasmic localization. J Biol
Chem. 286:7629–7640. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yeung F, Hoberg JE, Ramsey CS, Keller MD,
Jones DR, Frye RA and Mayo MW: Modulation of NF-kappaB-dependent
transcription and cell survival by the SIRT1 deacetylase. EMBO J.
23:2369–2380. 2004. View Article : Google Scholar : PubMed/NCBI
|