Introduction
An estimated 1.7 million individuals are diagnosed
with breast cancer and >450,000 individuals succumb to the
disease each year worldwide (1).
Breast cancer ranks sixth with regard to mortality among women in
Japan (2). Accordingly,
establishment of novel modalities for treatment against breast
cancer is urgently needed.
Human epidermal growth factor receptor 2 (HER2) is a
185-kDa transmembrane tyrosine kinase receptor and its signaling
promotes cell proliferation, differentiation and survival through
the RAS-RAF-MAPK and PI3K-Akt pathways (3,4). HER2
overexpression at the gene or protein level is observed in 20–25%
of human breast cancers, and its overexpression is associated with
a poor clinical outcome (5).
Trastuzumab, a monoclonal antibody specific to the extracellular
domain of HER2, is a promising tool for treating breast cancers
with HER2 overexpression (6). In a
phase III clinical trial, the combined treatment of trastuzumab and
paclitaxel (PTX) for metastatic HER2-positive breast cancer
elicited improved response, progression-free survival and overall
survival compared with chemotherapy without trastuzumab (6,7). The
mechanism underlying the antitumor effects of trastuzumab includes
the inhibition of HER2 downstream signaling pathways and activation
of the antibody-dependent cell-mediated cytotoxicity (ADCC)
response (8–10). However, the majority of patients who
have an initial response to trastuzumab acquire resistance to the
drug during prolonged treatment (6,7,9).
Recently, trastuzumab emtansine (T-DM1), a conjugate
of trastuzumab and derivative of maytansine 1 (DM1) was developed
as a new drug for anti-HER2-targeting therapy. T-DM1 binds to
cell-surface HER2 receptors and is delivered into lysosomes via
endocytosis where it is digested by lysosomal enzymes. The active
form of DM1 is released into the cytoplasm and inhibits the
assembly of microtubules (11–13).
The cytotoxic activity of T-DM1 towards breast and gastric cancer
cells was stronger than that of trastuzumab in vitro and
in vivo even if the tumor cells were resistant to
trastuzumab by PIK3CA mutation (11,14,15).
In phase III clinical studies, the efficacy of T-DM1 was found to
be superior to that of lapatinib and docetaxel in patients with
HER2-positive advanced breast cancers resistant to trastuzumab and
taxanes (16).
Trastuzumab or T-DM1 have been used for the
treatment of breast cancers with HER2 overexpression. To determine
indications for trastuzumab or T-DM1 therapy, the extent of HER2
expression in breast cancer cells was evaluated by
immuno-histochemistry (IHC) and gene amplification was determined
by fluorescence in situ hybridization (FISH) (7,17,18).
Breast cancers with a high HER2 expression or FISH positivity are
associated with a good response to trastuzumab or T-DM1 (7,11,16,18,19).
Thus, innovative modalities that enable breast cancer patients with
a low HER2 expression to receive trastuzumab or T-DM1 therapy are
required.
Combined treatment with gemcitabine (GEM) and PTX
produces longer progression-free survival and improved response
rates than PTX alone in metastatic breast cancer patients who
failed anthracycline-based chemotherapy (20,21).
GEM is a nucleoside analog that arrests cells in the S-phase in the
cell cycle and it is widely used as a standard therapeutic agent
for pancreatic cancer and metastatic breast cancer. By contrast,
PTX is an inhibitor of microtubule de-polymerization that induces
cell-cycle arrest in the G2-M phase. The improved therapeutic
efficacy of the combined therapy with GEM and PTX against breast
cancer appears to be based on their different cytotoxic mechanisms.
As most breast cancer patients express HER2 at low or moderate
levels, they have been treated with hormone therapy or
chemotherapy. If a low HER2 expression level in breast cancer was
to be enhanced by treatment with a considerable agent, the T-DM1
treatment could be applied by combination with that agent. We have
previously reported that GEM treatment induced the upregulation of
Wilms tumor 1 (WT1) in pancreatic cancer cells and enhanced the
efficacy of WT1-targeting immunotherapy against pancreatic cancer
(22). In the present study, we
aimed to demonstrate that treatment of low HER2-expressing breast
cancer cells with GEM enhanced their HER2 expression in
vitro. The antitumor effects of the combined treatment with GEM
and T-DM1 for low HER2-expressing breast cancer cells was
investigated.
Materials and methods
Cell lines and agents
MDA-MB-231 human breast cancer cells used in our
laboratory were confirmed to be original MDA-MB-231 by DNA short
tandem repeat analysis. MCF7 cells were obtained from RIKEN BRC
through the National Bio-Resource Project of the MEXT (Ibaraki,
Japan). The BT-20 cells were provided from Dr Shigeo Koido, from
Jikei University. GEM was purchased from Eli Lilly Japan (Kobe,
Japan). 5-Fluorouracil (5-FU) was purchased from Kyowa Hakko Kirin
Co. (Tokyo, Japan) and PTX was obtained from Sigma-Aldrich (St.
Louis, MO, USA). The NF-κB inhibitor BAY11-7082 was purchased from
Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Trastuzumab was
a gift from Chugai, Inc. (Tokyo, Japan) and T-DM1 was provided by
Genentech, Inc. (South San Francisco, CA, USA).
FACS analysis
To assess the HER2 expression level, the cells were
incubated with phycoerythrin (PE)-labeled anti-human HER2 (24D2) or
corresponding isotype control antibodies (BioLegend, San Diego, CA,
USA) in FACS buffer for 30 min at 4°C. After being washed, the
cells were analyzed using a MACSQuant Analyzer (Miltenyi Biotech
K.K., Bergisch Gladbach, Germany). To assess the T-DM1 binding
level of the MCF7 cells, 1×106 cells were incubated with
10 µg/ml T-DM1 at 37°C for 1 h. After being washed, the
cells were incubated with PE-labeled anti-human IgG (HP6017) or
corresponding isotype control antibodies (both from BioLegend) in
FACS buffer for 30 min at 4°C. The cells were analyzed using a
MACSQuant Analyzer (Miltenyi Biotech K.K.). Prior to using the
analyzer, 4 µg/ml propidium iodide (PI) (Sigma-Aldrich) was
added to the sample to exclude dead cells. The mean fluorescence
intensity (MFI) of HER2 and human IgG was analyzed using
MACSQuantify software.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the cells using TRIzol
reagent (Life Technologies, Carlsbad, CA, USA) followed by
phenol-chloroform extraction and isopropanol precipitation or the
Micro-to-Midi Total RNA Purification System (Life Technologies).
cDNA was synthesized from 500 ng of the total RNA using the
PrimeScript RT reagent kit (Takara Bio, Inc., Otsu, Shiga, Japan)
and the GeneAmp PCR System 9700 (Applied Biosystems). For RT-qPCR
detection of HER2 and 18S rRNA, 5 ng of cDNA was amplified using
SYBR Premix Ex Taq II (Takara Bio, Inc.) and the 7300
Real-Time PCR System (Applied Biosystems). The PCR conditions
consisted of an initial denaturation step (95°C for 30 sec)
followed by 40 cycles (95°C for 5 sec and 62°C for 31 sec) and a
dissociation step. The primer sequences (Operon Biotechnologies
K.K., Tokyo, Japan) used were: HER2 5′-TCCTGTGTGGAC CTGGAT-3′
(forward), and 5′-TGCCGTCGCTTGATGAG-3′ (reverse); and 18S rRNA
5′-CGGCTACCACATCCAAGGAA-3′ (forward), and 5′-GCTGGAATTACCGCGGCT-3′
(reverse). The data were analyzed using the comparative ΔΔCt method
by calculating the difference between the threshold cycle (CT)
values of the target and the reference genes for each sample and
then comparing the ΔCT values of each drug treatment to the
untreated group.
Immunoblot analysis
The cells were homogenized in RIPA buffer (20 mM
Tris-HCl pH 7.5, 150 mM NaCl, 0.1% deoxycholic acid, 1 mM EDTA,
0.1% SDS, 1% NP-40) with 100 mM PMSF, 1 mg/ml leupeptin, 1 mg/ml
aprotinin and 0.9 M Na3VO4, and the protein
concentrations were analyzed using the Pierce BCA protein assay kit
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). Protein samples
(10 µg) were separated by electrophoresis in 7.5% sodium
dodecyl sulfate-polyacrylamide gels (ATTO, Tokyo, Japan) and
transferred to polyvinylidene difluoride membranes (Bio-Rad
Laboratories, Hercules, CA, USA). After being blocked with 3%
non-fat milk and 3% bovine serum albumin for 1 h, the membrane was
treated with Abs directed against HER2 (1:1,000) (e2-4001 + 3B5;
Thermo Fisher Scientific, Inc.) and GAPDH (1:600,000) (2D4A7; Abcam
Inc., Cambridge, UK) and then with secondary antibodies (Cell
Signaling Technology Inc., Danvers, MA, USA) conjugated to
horseradish peroxidase. Chemi-Lumi One Super (Nakalai Tesque, Inc.,
Kyoto, Japan) was used for chemiluminescence detection.
Estimation of the antiproliferative
effects of different agents by in vitro cell growth assays
MCF7 cells in the culture dishes were treated with
GEM (0, 10 and 30 ng/ml) for 2 h, PTX (0, 5.6 and 16.7 ng/ml) for
24 h or 5-FU (0, 10 or 40 µg/ml) for 1.5 h. After washing
the cells with phosphate-buffered saline (PBS), they were incubated
in a medium containing T-DM1 (0, 10 and 30 µg/ml) for 96 h.
Identical numbers of trypan blue non-stained cells were seeded in
96-well plates (3×103/well). After a 96-h incubation,
cell growth was examined by spectrophotometry using Cell Counting
Kit-8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan).
Fluorescence was measured at 450 nm using an iMark microplate
absorbance reader (Bio-Rad Laboratories).
Statistical analysis
Data are presented as the mean ± standard deviation
(SD). Comparisons between the untreated control and drug-treated
groups were performed by the unpaired Student’s or Welch’s t-tests
for two independent groups and the Dunnett or Bonferroni-Dunn
methods for multiple-group comparisons. P<0.05 was considered to
indicate a statistically significant result. Statistical analyses
were performed using Microsoft Office Excel 2007 (Microsoft
Corporation, Redmond, WA, USA) with the add-in software Statcel 3
(OMS Publishing Inc., Saitama, Japan).
Results
GEM treatment increases HER2 expression
in breast cancer cells with low HER2 expression
MCF7, MDA-MB-231 and BT-20 human breast cancer cells
have low HER2 expression levels. Alterations in HER2 expression in
these cell lines by treatment with GEM or PTX was examined.
HER2-expression was significantly enhanced by GEM treatment,
although the extent of HER2 upregulation was different among the
three cell lines (Fig. 1A). PTX
treatment induced a low and moderate HER2 upregulation in the
MDA-MB-231 and MCF7 cells, respectively, whereas HER2 was
downregulated in BT-20 cells (Fig.
1B). HER2 mRNA in GEM-treated MCF7 cells significantly
increased when the cells were treated with 30 or 100 ng/ml GEM
(Fig. 2A). The results from the
immunoblot analysis showed that HER2 protein expression in
GEM-treated MCF7 cells increased (Fig.
2B). Although HER2 expression in PTX (50 ng/ml)- or 5-FU (40
µg/ml)-treated MCF7 cells increased 3.2- or 3-fold,
respectively, a larger increase, i.e., 4.6-fold, was identified
with GEM (100 ng/ml) treatment (Fig.
2C).
 | Figure 1GEM and PTX induce HER2 upregulation
in three human breast cancer cell lines. (A) The cells were
untreated (upper panel) or treated (middle panel) with GEM (100
ng/ml) for 2 h. After being washed with PBS, the cells were
incubated for 48 h. HER2 expression was examined by flow cytometry.
The gray and black histogram profiles indicate the isotype control
and HER2 expression, respectively. Alterations in the HER2
expression (mean ± SD, n=3) are shown in graphs (lower panel). The
values indicate the Δ MFI of HER2, such as the MFI of HER2 minus
that of the isotype control of the samples. *P<0.05
and **P<0.01 vs. the untreated samples. (B) The cells
were untreated (upper panel) or treated (middle panel) with PTX (50
ng/ml) for 24 h. The experimental procedures were the same as in
(A). Alterations in HER2 expression (mean ± SD, n=3) are shown in
the graphs (lower panel). The values indicate the Δ MFI of HER2,
such as the MFI of HER2 minus that of the isotype controls of the
samples. *P<0.05 and **P<0.01 vs. the
untreated samples. GEM, gemcitabine; PTX, paclitaxel; HER2, human
epidermal growth factor receptor 2; PBS, phosphate-buffered saline;
PE, phycoerythrin; MFI, mean fluorescence intensity. |
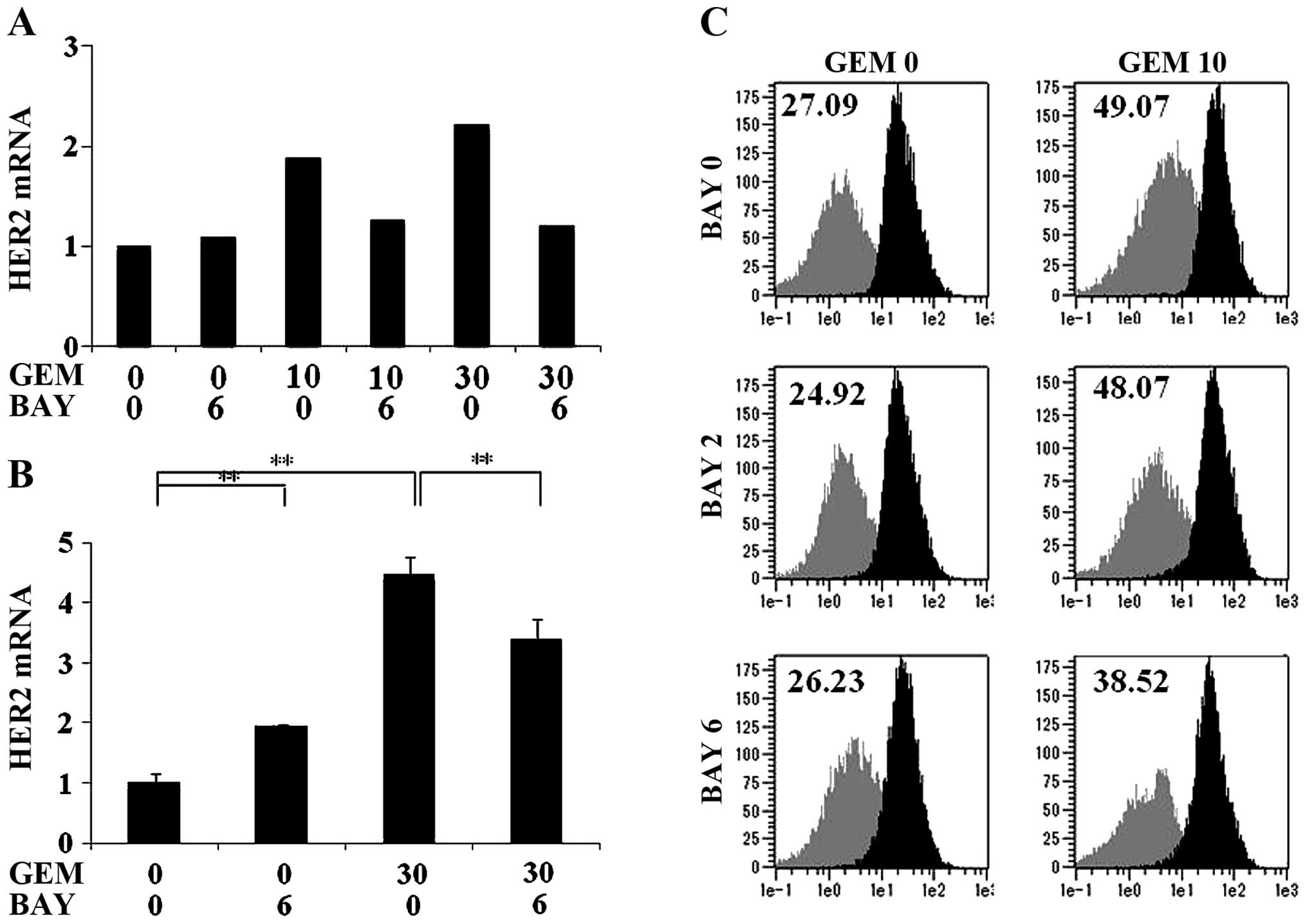
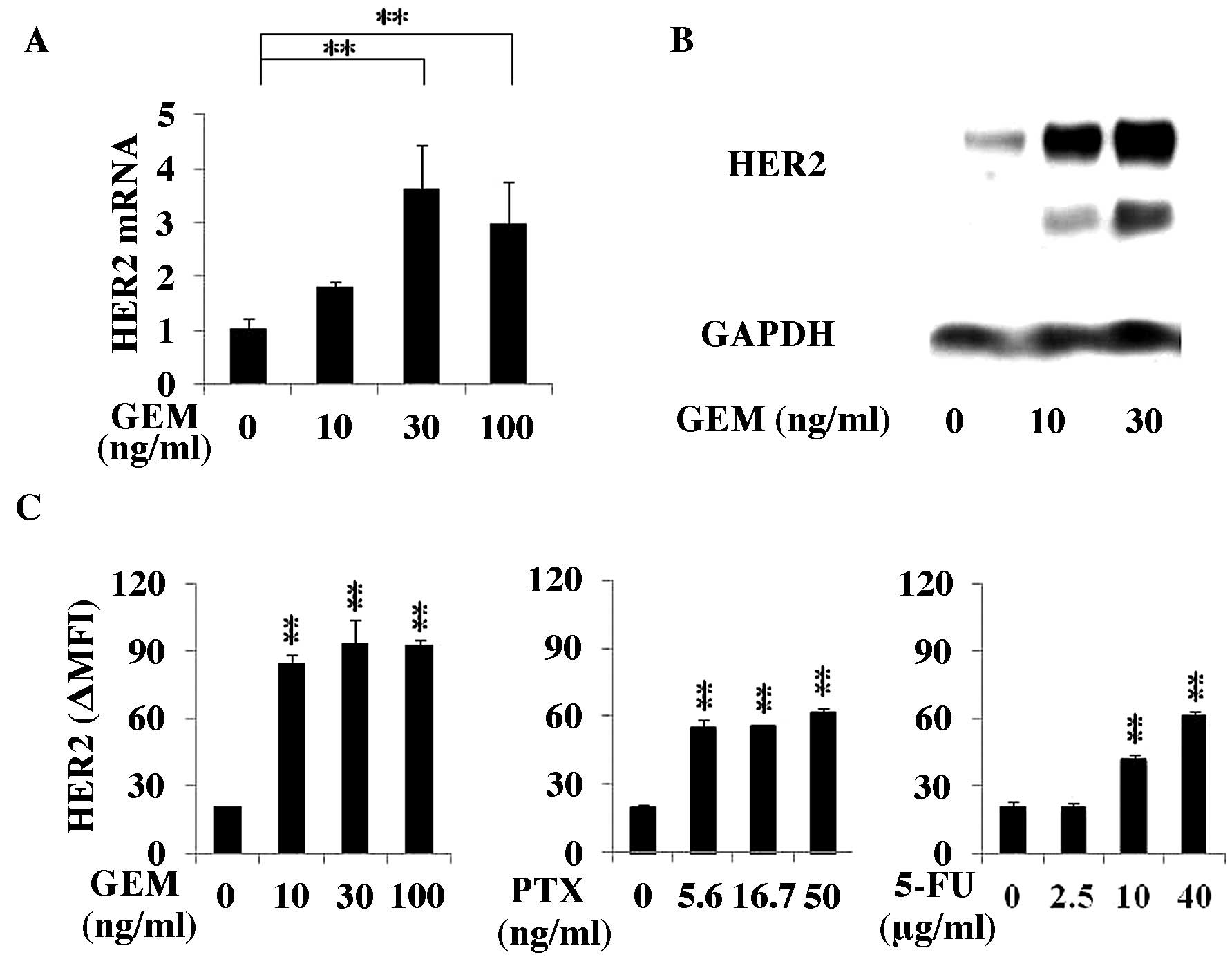 | Figure 2GEM treatment enhanced HER2 expression
of the MCF7 cells at the mRNA and protein levels. (A) MCF7 cells
were treated with GEM (0, 10, 30 or 100 ng/ml) for 2 h, washed with
PBS and incubated for another 48 h in fresh medium. The HER2 mRNA
expression in the cells was analyzed using RT-qPCR (n=3). (B) The
MCF7 cells were treated with GEM (0, 10, 30 or 100 ng/ml) for 2 h,
washed with PBS and incubated for another 48 h in fresh medium.
HER2 and GAPDH protein expression was examined by immunoblot
analysis. (C) MCF7 cells were treated with GEM for 2 h, PTX for 24
h or 5-FU for 1.5 h, washed with PBS and cultured for 48 h in fresh
medium. The HER2 expression was analyzed by flow cytometry. The
values indicate the Δ MFI of HER2, such as the MFI of HER2 minus
that of the isotype control of the samples (n=3).
*P<0.05 and **P<0.01 vs. the untreated
samples. GEM, gemcitabine; HER2, human epidermal growth factor
receptor 2; PBS, phosphate-buffered saline; MFI, mean fluorescence
intensity. |
GEM-mediated HER2 upregulation is
inhibited by treatment with an NF-κB inhibitor
The upregulation of HER2 mRNA induced by GEM
treatment was significantly inhibited by treatment with the NF-κB
inhibitor BAY11-7082 (Fig. 3A and
B). Fig. 3A shows the result
obtained by prolonged treatment of BAY11-7082 with 2 h GEM
treatment. Fig. 3B shows the result
obtained by 2 h simultaneous treatment of GEM and BAY11-7082. The
increase of the HER2 protein expression on the cell surface induced
by GEM treatment was mildly suppressed by prolonged treatment with
6 µM BAY11-7082 (Fig.
3C).
T-DM1 binding to HER2 on MCF7 cells is
increased by GEM treatment
As HER2 expression was upregulated on GEM-treated
breast cancer cells, we examined whether T-DM1 binding to HER2
increases on GEM-treated MCF7 cells. T-DM1 binding to HER2 on the
MCF7 cells was increased following HER2 upregulation by GEM
treatment (Fig. 4A and B). As shown
in Fig. 4A, HER2 expression was
increased 3.95-fold and T-DM1 binding was increased 3.62-fold by
GEM treatment (Fig. 4A), indicating
that the extent of the increase in HER2 expression and T-DM1
binding was similar. T-DM1 binding to HER2 on MCF7 cells treated
with GEM (10 ng/ml) increased at 3.56-fold compared with the
untreated MCF7 cells (Fig. 4B).
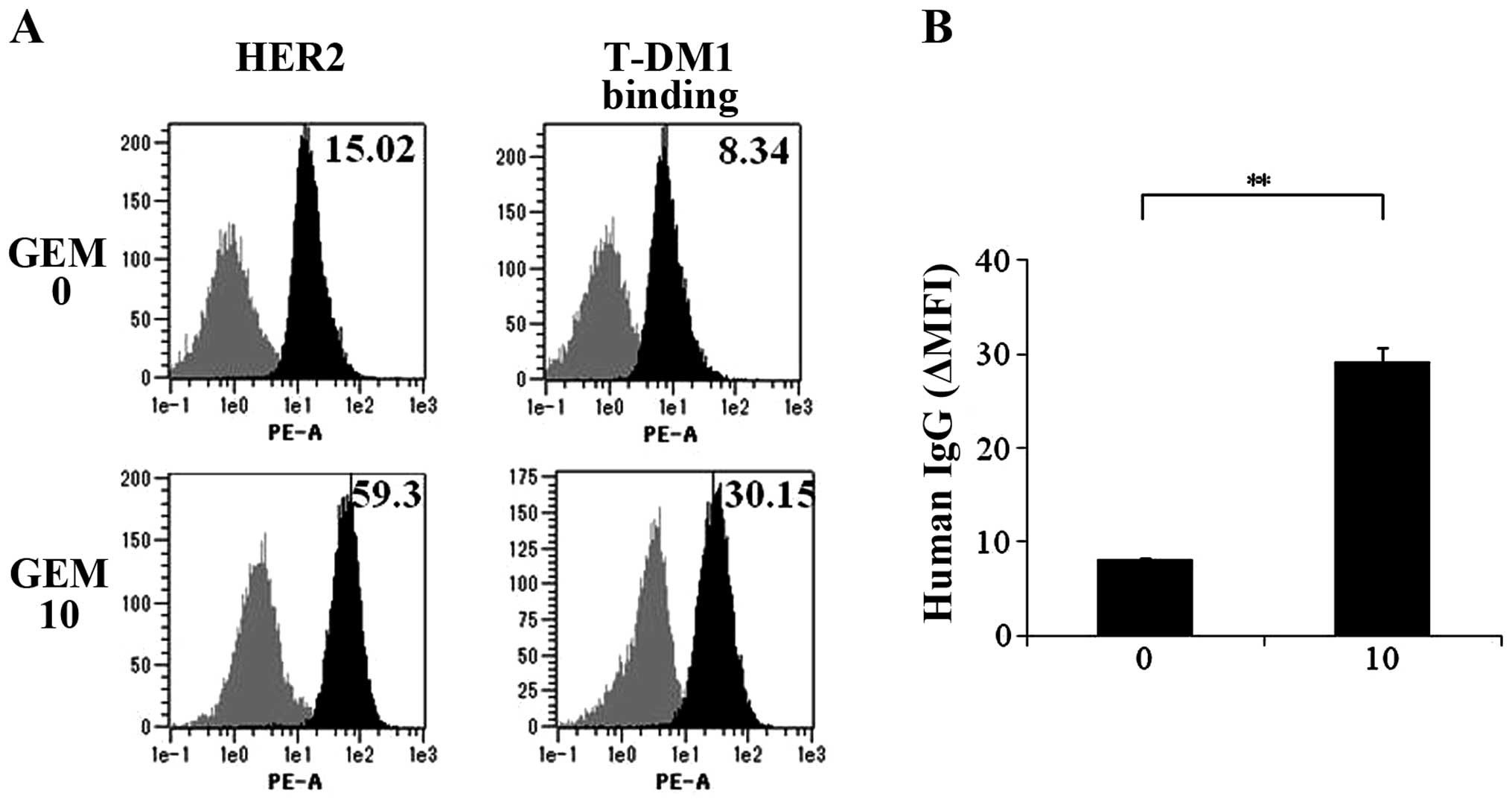 | Figure 4T-DM1 binding to the MCF7 cells is
increased by GEM treatment. (A) MCF7 cells were untreated or
treated with GEM (10 ng/ml) for 2 h, washed with PBS and incubated
in fresh medium for 48 h. HER2 expression of the cells was examined
by flow cytometry. Additionally, untreated or GEM-treated cells
were detached and treated with T-DM1 (10 µg/ml) for 1 h. The
cells were analyzed for T-DM1 binding by flow cytometry using
PE-labeled anti-human IgG. Upper left panel, HER2 expression of
untreated MCF7 cells. Upper right panel, T-DM1 binding to untreated
MCF7 cells. Lower left panel, HER2 expression on GEM-treated MCF7
cells. Lower right panel, T-DM1 binding to GEM-treated MCF7 cells.
Grey and black areas indicate the isotype control and specific
antibody, respectively. Values indicate Δ MFI for HER2, such as the
MFI for HER2 minus that of the isotype control for the samples. (B)
As described in (A), the untreated and GEM-treated MCF7 cells were
incubated with T-DM1 for 1 h, and the binding of T-DM1 was examined
by flow cytometry using PE-labeled anti-human IgG. The Δ MFI of
human IgG was calculated as the MFI of PE-labeled anti-human IgG
minus that of the isotype control of the samples (n=3),
**P<0.01. T-DM1, trastuzumab emtansine; GEM,
gemcitabine; PBS, phosphate-buffered saline; MFI, mean fluorescence
intensity. |
Combined treatment with GEM and T-DM1
synergistically inhibits MCF7 cell proliferation
When the MCF7 cells were pre-treated with 10 ng/ml
GEM, 10 µg/ml T-DM1 treatment produced marked synergistic
antiproliferation effects on the MCF7 cells (Fig. 5). No synergistic effects were
observed when the MCF7 cells were treated with PTX and T-DM1. Low
synergism was observed following the combined treatment with 5-FU
(10 µg/ml) and T-DM1 (10 µg/ml).
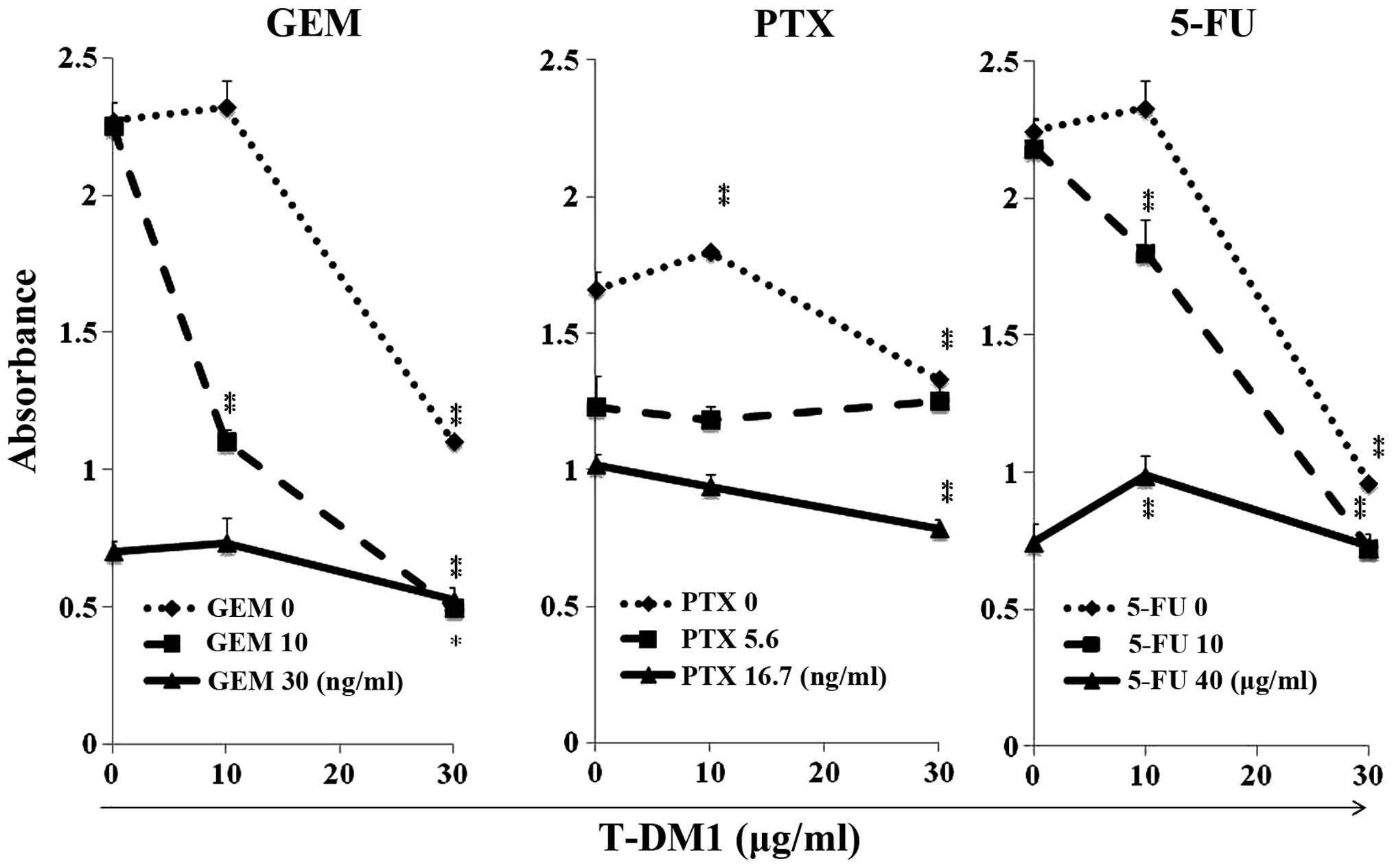 | Figure 5Combined treatment with GEM and T-DM1
synergistically inhibited the growth of MCF7 cells. MCF7 cells were
treated with GEM for 2 h, PTX for 24 h or 5-FU for 1.5 h. The cells
were washed with PBS and incubated in medium containing T-DM1 (0,
10 or 30 µg/ml) for 96 h. The cells were detached and an
identical number of trypan-blue non-stained cells was seeded into
the 96-well plates (3×103 cells/well) and incubated for
96 h. Cell growth was examined by spectrophotometry. Vertical axis,
OD; horizontal axis, concentration of T-DM1 (µg/ml).
*P<0.05 and **P<0.01 vs. the untreated
samples with T-DM1 at each GEM, PTX or 5-FU concentration. GEM,
gemcitabine; T-DM1, trastuzumab emtansine; PTX, paclitaxel; 5-FU,
5-fluorouracil; PBS, phosphate-buffered saline. |
Discussion
In the present study, we found that treatment with
GEM or PTX enhanced the HER2 expression of breast cancer cells with
a low HER2 expression. GEM had stronger potency for HER2
upregulation than PTX, suggesting that modulation of the DNA
synthesis may be more associated with HER2 upregulation. An
increase of HER2 expression by GEM was induced at the
transcriptional level and it appears to be at least partially
regulated by NF-κB signaling. Hernández-Vargas et al
reported that the NF-κB pathway was activated in GEM-treated breast
cancer cells to induce GEM resistance (23). NF-κB has been reported to play a
critical role in HER2 overexpression in breast cancer (24). Cao et al reported that
long-term radiation treatment for breast cancer activated NF-κB,
resulting in accelerated NF-κB signaling to the HER2 promoter for
HER2 mRNA transactivation (25). It
has been reported that GEM has a stronger NF-κB activating ability
than PTX or 5-FU (23). Together, a
higher HER2 upregulation in breast cancer cells by GEM treatment
may be the result of higher NF-κB activation than PTX
treatment.
T-DM1 is a conjugate of trastuzumab and the
cytotoxic agent emtansine (12,13).
In addition to the cytotoxic activity of DM1, T-DM1 has two other
antitumor activities including functioning as a monoclonal antibody
that blocks intracellular signal transduction and ADCC. The
formation of HER2 homodimers or HER2 and HER1/3/4 heterodimers on
tumor cells accelerates the phosphorylation process of
RAS/RAF/MEK/MARK and PI3K/AKT/mTOR without ligand binding,
resulting in the promotion of tumorigenesis (26). It has been shown that trastuzumab
inhibits the PI3K/AKT/mTOR pathway mediated by PTEN (27). Mutation of phosphoinositide 3-kinase
(PI3K) was found to be closely associated with trastuzumab
resistance (28) and 42% of
HER2-enriched breast cancers have a PI3K mutation (29). It may be possible that T-DM1
treatment has less resistance to breast cancers with PI3K mutation
than trastuzumab, and an increase in HER2 expression and the
resultant increase in T-DM1 binding to breast cancer cells may be
beneficial for treating breast cancers with PIK3CA mutations.
Although it is possible that HER2 upregulation by
GEM enhances the ADCC activity of T-DM1, we did not observe
enhanced trastuzumab or T-DM1-mediated ADCC activity towards
GEM-treated breast cancer cells. Since the 51Chromium
(51Cr) release assay was not appropriate for measuring
the ADCC activity against GEM-treated target cells due to the large
spontaneous release of 51Cr, enhanced ADCC activity by
GEM treatment was demonstrated by another appropriate modality.
Resistance to ADCC has been induced by long-term treatment with
trastuzumab, and a possible mechanism involved the downregulation
of the extracellular domain of HER2 (30). Notably, lapatinib, a HER2 tyrosine
kinase inhibitor, accumulated HER2 and potentiated
trastuzumab-dependent cell cytotoxicity (31). GEM treatment may also recover the
HER2 expression of breast cancer cells with HER2 downregulation and
overcome resistance to trastuzumab-mediated ADCC.
It was confirmed that the amount of T-DM1 binding to
HER2 on the breast cancer cells was increased along with the HER2
upregulation induced by GEM treatment, and the extent of the
increase in HER2 expression and T-DM1 binding was almost identical.
These results suggest that T-DM1 successfully bound to HER2
molecules that were upregulated by GEM treatment. The more T-DM1
that binds to cell surface HER2, the more active DM1 is generated
in the cells (11–13).
Significant synergistic antiproliferative effects
between GEM and T-DM1 are based on high HER2 upregulation induced
by GEM and result in the enhancement of T-DM1 binding. By contrast,
synergistic antiproliferative effects were not observed in PTX and
T-DM1-treated MCF7 cells although HER2 upregulation was induced by
PTX treatment. One possible reason for this result may be that PTX
is an inhibitor of microtubule de-polymerization, which is similar
to DM1, while GEM is an inhibitor of DNA synthesis. It is
conceivable that the combined treatment with chemotherapeutic
agents with different mechanisms may be more cytotoxic. Although
synergistic antiproliferative effects were observed between 5-FU
(10 µg/ml) and T-DM1 (10 µg/ml), synergism was lower
than that between GEM and T-DM1, possibly due to a lower HER2
upregulation by 5-FU compared with GEM.
A considerable number of breast cancer patients have
been unable to receive therapeutic benefits by T-DM1 treatment due
to low HER2 expression in their breast cancer tissues.
Pre-treatment with GEM may overcome this limitation of T-DM1
therapy by inducing HER2 upregulation and enabling a low
HER2-expressing breast cancer patients to be candidates for T-DM1
therapy. Experiments involving HER2 upregulation in vivo by
GEM treatment are now being performed using immune-deficient mice
transplanted with human low HER2-expressing breast cancer
cells.
References
|
1
|
Cancer statistics, World Cancer Research
Fund International. http://www.wcrf.org/cancer_statistics/world_cancer_statistics.php.
http://www.wcrf.org/cancer_statistics/cancer_facts/women-breast-cancer.php.
Accessed: date?
|
|
2
|
Center for Cancer Control and Information
Services, National Cancer Center, Japan. Vital Statistics Japan.
Ministry of Health, Labour and Welfare; 2013
|
|
3
|
Hudis CA: Trastuzumab - mechanism of
action and use in clinical practice. N Engl J Med. 357:39–51. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yarden Y and Sliwkowski MX: Untangling the
ErbB signalling network. Nat Rev Mol Cell Biol. 2:127–137. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Slamon DJ, Clark GM, Wong SG, Levin WJ,
Ullrich A and McGuire WL: Human breast cancer: Correlation of
relapse and survival with amplification of the HER-2/neuoncogene.
Science. 235:177–182. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Slamon DJ, Leyland-Jones B, Shak S, Fuchs
H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M,
et al: Use of chemotherapy plus a monoclonal antibody against HER2
for metastatic breast cancer that overexpresses HER2. N Engl J Med.
344:783–792. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seidman AD, Fornier MN, Esteva FJ, Tan L,
Kaptain S, Bach A, Panageas KS, Arroyo C, Valero V, Currie V, et
al: Weekly trastuzumab and paclitaxel therapy for metastatic breast
cancer with analysis of efficacy by HER2 immunophenotype and gene
amplification. J Clin Oncol. 19:2587–2595. 2001.PubMed/NCBI
|
|
8
|
Nahta R and Esteva FJ: Herceptin:
Mechanisms of action and resistance. Cancer Lett. 232:123–138.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nahta R, Yu D, Hung MC, Hortobagyi GN and
Esteva FJ: Mechanisms of disease: Understanding resistance to
HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol.
3:269–280. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lewis GD, Figari I, Fendly B, Wong WL,
Carter P, Gorman C and Shepard HM: Differential responses of human
tumor cell lines to anti-p185HER2 monoclonal antibodies. Cancer
Immunol Immunother. 37:255–263. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Burris HA III, Tibbitts J, Holden SN,
Sliwkowski MX and Lewis Phillips GD: Trastuzumab emtansine (T-DM1):
A novel agent for targeting HER2+ breast cancer. Clin
Breast Cancer. 11:275–282. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
LoRusso PM, Weiss D, Guardino E, Girish S
and Sliwkowski MX: Trastuzumab emtansine: A unique antibody-drug
conjugate in development for human epidermal growth factor receptor
2-positive cancer. Clin Cancer Res. 17:6437–6447. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Barok M, Joensuu H and Isola J:
Trastuzumab emtansine: Mechanisms of action and drug resistance.
Breast Cancer Res. 16:2092014. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Barok M, Tanner M, Köninki K and Isola J:
Trastuzumab-DM1 causes tumour growth inhibition by mitotic
catastrophe in trastuzumab-resistant breast cancer cells in vivo.
Breast Cancer Res. 13:R462011. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barok M, Tanner M, Köninki K and Isola J:
Trastuzumab-DM1 is highly effective in preclinical models of
HER2-positive gastric cancer. Cancer Lett. 306:171–179. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Verma S, Miles D, Gianni L, Krop IE,
Welslau M, Baselga J, Pegram M, Oh DY, Diéras V, Guardino E, et al
EMILIA Study Group: Trastuzumab emtansine for HER2-positive
advanced breast cancer. N Engl J Med. 367:1783–1791. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cox G, Vyberg M, Melgaard B, Askaa J,
Oster A and O’Byrne KJ: Herceptest: HER2 expression and gene
amplification in non-small cell lung cancer. Int J Cancer.
92:480–483. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wolff AC, Hammond ME, Hicks DG, Dowsett M,
McShane LM, Allison KH, Allred DC, Bartlett JM, Bilous M,
Fitzgibbons P, et al: College of American Pathologists:
Recommendations for human epidermal growth factor receptor 2
testing in breast cancer: American Society of Clinical
Oncology/College of American Pathologists clinical practice
guideline update. Arch Pathol Lab Med. 138:241–256. 2014.
View Article : Google Scholar :
|
|
19
|
Dawood S, Broglio K, Buzdar AU, Hortobagyi
GN and Giordano SH: Prognosis of women with metastatic breast
cancer by HER2 status and trastuzumab treatment: An
institutional-based review. J Clin Oncol. 28:92–98. 2010.
View Article : Google Scholar :
|
|
20
|
Albain KS, Nag SM, Calderillo-Ruiz G,
Jordaan JP, Llombart AC, Pluzanska A, Rolski J, Melemed AS,
Reyes-Vidal JM, Sekhon JS, et al: Gemcitabine plus paclitaxel
versus paclitaxel monotherapy in patients with metastatic breast
cancer and prior anthracycline treatment. J Clin Oncol.
26:3950–3957. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aogi K, Yoshida M, Sagara Y, Kamigaki S,
Okazaki M, Funai J, Fujimoto T, Toi M, Saeki T and Takashima S: The
efficacy and safety of gemcitabine plus paclitaxel combination
first-line therapy for Japanese patients with metastatic breast
cancer including triple-negative phenotype. Cancer Chemother
Pharmacol. 67:1007–1015. 2011. View Article : Google Scholar
|
|
22
|
Takahara A, Koido S, Ito M, Nagasaki E,
Sagawa Y, Iwamoto T, Komita H, Ochi T, Fujiwara H, Yasukawa M, et
al: Gemcitabine enhances Wilms’ tumor gene WT1 expression and
sensitizes human pancreatic cancer cells with WT1-specific
T-cell-mediated antitumor immune response. Cancer Immunol
Immunother. 60:1289–1297. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hernández-Vargas H, Rodríguez-Pinilla SM,
Julián-Tendero M, Sánchez-Rovira P, Cuevas C, Antón A, Ríos MJ,
Palacios J and Moreno-Bueno G: Gene expression profiling of breast
cancer cells in response to gemcitabine: NF-kappaB pathway
activation as a potential mechanism of resistance. Breast Cancer
Res Treat. 102:157–172. 2007. View Article : Google Scholar
|
|
24
|
Cao Y, Luo JL and Karin M: IkappaB kinase
alpha kinase activity is required for self-renewal of
ErbB2/Her2-transformed mammary tumor-initiating cells. Proc Natl
Acad Sci USA. 104:15852–15857. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cao N, Li S, Wang Z, Ahmed KM, Degnan ME,
Fan M, Dynlacht JR and Li JJ: NF-kappaB-mediated HER2
overexpression in radiation-adaptive resistance. Radiat Res.
171:9–21. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tzahar E, Waterman H, Chen X, Levkowitz G,
Karunagaran D, Lavi S, Ratzkin BJ and Yarden Y: A hierarchical
network of interreceptor interactions determines signal
transduction by Neu differentiation factor/neuregulin and epidermal
growth factor. Mol Cell Biol. 16:5276–5287. 1996.PubMed/NCBI
|
|
27
|
Spector NL and Blackwell KL: Understanding
the mechanisms behind trastuzumab therapy for human epidermal
growth factor receptor 2-positive breast cancer. J Clin Oncol.
27:5838–5847. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
O’Brien NA, Browne BC, Chow L, Wang Y,
Ginther C, Arboleda J, Duffy MJ, Crown J, O’Donovan N and Slamon
DJ: Activated phosphoinositide 3-kinase/AKT signaling confers
resistance to trastuzumab but not lapatinib. Mol Cancer Ther.
9:1489–1502. 2010. View Article : Google Scholar
|
|
29
|
Cancer Genome Atlas Network: Comprehensive
molecular portraits of human breast tumours. Nature. 490:61–70.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yoshida R, Tazawa H, Hashimoto Y, Yano S,
Onishi T, Sasaki T, Shirakawa Y, Kishimoto H, Uno F, Nishizaki M,
et al: Mechanism of resistance to trastuzumab and molecular
sensitization via ADCC activation by exogenous expression of
HER2-extracellular domain in human cancer cells. Cancer Immunol
Immunother. 61:1905–1916. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Scaltriti M, Verma C, Guzman M, Jimenez J,
Parra JL, Pedersen K, Smith DJ, Landolfi S, Ramon y Cajal S,
Arribas J, et al: Lapatinib, a HER2 tyrosine kinase inhibitor,
induces stabilization and accumulation of HER2 and potentiates
trastuzumab-dependent cell cytotoxicity. Oncogene. 28:803–814.
2009. View Article : Google Scholar
|