Introduction
Resveratrol
(trans-3,4′,5-trihydroxy-trans-stilbene) is a
naturally-occurring polyphenolic stilbene found in the skin of red
grapes, various other fruits and the roots of the Polygonum
cuspidatum and is an important constituent of the Chinese and
Japanese folk medicine (1). When
used at low concentrations, resveratrol has cytoprotective
activity, which is mostly attributed to its antioxidant properties
(2). At higher concentrations,
resveratrol possesses anticancer activity by interfering with
various cellular events associated with initiation, promotion and
progression of multistage carcinogenesis (2). This anticancer property was observed
in both in vitro and in vivo models of the tumor cell
lines (3,4). In addition, resveratrol has been
reported to have diverse effects on cellular signaling pathways,
such as downregulation of angiogenesis associated genes (3), activation of apoptotic mechanisms
(5) and induction of cell cycle
arrest (6). Furthermore,
resveratrol was found to sensitize resistant tumor cell lines to a
variety of chemotherapeutic agents, such as paclitaxel (7), thalidomide and bortezomib (8).
Single-strand DNA breaks of the cleavable complex
are reversible, yet these lesions may be converted into
irreversible double-strand breaks during the DNA synthesis,
following collision with the replication complexes. Double-strand
breaks, which are recognized as the lethal lesions, activate the
DNA damage response signal pathways. The severity and persistence
of such damage determines the eventual outcome of a cell. In
response to the DNA-damaging agents, the cells activate the cell
cycle checkpoints and a complex response network involving the DNA
damage sensors, signal transducers and effector pathways, which
influence the cellular decision among the cell cycle arrest, the
DNA repair, the apoptosis induction or other cell death modalities
(9,10). Ataxia telangiectasia mutated (ATM)
and/or ataxia telangiectasia-Rad3-related (ATR) kinases are key
players in the response to DNA damage. These kinases are dependent
on DNA damage and phosphorylate downstream effector kinases, such
as checkpoint kinases 1 (Chk1 and Chk2). Thus, these kinases relay
and amplify the damage signal and effector proteins that control
the cell cycle progression, chromatin restructuring and DNA
repair.
In view of the great chemotherapeutic potential of
resveratrol, many analogues of resveratrol have been synthesized by
swapping the benzene rings to the aromatic heterocycles with the
aim of generating novel resveratrol analogs with improved
anticancer activities. Previously, we have reported a study in
which a class of resveratrol analogues, styrylquinazolines, was
found to inhibit COX-2-induced prostaglandin E2
(PGE2) production (11).
In this follow-up study, we examine the antiproliferative effects
and the underlying molecular mechanism of styrylquinazolines in
HeLa human cervical cancer cells.
Materials and methods
Chemical and reagents
Styrylquinazoline derivatives (Table I) were prepared as previously
described (11). RPMI-1640 medium,
fetal bovine serum (FBS), penicillin and streptomycin were obtained
from Life Technologies Inc. (Grand Island, NY, USA). Cyclin B1,
p53, p21 and β-actin monoclonal antibodies were purchased from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Cdk1,
phospho-Cdk1 (Tyr15), cell division cycle 25C (Cdc25C),
phospho-Cdc25C (Ser216), Chk1, phospho-Chk1 (Ser345), Chk2,
phospho-Chk2 (Thr68), ATM, phospho-ATM (Ser1981), ATR, phospho-ATR
(Ser428) and phospho-p53 (Ser15) antibodies were purchased from
Cell Signaling Technology, Inc. (Beverly, MA, USA). Caffeine and
lactacystin were purchased from Sigma (St. Louis, Mo, USA).
 | Table ICytotoxic activity of resveratrol and
its styrylquinazoline analogues on the proliferation of various
cancer cells in vitro. |
Table I
Cytotoxic activity of resveratrol and
its styrylquinazoline analogues on the proliferation of various
cancer cells in vitro.
 |
|---|
| R1 | R2 | R3 |
IC50(μM)a
|
|---|
| HeLa | HL-60 | A549 |
|---|
| H | OH | OH | 49.61±3.01 | 21.54±1.72 | 36.88±3.01 |
| OH | H | OH | 29.12±1.72 | 14.39±2.69 | 22.57±1.89 |
| OH | OH | OH | 21.44±2.25 | 13.52±2.16 | 22.68±3.52 |
|
OCH3 | H | OH | 95.04±6.97 | 53.68±0.86 | 82.44±5.82 |
|
OCH3 | OH | OH | 82.68±5.79 | 64.18±3.72 | 91.44±7.65 |
|
OCH3 | OH |
OCH3 | 53.25±4.10 | 30.65±2.15 | 49.62±3.65 |
| H |
OCOCH3 |
OCOCH3 | 66.87±4.55 | 36.18±6.41 | 63.09±6.43 |
|
OCH3 |
OCOCH3 |
OCOCH3 | 36.55±3.84 | 24.32±4.92 | 35.46±4.90 |
|
OCOCH3 |
OCOCH3 |
OCOCH3 | 10.25±1.86 | 10.59±1.54 | 13.75±1.88 |
| Resveratrol | | | 136.57±7.32 | 66.90±4.85 | 83.32±7.52 |
Cell culture and sample treatment
HeLa human cervical carcinoma, HL-60 human
promyelocytic leukemia and A549 human lung adenocarcinoma cell
lines were obtained from the Korean Cell Line Bank (Seoul, Korea).
The cells were grown at 37°C in RPMI-1640 medium supplemented with
10% FBS, penicillin (100 U/ml) and streptomycin sulfate (100
μg/ml) in a humidified atmosphere of 5% CO2. The
cells were incubated with 8-AEDQ (8 μM) for various times
(5, 10, 15, 20 or 25 h).
Cytotoxicity test
Cytotoxicity was assessed by an MTT assay. Briefly,
the cells (5×104 cells/ml) were seeded in each well
containing 100 μl of the RPMI medium supplemented with 10%
FBS in 96-well plates. After 24 h, various concentrations of 8-ADEQ
were added. After 48 h, 50 μl of MTT [5 mg/ml stock solution
in phosphate-buffered saline (PBS)] was added, and the plates were
incubated for an additional 4 h. The medium was discarded and the
formazan blue, which was formed in the cells, was dissolved with
100 μl dimethyl sulfoxide (DMSO). The optical density was
measured at 540 nm.
Trypan blue assay
The in vitro growth inhibition effect of
8-ADEQ on the HeLa cells was determined by a trypan blue dye
exclusion. The reduction in the viable cell number was assessed for
each 3 days. The HeLa cells were grown at 37°C in RPMI medium
supplemented with 10% FBS, penicillin (100 U/ml) and streptomycin
sulfate (100 μg/ml) in a humidified atmosphere of 5%
CO2. The cells were seeded at a concentration of
1×105 cells/ml and were maintained for logarithmic
growth and incubated for 1–3 days with 8-ADEQ at various
concentrations. 8-ADEQ dissolved in DMSO was added to the medium in
serial dilution (the final DMSO concentration in all the assays did
not exceed 0.1%). The cells were loaded on a hemocytometer and the
viable cell number was determined based on the exclusion of the
trypan blue dye.
Flow cytometric cell cycle analysis
The HeLa cells were incubated until 70–80%
confluency. After treatment with or without 8-ADEQ, the cells were
harvested and fixed in 70% ethanol for 1 h at −20°C. After being
washed with PBS, the cells were labeled with propidium iodide (PI)
(50 μg/ml) in the presence of RNase A (100 μg/ml) and
were incubated at room temperature in the dark for 30 min and were
analyzed using the fluorescence-activated cell sorting (FACS)
cater-plus flow cytometry (Becton-Dickinson, Co., Heidelberg,
Germany).
Protein extraction and western blot
analysis
The HeLa cells were collected by centrifugation and
washed once with PBS. The washed cell pellets were resuspended in
extraction lysis buffer (50 mM HEPES pH 7.0, 250 mM NaCl, 5 mM
EDTA, 0.1% Nonidet P-40, 1 mM PMSF, 0.5 m MDTT, 5 mM Na fluoride
and 0.5 mM Na orthovanadate) containing 5 μg/ml each of
leupeptin and aprotinin then incubated for 20 min at 4°C. The cell
debris was removed by microcentrifugation, followed by quick
freezing of the supernatants. The protein concentration was
determined using the Bio-Rad protein assay reagent according to the
manufacturer’s instruction. The cellular protein from the treated
and untreated cell extracts was electroblotted onto a PVDF membrane
following separation on a 10–12% SDS-polyacrylamide gel
electrophoresis. The immunoblot was incubated overnight with a
blocking solution (5% skim milk) at 4°C, followed by incubation
overnight with a primary antibody. The blots were washed four times
with Tween-20/Tris-buffered saline (T/TBS) and incubated with a
1:1,000 dilution of horseradish peroxidase-conjugated secondary
antibody for 2 h at room temperature. The blots were washed again
three times with T/TBS and then developed by enhanced
chemiluminescence (GE Healthcare, Milwaukee, WI, USA).
Small interfering RNA transfection
RNA interference of ATM, ATR and p53 was performed
using 21-bp (including a 2-deoxynucleotide overhang) siRNA duplexes
purchased from Invitrogen (Carlsbad, CA, USA). The coding strand
for ATM, ATR and p53 siRNA were: 5′-GCG CCU GAU UCG AGA UCC UTT-3′;
5′-CCU CCG UGA UGU UGC UUG ATT-3′; and 5′-UCA CCU CAU CCA UUG CUU
GGG ACG G-3′, respectively. For the transfection, the HeLa cells
were seeded in 6-well plates and were transfected at 30% confluence
with 100 nM siRNA duplexes using Lipofectamine™ RNAiMAX
(Invitrogen) according to the manufacturer’s recommendations. The
cells were transfected with a control non-specific siRNA duplex
(5′-ACU CUA UCU GCA CGC UGA CUU-3′; Invitrogen) are were used as
controls for direct comparison. After 48 h of transfection, the
cells were treated with or without 8-ADEQ for 25 h. Both floating
and adherent cells were collected, washed with PBS and processed
for analysis of the cell cycle distribution.
Statistical analysis
Results are expressed as the mean ± SDs of
triplicate experiments. Statistically significant values were
compared using the ANOVA and the Dunnett’s post-hoc test,
and P-values of <0.05 were considered to indicate a
statistically significant result.
Results
Inhibition of cancer cell growth by
styrylquinazoline derivatives
To assess the inhibitory effects of resveratrol or
styrylquinazoline derivatives on cancer cell viability, we first
determined the cytotoxicities of these compounds using the MTT
assay. As shown in Table I, the
styrylquinazoline derivatives showed varying degrees of
cytotoxicity, as assessed using their IC50 values.
8-ADEQ showed dose-dependent cytotoxic effects in three cancer
cells and was the most potent in comparison to other
styrylquinazolines and to resveratrol. Among the tested cancer cell
lines, human cervical carcinoma HeLa cells were the most sensitive
to 8-ADEQ. The cell numbers were counted using trypan blue dye
exclusion assays following treatment with 8-ADEQ at various
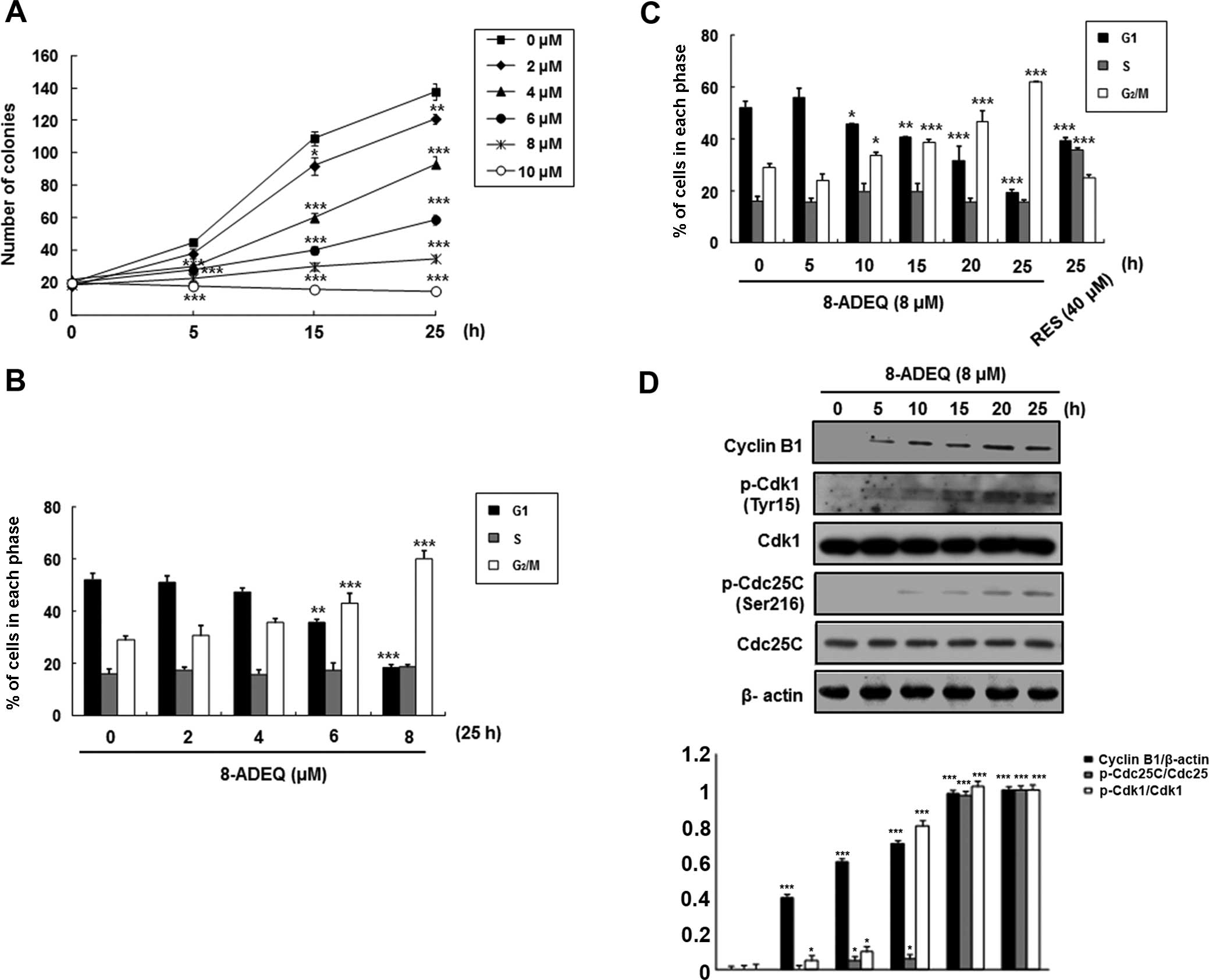
concentrations and times. Exponentially growing HeLa cultures
exhibited growth inhibition when treated with 8-ADEQ at
concentrations of 2–10 μM over the experimental period
(Fig. 1A). At 8 μM, 8-ADEQ
had a cytostatic effect, yet at 10 μM it had a cytocidal
effect. Therefore, this concentration was used throughout the
present study. Further experiments were performed using HeLa cells
to evaluate the effect of 8-ADEQ on the cell cycle arrest and to
identify the mechanism involved.
Effects of 8-ADEQ on the cell cycle
arrest and on the expression of the cell cycle regulatory proteins
in HeLa cells
To investigate whether 8-ADEQ affects cell cycle
regulation, the HeLa cells were cultured with 8-ADEQ at
concentrations of 8 μM for 25 h and then the DNA contents
were analyzed by DNA flow cytometric analysis. As shown in Fig. 1B and C, 8-ADEQ induced the cell
cycle arrest at the G2/M phase in a concentration- and
time-dependent manner and a concomitant decrease of the cells in
the G1 phase. It is well known that the complex of
cyclin-dependent kinase 1 (Cdk1; also known as Cdc2) and cyclin B1
is important for cell cycle entry into mitosis in most organisms
(12,13). We first determined whether 8-ADEQ
affected the expression of these G2/M-related proteins
in the cells treated with 8-ADEQ (8 μM) for indicated times.
Under this condition, it was found that 8-ADEQ upregulated cyclin
B1 protein level but did not affect the Cdk1 levels (Fig. 1D). Although phosphorylation of Cdk1
at Thr161 is essential for activation of the Cdk1-cyclin B1 kinase
complex, reversible phosphorylations at Thr14 and Tyr15 of Cdk1
suppress its kinase activity (14).
Dephosphorylation of Thr14 and Tyr15 of Cdk1, and hence activation
of the Cdk1-cyclin B1 kinase complex, is catalyzed by dual
specificity phosphatases Cdc25B and Cdc25C, and this reaction is
believed to be a rate-limiting step for entry into mitosis
(15). To gain insights into the
mechanism of cell cycle arrest upon treatment with 8-ADEQ,
phosphorylations of Cdk1 and Cdc25C proteins were compared by
immunoblotting, using lysates from control and 8-ADEQ (8
μM)-treated cells for 25 h. Under this condition, 8-ADEQ
substantially increased phosphorylation of Cdk1 (Tyr15) and Cdc25C
(Ser216) (Fig. 1D). Collectively,
these results indicate that the 8-ADEQ-mediated G2/M
phase cell cycle arrest is associated with the increased cyclin B1
protein levels and phosphorylations of Cdk1 and Cdc25C.
Effects of 8-ADEQ on the phosphorylation
of Chk1/Chk2 and p53 that regulate G2/M transition
Considering that Chk1 and Chk2 have been implicated
in the phosphorylation (Ser216) of Cdc25C (16) and are activated by phosphorylation
on Ser345 and Thr68, respectively (17), we evaluated the degree of
phosphorylation of these intermediaries of the DNA damage
checkpoints on western blot analysis using whole extracts from the
control or the 8-ADEQ-treated cells. In 8-ADEQ-treated HeLa cells,
p-Chk1 (Ser345) or p-Chk2 (Thr68) showed an increase over the
control, which was evident as early as 5 h after the 8-ADEQ
treatment and increased for the duration of the experiment
(Fig. 2A). However, the levels of
Chk1 or Chk2 protein were not affected by the 8-ADEQ treatment.
Since these checkpoint kinases act upstream of p53 (18), we examined the phosphorylation
status of p53 by western blot analysis. The phophoryation of p53
(Ser15) had increased after the 8-ADEQ treatment and was then
sustained until 25 h (Fig. 2A),
though 8-ADEQ did not influence the expression level of p53. The
tumor-suppressor p53 regulates the DNA damage-induced cell cycle
arrest (19) by directly
stimulating the expression of p21CIP1/WAF1, an inhibitor
of cyclin-dependent kinases (Cdks). For this reason, the HeLa cells
were treated with 8-ADEQ (8 μM) at different time intervals,
and then the expression of p21CIP1/WAF1 was determined
by western blot analysis. Accordingly, the p21CIP1/WAF1
expression had increased in a time-dependent manner as well,
resulting from transcriptional regulation by p53 (Fig. 2A). The p21CIP1/WAF1
protein was induced by both p53-dependent and p53-independent
mechanisms following the anticancer agent treatment (20). As shown in Fig. 2A, the increased
p21CIP1/WAF1 protein level after the 8-ADEQ treatment
did not parallel the expression level of p53 in HeLa cells, a cell
lineage with the wild-type p53 gene. Therefore, in order to
determine whether p53 is involved in the 8-ADEQ-induced
G2/M arrest, the p53 genes were knocked down by
p53-specific siRNAs. As shown in Fig.
2B, p53 siRNA significantly reduced the expression levels of
p53 protein in the 8-ADEQ-treated HeLa cells. Fig. 2C shows the p53 siRNA-transfected and
control siRNA cells treated with 8-ADEQ, and their cell cycle
distribution was determined after 25 h. Treatment of the control
siRNA-transfected cells with 8-ADEQ resulted in a ~3-fold increase
in the G2/M phase cells from 19.5 to 55%. However, the
8-ADEQ-induced G2/M arrest response was significantly
decreased in cells transfected with p53 siRNA from 55 to 32.5%.
These results indicated that p53 is involved in the 8-ADEQ-induced
G2/M arrest.
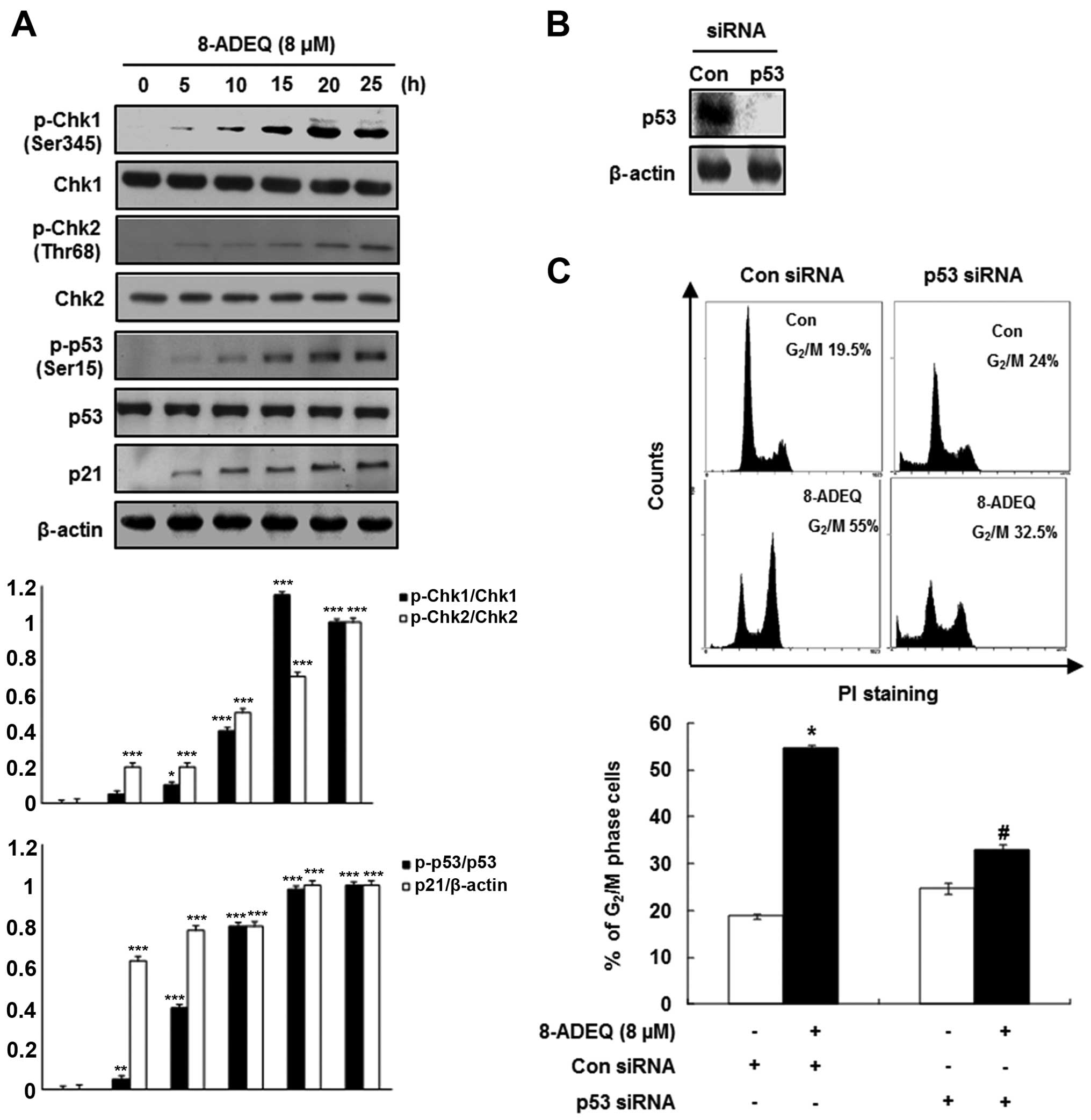 | Figure 2Effects of 8-ADEQ on the
phosphorylation of Chk1/2 and p53 and p21CIP1/WAF1
expression and G2/M cell cycle in HeLa cells. (A) The
HeLa cells were treated with or without 8-ADEQ (8 μM) for
the indicated time and were then harvested. The expression levels
of p-Chk1, Chk1, p-Chk2, Chk2, p-p53, p53 and p21 were examined
using western blot analysis, as described in Materials and methods.
β-actin was used as an internal control. The experiments were
repeated three times and similar results were obtained. A
densitometric analysis was performed using Bio-Rad Quantity
one® software. (B) The HeLa cells were transfected with
either p53 siRNA or non-specific control siRNA for 24 h. The
expression levels of p53 were examined using western blot analysis.
β-actin was used as an internal control. (C) The HeLa cells were
transfected with either p53 siRNA or non-specific control siRNA for
24 h and were then treated with or without 8-ADEQ (8 μM) for
25 h. The cell cycle distribution was stained with PI and detected
by flow cytometry as described in Materials and methods. Data are
presented as the means ± SD of three independent experiments.
*P<0.05, **P<0.01,
***P<0.001 vs. the control group,
#P<0.05 vs. the 8-ADEQ-treated group. PI, propidium
iodide. |
Effect of 8-ADEQ on DNA damage response
in HeLa cells
In order to determine whether growth inhibition via
cell cycle arrest by 8-ADEQ is associated with the DNA damage, we
performed western blot analysis for H2A.X, ATM and ATR
phosphorylations in the 8-ADEQ-treated HeLa cells. The histone H2A
variant H2A.X specifically controls the recruitment of the DNA
repair proteins to the sites of the DNA damage and this activity is
phosphorylation dependent (21). As
shown in Fig. 3A, immunoblotting of
HeLa lysates showed a clear and steady increase in phosphorylation
(Ser129) of H2A.X (γ-H2A.X) level from 5 to 25 h after the 8-ADEQ
treatment. Based on the results of γ-H2A.X, we expected that the
8-ADEQ treatment causes the DNA double-strand breaks. We
hypothesized that the 8-ADEQ-induced DNA damage is sensed by
members of the phosphoinositide 3-kinase-related kinases (ATM and
ATR) for the early signal transmission through the cell cycle
checkpoint (22). As shown in
Fig. 3A, 8-ADEQ treatment (8
μM) affected phosphorylation of ATM/ATR, which was increased
at 5 h and subsequently declined, yet no effect on the expression
of total ATM/ATR. To further confirm the 8-ADEQ-mediated cell cycle
arrest, we performed inhibitor or siRNA treatments to explore
critical molecular changes of the DNA damage pathway. Pretreatment
of the HeLa cells with 10 mM caffeine, a known modulator of the DNA
damage checkpoint, abrogated 8-ADEQ-induced G2/M phase
accumulation from 49.7 to 25.5% (Fig.
3B). To confirm whether ATM and ATR are the proximal kinases
responsible for the 8-ADEQ-mediated G2/M phase arrest,
we used siRNA technology to suppress the ATM/ATR protein
expression. Transfection of ATM and ATR siRNA revealed that ATM and
ATR expressions were almost completely suppressed (Fig. 3C). Subsequently, ATM/ATR-siRNA
transfected and control cells were treated with 8-ADEQ (8
μM), and their cell cycle distribution was assessed after 25
h. Treatment of control siRNA-transfected cells with 8-ADEQ
resulted in a 2.3-fold increase of G2/M phase cells from
20.5 to 48% (Fig. 3D). The
8-ADEQ-induced G2/M phase arrest was partially but
significantly attenuated in the cells transfected with ATM/ATR
siRNA (ATM; from 48 to 35%, ATR; from 48 to 36.5%) (Fig. 3D). These results indicate that both
ATM and ATR play a pivotal role in the 8-ADEQ-induced
G2/M phase arrest.
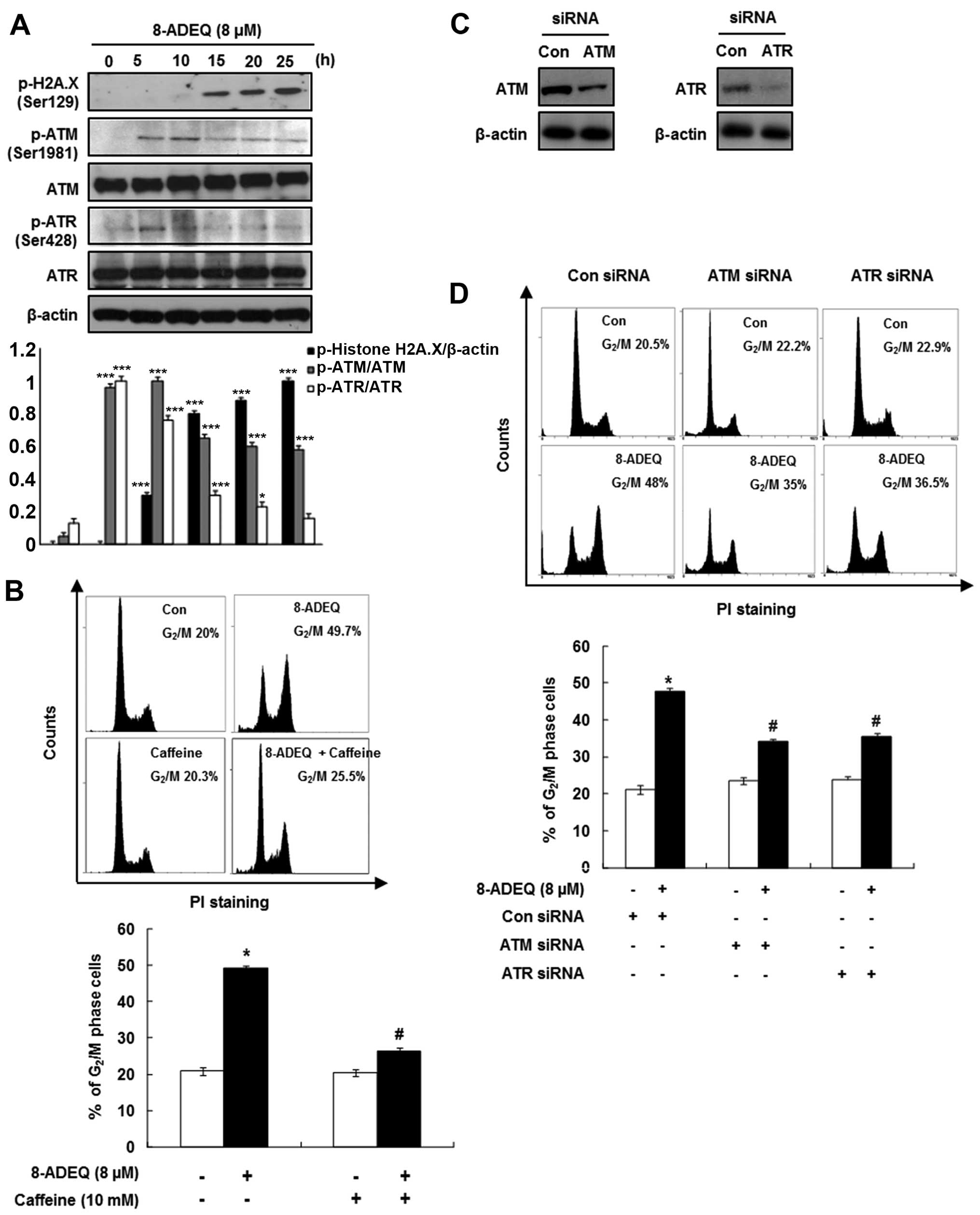 | Figure 3Effect of 8-ADEQ on ATM/ATR dependent
G2/M phase arrest in HeLa cells. (A) Expression levels
of p-H2A.X, p-ATM, ATM, p-ATR, ATR were examined using western blot
analysis as described in Materials and methods. β-actin was used as
an internal control. The experiments were repeated three times and
similar results were obtained. A densitometric analysis was
performed using Bio-Rad Quantity one® software. (B) The
HeLa cells were pretreated with or without caffeine (10 mM) 1 h,
treated for 25 h with 8-ADEQ (8 μM), and then cell cycle
distribution was analyzed with flow cytometry as described in
Materials and methods. (C) The HeLa cells were transfected with
ATM/ATR siRNA, or non-specific control siRNA for 24 h. The
expression levels of ATM/ATR were examined using western blot
analysis as described in Materials and methods. β-actin was used as
an internal control. (D) The HeLa cells were transfected with
ATM/ATR siRNA or non-specific control siRNA for 24 h and were then
treated with or without 8-ADEQ (8 μM) for 25 h. The cell
cycle distribution was stained with PI and detected by flow
cytometry as described in Materials and methods. Data are presented
as the means ± SDs of three independent experiments.
*P<0.05, **P<0.01,
***P<0.001 vs. the control group,
#P<0.05 vs. the 8-ADEQ-treated group. ATM, ataxia
telangiectasia mutated; ATR, ataxia telangiectasia-Rad3-related;
PI, propidium iodide. |
Discussion
Naturally-derived polyphenolic compounds, such as
resveratrol, have been demonstrated to possess cytostatic activity
against cervical cancer cells (23). With respect to resveratrol, we have
evaluated the chemopreventive effects of resveratrol derivatives,
which exerted more cytotoxic effects on various cancer cells than
resveratrol. A series of styrylquinazoline derivatives were
evaluated for cytotoxicity against three cancer cells and showed
that 3′,4′,8-acetylated or hydroxyl styrylquinazolines showed a
better cytotoxicity profile. Among these styrylquinazoline analogue
compounds, (E)-8-acetoxy-2-[2-(3,4-diacetpxyphenyl)
ethenyl]-quinazoline (8-ADEQ) demonstrated the lowest
IC50 values in three cancer cell lines tested and was
chosen for further studies on the mechanism of action in human
cervical carcinoma HeLa cells.
Resveratrol is an antioxidant and has been shown to
inhibit various stages of tumor development (24). Cancer preventive and anticancer
activities of resveratrol have been observed, where it prevents
skin tumorigenesis in a mouse model (25) and inhibits growth and induces
apoptosis as well as cell cycle arrest (S or G2 phase)
in various human cancer cell lines (26,27).
The present study revealed that 8-ADEQ treatment concentration- and
time-dependently arrested HeLa cells in the G2/M phase
of the cell cycle. To further determine whether 8-ADEQ-induced
G2/M phase arrest would be irreversible, cell cycle
arrest was examined after being washed with 8-ADEQ-free media.
Following treatment with 8-ADEQ (8 μM) for 25 h, media in
the cells were changed to 8-ADEQ-free media and then incubated for
12 h. The 8-ADEQ-induced G2/M phase arrest was not
prevented after washing the cells with 8-ADEQ-free media in
comparison to the only 8-ADEQ-treated cells. This indicates that
8-ADEQ-induced cell cycle arrest is irreversible (data not shown).
In several species, inactivation of the Cdk1/cyclin B1 kinase
complex is the main stream of the blocking progression from the
G2 phase into mitosis, which is induced by DNA damage
and replication checkpoints (28).
In 8-ADEQ-treated cells, a marked upregulation in the cyclin B1
level and an accumulation of phosphorylated Cdk1 (Tyr15) were
accompanied by cell cycle arrest, which suggested that the cells
had downregulated the kinase function of Cdk1 in response to
incubation with the 8-ADEQ. This is accomplished in part by
maintaining the Cdc25 phosphatase in a phosphorylated form (Ser216)
that offers a binding site for 14-3-3 proteins and results in
degradation of Cdc25C protein (29). Moreover, the Cdc25C protein
degradation is significant to 8-ADEQ-induced G2/M arrest
since a Cdc25C-specific proteosome inhibitor, lactacystin,
treatment (data not shown) abrogated these cell cycle arrests.
Notably, treatment with 8-ADEQ did not influence the expression
level of Cdc25C at 25 h, whereas it significantly reduced this
protein level at 48 h suggesting that G2/M arrest by
8-ADEQ is mediated by the level of Cdc25C proteins at late time
(data not shown). Further investigations should provide a thorough
analysis of the Cdc25C expression at late time within the
8-ADEQ-treated cells. The activation of Cdc25C is controlled by its
inhibitory phosphorylation on Ser216 residue by checkpoint kinases
Chk1 and Chk2 in response to the DNA damage by the IR and/or UV
light and by interference with the DNA replication (30). Although Chk1 and Chk2 kinases are
structurally distinct, they are functionally related and
phosphorylate an overlapping pool of cellular substrates (17). Chk1 is activated by ATR, whereas
Chk2 is activated by ATM-dependent phosphorylation (31). The results of the present study
indicate that phosphorylation of Chk1 (Ser345) and Chk2 (Thr68) was
very low in control HeLa cells, but increased time-dependently upon
8-ADEQ treatment. The time course for phosphorylation of Cdc25C
treated with 8-ADEQ mirrored those of the Chk1 and Chk2
phosphorylation.
Many forms of post-translational modifications have
been implicated in the regulation of p53 activity. In particular,
phosphorylation of p53 has been studied extensively and was shown
to be involved in the activation of p53 in response to genotoxic
and other forms of stress (32). By
and large, activation of p53, which is activated by genotoxic
stress, may trigger the onset of DNA repair and leads to the
G2 cell cycle arrest (13). In the present study, we demonstrated
that 8-ADEQ induced the phosphorylation of p53 at Ser15, which was
suggested to play a positive role on the p53 activation.
Furthermore, silencing p53 expression substantially prevented the
cells from undergoing G2/M arrest, which suggested that
p53 plays an important role for the 8-ADEQ-induced cell cycle. The
p21CIP1/WAF1 protein is a universal inhibitor of CDKs
and is mainly regulated by the p53 tumor-suppressor protein
(33). Additionally, our data
suggest that p21CIP1/WAF1 upregulation by 8-ADEQ may be
involved in a p53-dependent pathway.
Several studies have shown that resveratrol induces
DNA damage in many human cancer cell lines (34) and that it is capable of binding to
the DNA and cleave or damage the DNA in a Ca2+-dependent
pathway (35). H2A.X is a variant
form of histone H2A that is phosphorylated at Ser139 by ATM or ATR,
and marks an early event in response to DNA damage (36). The present study indicated that
8-ADEQ induces selective phosphorylation of H2A.X at Ser139 in the
HeLa cells.
The proximal transducer kinases ATM and ATR, both
possess the functional sensors of the DNA damage. ATM is a
phosphoinositide 3-kinase-related kinase that plays an important
role in cell proliferation and DNA repair (37). The DNA damage check points are
predominantly associated with the activation of the ATM, whereas
the ATR is activated by stalling of the replication fork induced by
UV damage, nucleotide imbalance and the DNA cross-linking (38). During this process, the ATM
undergoes autophosphorylation on Ser1981 and is recruited to sites
of the DNA damage where it initiates a series of signaling cascades
by phosphorylating multiple DNA damage response and cell cycle
proteins including Chk1 (Ser345 and Ser317) and Chk2 (Thr68)
(39). The DNA damage activates the
ATM/ATR kinases, initiating two parallel cascades that inactivate
Cdk1-cyclin B, a critical complex in regulation of the
G2/M transition (40,41).
The first cascade involves the key player, p53. Phosphorylation of
p53 dissociates it from the MDM2 and hence activates its
transcriptional activity. The p53-mediated downstream gene
expressions, such as 14-3-3, GADD45 and p21CIP1/WAF1,
inhibits the Cdk1-cyclin B complex. The second cascade involves
phosphorylation and inactivation of Cdc25 phosphatases by Chk
kinases, and the inactivated Cdc25 phosphatases is no longer able
to activate Cdk1 (42). Studies
have indicated that Cdc25A is a critical regulator of the
G1/S phase transition, whereas Cdc25B and Cdc25C are
predominantly expressed in G2 and M phases to regulate
entry of the cells into the M phase by dephosphorylation/activation
of the Cdk1-cyclin B complex (28).
Our results clearly demonstrate that 8-ADEQ treatment activates ATM
and ATR by respective phosphorylation of Ser1981 and Ser428
residues. This finding is in line with a previous study wherein
resveratrol induced cell cycle arrest via
ATM/ATR-Chk1/2-Cdc25C-Cdk1 pathway in ovarian cancer cells
(37). To show the involvement of
ATM/ATR in 8-ADEQ-mediated cell cycle arrest, we selectively
silenced the expression of these proteins by ATM/ATR-specific
siRNAs. Silencing ATM/ATR expression prevented the 8-ADEQ-treated
cells from undergoing G2/M arrest. Moreover, caffeine
has been shown to inhibit ATM/ATR kinase activities (43). We demonstrated that inhibition of
ATM/ATR by caffeine markedly attenuated the 8-ADEQ-induced
G2/M arrest (Fig.
3B).
In conclusion, the present observations support the
idea that 8-ADEQ treatment inhibits the proliferation of human
cervical cancer HeLa cells by DNA damage-mediated G2/M
cell cycle arrest. This is mediated by the activation of both
Chk1/2-Cdc25 and p53-p21CIP1/WAF1 via ATM/ATR pathways.
Accordingly, we conclude that 8-ADEQ appears to have potential in
the treatment of cervical cancer.
Acknowledgments
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the ministry of Education, Science and Technology
(nos. 2011-0023407 and 2011-0030724).
References
|
1
|
Brisdelli F, D’Andrea G and Bozzi A:
Resveratrol: A natural polyphenol with multiple chemopreventive
properties. Curr Drug Metab. 10:530–546. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fauconneau B, Waffo-Teguo P, Huguet F,
Barrier L, Decendit A and Merillon JM: Comparative study of radical
scavenger and antioxidant properties of phenolic compounds from
Vitis vinifera cell cultures using in vitro tests. Life Sci.
61:2103–2110. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim AL, Zhu Y, Zhu H, Han L, Kopelovich L,
Bickers DR and Athar M: Resveratrol inhibits proliferation of human
epidermoid carcinoma A431 cells by modulating MEK1 and AP-1
signalling pathways. Exp Dermatol. 15:538–546. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Baur JA and Sinclair DA: Therapeutic
potential of resveratrol: The in vivo evidence. Nat Rev Drug
Discov. 5:493–506. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shankar S, Singh G and Srivastava RK:
Chemoprevention by resveratrol: Molecular mechanisms and
therapeutic potential. Front Biosci. 12:4839–4854. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alao JP: The regulation of cyclin D1
degradation: Roles in cancer development and the potential for
therapeutic invention. Mol Cancer. 6:242007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aggarwal BB, Bhardwaj A, Aggarwal RS,
Seeram NP, Shishodia S and Takada Y: Role of resveratrol in
prevention and therapy of cancer: Preclinical and clinical studies.
Anticancer Res. 24:2783–2840. 2004.PubMed/NCBI
|
|
8
|
Bhardwaj A, Sethi G, Vadhan-Raj S,
Bueso-Ramos C, Takada Y, Gaur U, Nair AS, Shishodia S and Aggarwal
BB: Resveratrol inhibits proliferation, induces apoptosis, and
overcomes chemoresistance through down-regulation of STAT3 and
nuclear factor-kappaB-regulated antiapoptotic and cell survival
gene products in human multiple myeloma cells. Blood.
109:2293–2302. 2007. View Article : Google Scholar
|
|
9
|
Shimizu T, Nakazato T, Xian MJ, Sagawa M,
Ikeda Y and Kizaki M: Resveratrol induces apoptosis of human
malignant B cells by activation of caspase-3 and p38 MAP kinase
pathways. Biochem Pharmacol. 71:742–750. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
de la Lastra CA and Villegas I:
Resveratrol as an anti-inflammatory and anti-aging agent:
Mechanisms and clinical implications. Mol Nutr Food Res.
49:405–430. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Park JH, Min HY, Kim SS, Lee JY, Lee SK
and Lee YS: Styrylquinazolines: A new class of inhibitors on
prostaglandin E2 production in
lipopolysaccharide-activated macrophage cells. Arch Pharm.
337:20–24. 2004. View Article : Google Scholar
|
|
12
|
Molinari M: Cell cycle checkpoints and
their inactivation in human cancer. Cell Prolif. 33:261–274. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Taylor WR and Stark GR: Regulation of the
G2/M transition by p53. Oncogene. 20:1803–1815. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Harvey SL, Charlet A, Haas W, Gygi SP and
Kellogg DR: Cdk1-dependent regulation of the mitotic inhibitor
Wee1. Cell. 122:407–420. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Senderowicz AM and Sausville EA:
Preclinical and clinical development of cyclin-dependent kinase
modulators. J Natl Cancer Inst. 92:376–387. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou BB and Bartek J: Targeting the
checkpoint kinases: Chemosensitization versus chemoprotection. Nat
Rev Cancer. 4:216–225. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bartek J and Lukas J: Chk1 and Chk2
kinases in checkpoint control and cancer. Cancer Cell. 3:421–429.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ahn J, Urist M and Prives C: Questioning
the role of checkpoint kinase 2 in the p53 DNA damage response. J
Biol Chem. 278:20480–20489. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vousden KH and Lu X: Live or let die: The
cell’s response to p53. Nat Rev Cancer. 2:594–604. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu S, Bishop WR and Liu M: Differential
effects of cell cycle regulatory protein p21WAF1/Cip1 on
apoptosis and sensitivity to cancer chemotherapy. Drug Resist
Updat. 6:183–195. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kinner A, Wu W, Staudt C and Iliakis G:
Gamma-H2AX in recognition and signaling of DNA double-strand breaks
in the context of chromatin. Nucleic Acids Res. 36:5678–5694. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Matsuoka S, Huang M and Elledge SJ:
Linkage of ATM to cell cycle regulation by the Chk2 protein kinase.
Science. 282:1893–1897. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Signorelli P and Ghidoni R: Resveratrol as
an anticancer nutrient: Molecular basis, open questions and
promises. J Nutr Biochem. 16:449–466. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Das DK, Sato M, Ray PS, Maulik G, Engelman
RM, Bertelli AA and Bertelli A: Cardioprotection of red wine: Role
of polyphenolic antioxidants. Drugs Exp Clin Res. 25:115–120.
1999.PubMed/NCBI
|
|
25
|
Nawroth R, Poell G, Ranft A, Kloep S,
Samulowitz U, Fachinger G, Golding M, Shima DT, Deutsch U and
Vestweber D: VE-PTP and VE-cadherin ectodomains interact to
facilitate regulation of phosphorylation and cell contacts. EMBO J.
21:4885–4895. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ahmad N, Adhami VM, Afaq F, Feyes DK and
Mukhtar H: Resveratrol causes WAF-1/p21-mediated
G1-phase arrest of cell cycle and induction of apoptosis
in human epidermoid carcinoma A431 cells. Clin Cancer Res.
7:1466–1473. 2001.PubMed/NCBI
|
|
27
|
Zhang P, Li H, Wu ML, Chen XY, Kong QY,
Wang XW, Sun Y, Wen S and Liu J: c-Myc downregulation: A critical
molecular event in resveratrol-induced cell cycle arrest and
apoptosis of human medulloblastoma cells. J Neurooncol. 80:123–131.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou BB and Elledge SJ: The DNA damage
response: Putting checkpoints in perspective. Nature. 408:433–439.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Peng CY, Graves PR, Thoma RS, Wu Z, Shaw
AS and Piwnica-Worms H: Mitotic and G2 checkpoint control:
Regulation of 14-3-3 protein binding by phosphorylation of Cdc25C
on serine-216. Science. 277:1501–1505. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reinhardt HC and Yaffe MB: Kinases that
control the cell cycle in response to DNA damage: Chk1, Chk2, and
MK2. Curr Opin Cell Biol. 21:245–255. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shiloh Y: ATM and related protein kinases:
Safeguarding genome integrity. Nat Rev Cancer. 3:155–168. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yu J and Zhang L: The transcriptional
targets of p53 in apoptosis control. Biochem Biophys Res Commun.
331:851–858. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Graña X and Reddy EP: Cell cycle control
in mammalian cells: Role of cyclins, cyclin dependent kinases
(CDKs), growth suppressor genes and cyclin-dependent kinase
inhibitors (CKIs). Oncogene. 11:211–219. 1995.PubMed/NCBI
|
|
34
|
Athar M, Back JH, Tang X, Kim KH,
Kopelovich L, Bickers DR and Kim AL: Resveratrol: A review of
preclinical studies for human cancer prevention. Toxicol Appl
Pharmacol. 224:274–283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tay WM, da Silva GF and Ming LJ: Metal
binding of flavonoids and their distinct inhibition mechanisms
toward the oxidation activity of Cu2+-β-amyloid: Not
just serving as suicide antioxidants! Inorg Chem. 52:679–690. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mansilla S, Bataller M and Portugal J:
Mitotic catastrophe as a consequence of chemotherapy. Anticancer
Agents Med Chem. 6:589–602. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sahu RP, Batra S and Srivastava SK:
Activation of ATM/Chk1 by curcumin causes cell cycle arrest and
apoptosis in human pancreatic cancer cells. Br J Cancer.
100:1425–1433. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Goel A, Kunnumakkara AB and Aggarwal BB:
Curcumin as ‘Curecumin’: From kitchen to clinic. Biochem Pharmacol.
75:787–809. 2008. View Article : Google Scholar
|
|
39
|
van Vugt MA and Medema RH: Getting in and
out of mitosis with Polo-like kinase-1. Oncogene. 24:2844–2859.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Iliakis G, Wang Y, Guan J and Wang H: DNA
damage checkpoint control in cells exposed to ionizing radiation.
Oncogene. 22:5834–5847. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang J, Yu Y, Hamrick HE and
Duerksen-Hughes PJ: ATM, ATR and DNA-PK: Initiators of the cellular
genotoxic stress responses. Carcinogenesis. 24:1571–1580. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shieh SY, Ahn J, Tamai K, Taya Y and
Prives C: The human homologs of checkpoint kinases Chk1 and Cds1
(Chk2) phosphorylate p53 at multiple DNA damage-inducible sites.
Genes Dev. 14:289–300. 2000.PubMed/NCBI
|
|
43
|
Zeng Y, Forbes KC, Wu Z, Moreno S,
Piwnica-Worms H and Enoch T: Replication checkpoint requires
phosphorylation of the phosphatase Cdc25 by Cds1 or Chk1. Nature.
395:507–510. 1998. View
Article : Google Scholar : PubMed/NCBI
|