Introduction
Gastric cancer (GC) is a malignant tumor arising
from the lining of the stomach. There has been a significant
decrease in the incidence and mortality of this cancer type over
several decades. However, GC remains the fourth most common cancer
and the second most common cause of cancer death worldwide
(1). The American Cancer Society
estimates that ~22,220 cases of GC are to be diagnosed (13,730
males and 8,490 females), and ~10,990 individuals may succumb to GC
(6,720 males and 4,270 females) in the US in 2014. Surgery at early
stage is the most common treatment modality. However, most GC
patients present with an advanced stage of the disease, except in
countries with national screening programs such as Japan and Korea
(2), where early-stage tumors are
usually asymptomatic (2).
MHY-449, also known as
(±)-(R*)-5-methoxy-11-methyl-2-((R*)-2-methyloxiran-2-yl)-1,2-dihydrobenzofuro[4,5-b][1,8]
naphthyridin-6(11H)-one, was designed and synthesized based on the
chemical structure of psorospermin with a xanthone template and
acronycine derivatives, with an acridone template (3). The cytotoxicity of MHY-449 was
assessed against five human cancer cell lines including prostate
cancer cell lines (LNCaP, DU145, and PC3) and breast cancer cell
lines (MCF-7/ADR and MCF-7) (3,4).
MHY-449 has cytotoxicity against human prostate and breast cancer
cells. It suppressed cell growth by inducing G2/M phase cell cycle
arrest in MCF-7/ADR cells and proliferation of
androgen-independent, p53-null, and phosphatase and tensin homolog
(PTEN)-negative PC3 cells by inducing apoptosis (3,4). In
addition, MHY-449 effectively induced G2/M phase cell cycle arrest
and apoptosis in HCT116 human colon cancer cells, thereby
suppressing their growth (5). This
study was designed to evaluate the cytotoxic effects and underlying
molecular mechanisms of action of MHY-449 against GC cells, which
have yet to be adequately evalutated.
Materials and methods
Chemicals
The simplified code name and structure of MHY-449
used in this study is shown in Fig.
1. The method for the design and synthesis of this compound was
previously described (3). MHY-449
was dissolved in dimethyl sulfoxide (DMSO), stored at −20°C prior
to the experiments, and working dilutions were prepared in culture
medium. The maximum concentration of DMSO did not exceed 0.1% (v/v)
in the treatment range, where there was no influence on cell
growth. DMSO and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide (MTT) were obtained from Amresco LLC (Solon,
OH, USA). Propidium iodide (PI), N-acetyl-L-cysteine (NAC), and the
monoclonal antibody against β-actin were purchased from
Sigma-Aldrich Co. LLC (St. Louis, MO, USA). Antibodies specific for
Fas, Fas-ligand (FasL), caspase-3, 8, and 9, poly(ADP-ribose)
polymerase (PARP), B-cell lymphoma 2 (Bcl-2), Bcl-2-associated X
protein (BAX), p53, p21WAF1/CIP1, and
p27KIP1, as well as Z-VAD-FMK, were obtained from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Cell culture and cell viability
assay
The AGS human GC cell line was cultured in RPMI-1640
(GE Healthcare Life Sciences, Logan, UT, USA) supplemented with 10%
fetal bovine serum (FBS; GE Healthcare Life Sciences), 100 U/ml
penicillin, and 100 µg/ml streptomycin (GE Healthcare Life
Sciences) at 37°C in humidified 5% CO2. Cell viability
was determined using an MTT assay for which AGS cells were seeded
in a 24-well culture plate, cultured for 24 h in the growth medium,
and then treated with or without various reagents at the indicated
concentrations. The cells were incubated in the dark with 0.5 mg/ml
MTT at 37°C for 2 h. The formazan granules generated by the live
cells were dissolved in DMSO, and the absorbance at 540 nm was
monitored using a multi-well reader (Thermo Fisher Scientific,
Vantaa, Finland).
Nuclear staining with Hoechst 33342
The cells were stained with 4 µg/ml Hoechst
33342 (Life Technologies Corporation, Grand Island, NY, USA) at
37°C for 10 min. The cells were then observed under a fluorescence
microscope.
Annexin V staining
The cells were cultured under appropriate conditions
for 24 h, subsequently harvested, trypsinized, washed once with
cold phosphate-buffered saline (PBS), and suspended in 1X binding
buffer (BD Biosciences, San Jose, CA, USA). The cells were stained
with PI and Annexin V-fluorescein isothiocyanate (FITC) solution
(BD Pharmingen FITC Annexin V Apoptosis Detection kit I) at room
temperature for 15 min in the dark. The stained cells were analyzed
by flow cytometry within 1 h.
DNA fragmentation assay
The cells were lysed in a buffer containing 5 mM
Tris-HCl (pH 7.5), 5 mM ethylenediaminetetraacetic acid (EDTA), and
0.5% Triton X-100 for 30 min on ice. The lysates were vortexed and
cleared by centrifugation at 27,000 × g for 20 min. Fragmented DNA
in the supernatant was treated with RNase, followed by proteinase K
digestion, extraction with a phenol/chloroform/isoamyl alcohol
mixture (25:24:1, v/v/v), and isopropanol precipitation. DNA was
separated using a 1.6% agarose gel, stained with 0.1 µg/ml
ethidium bromide, and visualized using an ultraviolet source.
Flow cytometric analysis
The cells were cultured under the appropriate
conditions for 24 h, subsequently trypsinized, washed once with
cold PBS, and then fixed in 70% ethanol at −20°C overnight. The
fixed cells were stained with cold PI solution (50 µg/ml in
PBS) at 37°C for 30 min in the dark. Flow cytometric analysis was
performed using a Cytomic FC500 (Beckman Coulter, Istanbul,
Turkey). Sub-G1 populations were analyzed using WinCycle software
(Phoenix Flow Systems, San Diego, CA, USA).
Protein preparation and western blot
analysis
Total cells were lysed in lysis buffer containing 25
mM Tris (pH 7.5), 250 mM NaCl, 5 mM EDTA, 1% nonidet P-40, 100
µg/ml phenylmethylsulfonyl fluoride, and protease inhibitor
cocktail (Sigma-Aldrich Co. LLC). The lysates were subjected to
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF)
membranes. The membranes were probed with the relevant primary
antibodies overnight, and incubated with horseradish peroxidase
(HRP)-conjugated secondary antibodies (Santa Cruz Biotechnology,
Inc.), and then visualized using the enhanced chemiluminescence
(ECL) detection system (GE Healthcare, Piscataway, NJ, USA).
Caspase activity
The cells were collected and washed with cold PBS.
Total cells were incubated with the lysis buffer (R&D Systems,
Inc., Minneapolis, MN, USA) on ice for 10 min. The lyzed cells were
centrifuged at 10,000 × g for 1 min, and 100 µg of protein
was incubated with 2X reaction buffer and substrates of the
colorimetric tetrapeptides Z-DEVD, Z-IETD, and Ac-LEHD for
caspase-3, -8, and -9. The reaction mixtures were incubated at 37°C
for 2 h, and the enzyme-catalyzed release of p-nitroaniline (pNA)
was quantified at 405 nm using a multi-well reader (Thermo Fisher
Scientific).
Measurement of intracellular reactive
oxygen species (ROS)
The intracellular formation of reactive oxygen
species (ROS) was determined using 2′,7′-dichlorofluorescein
diacetate (DCF-DA; Molecular Probes, Eugene, OR, USA). The cells
were cultured under the appropriate conditions for 8 h,
subsequently trypsinized, and washed twice with cold PBS. The cells
were stained with 10 µM DCF-DA at 37°C for 30 min in the
dark. Fluorescence intensity was quantified using a Cytomic FC 500
(Beckman Coulter).
Statistical analysis
Data were presented as the mean ± standard deviation
(SD) of three separate experiments and analyzed by the Student’s
t-test. Means were considered significantly different at
*p<0.05 or **p < 0. 01.
Results
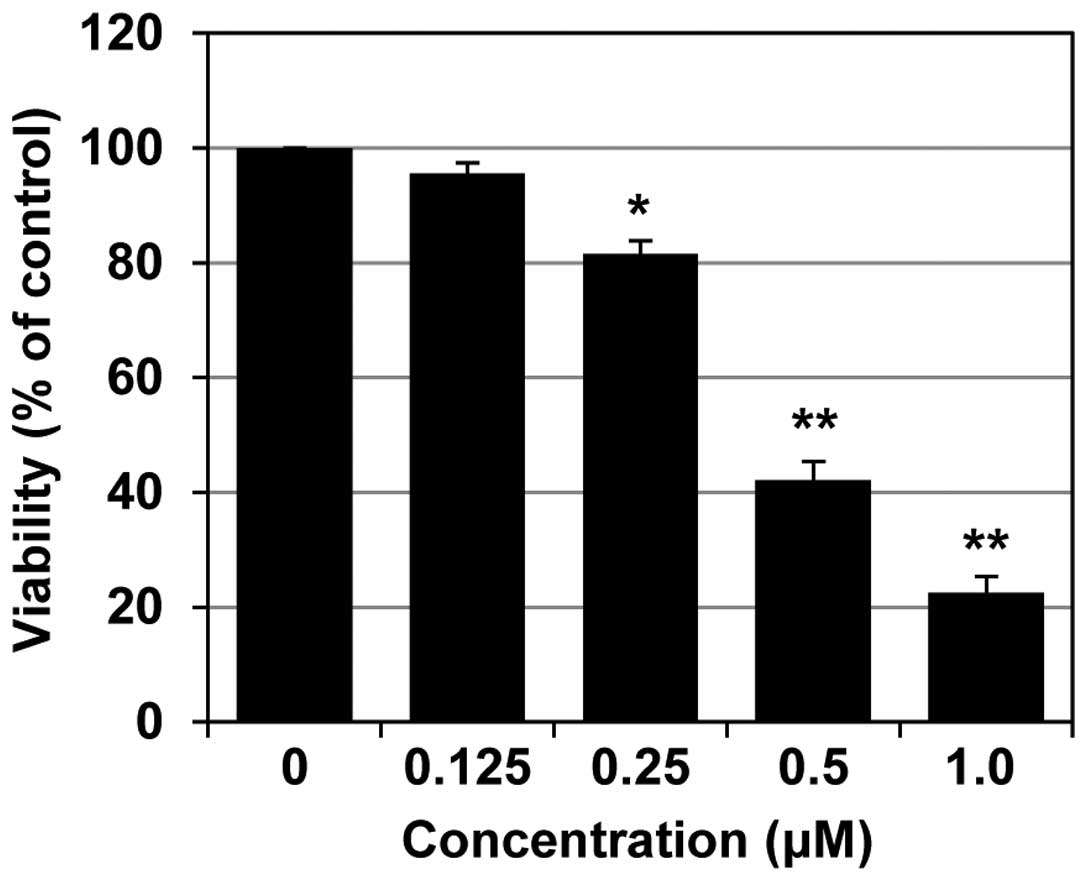
MHY-449 inhibits AGS cell growth
The inhibitory effect of MHY-449 against AGS cell
growth was first evaluated using the MTT assay. As shown in
Fig. 2, treatment of AGS cells with
0.125, 0.25, 0.5 and 1.0 µM MHY-449 for 24 h, significantly
reduced the cell viability at the two higher concentrations. The
half-maximal inhibitory concentration (IC50) of MHY-449
was ~0.4 µM at 24 h.
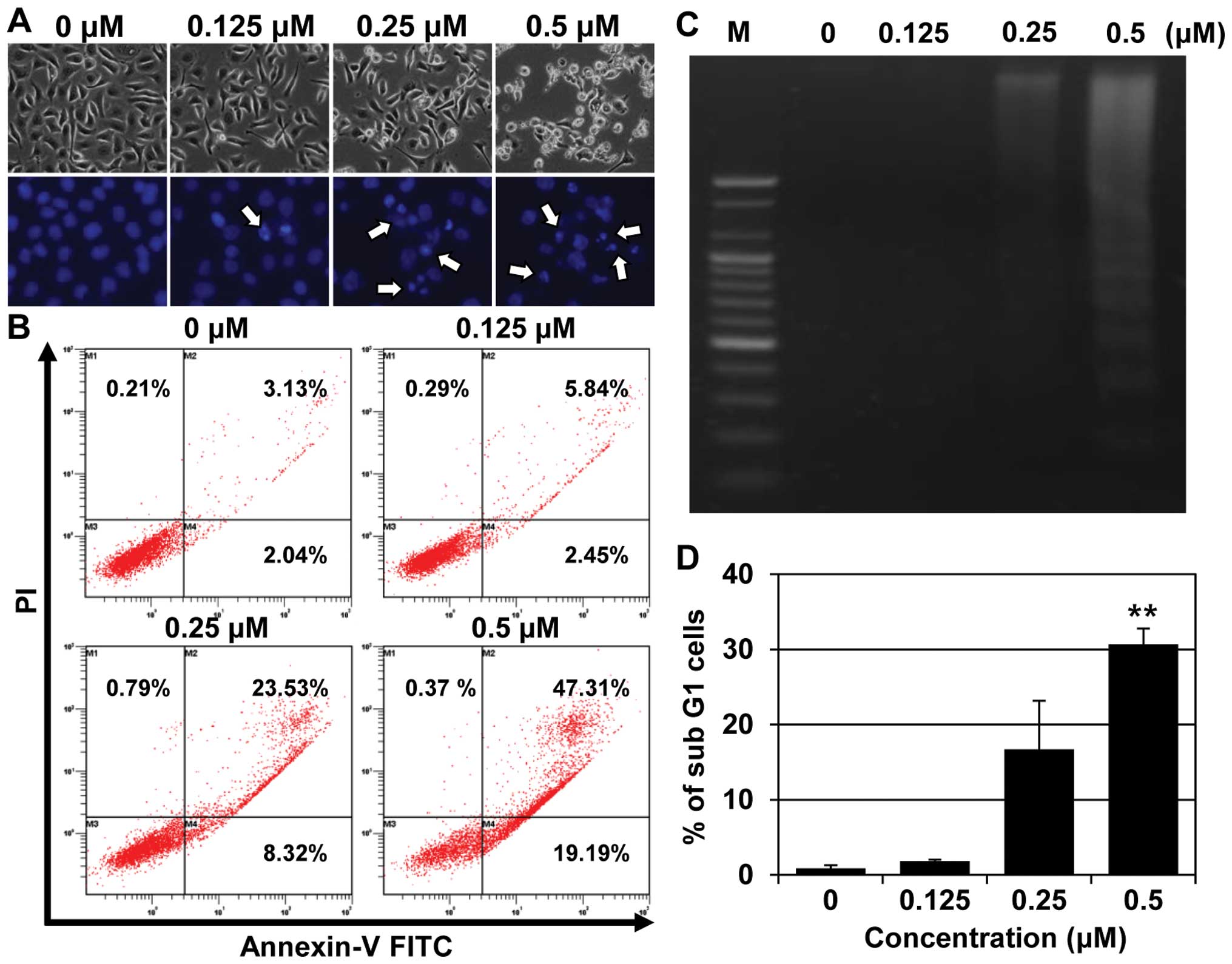
MHY-449 induces apoptosis in AGS
cells
We investigated whether the MHY-449-induced
inhibition of AGS cell growth was apoptosis-mediated. This was
achieved by analyzing the characteristics of cell death, including
nuclear morphological changes, by using flow cytometry and a DNA
fragmentation assay. The AGS cells treated with MHY-449 showed
characteristic features of cell shrinkage and rounding, as well as
the formation of apoptotic bodies compared with the untreated
control (Fig. 3A, upper panel). In
addition, a significant concentration-dependent reduction in cell
count was observed in MHY-449-treated cells.
Consistent with the results of the phase contrast
microscopy, Hoechst 33342 staining, confirmed the induction of
apoptosis in the AGS cells treated with MHY-449 for 24 h (Fig. 3A, lower panel). Control cells
exhibited normal, round nuclear morphology, while the
MHY-449-treated cells exhibited chromatin condensation and
fragmentation of nuclei, which are characteristics of apoptosis
(Fig. 3A, lower panel).
To confirm the MHY-449-induced cell death to be
apoptotic, we performed flow cytometric analysis using Annexin V
and PI staining. As shown in Fig.
3B, the proportion of late apoptotic cells (upper right
quadrant, Annexin V/PI-positive) increased from 3.13 to 47.31%
following 24 h exposure to 0.5 µM MHY-449. The results of
the flow cytometry also indicated that the MHY-449-triggered
apoptosis was concentration-dependent. Another biochemical method
commonly used to detect apoptosis is the fragmentation of genomic
DNA into multiples of 180 base pairs (bp), which produces a
laddering pattern with agarose gel electrophoresis. Treatment of
AGS cells with increasing concentrations of MHY-449 for 24 h
resulted in a concentration-dependent internucleosomal DNA
fragmentation (Fig. 3C). The effect
of MHY-449 on AGS cells was also examined using cell cycle
analysis. The cells treated with MHY-449 for 24 h showed a
concentration-dependent increase in the sub-G1 population (Fig. 3D). These findings suggested that
MHY-449 triggers the apoptosis of AGS cells.
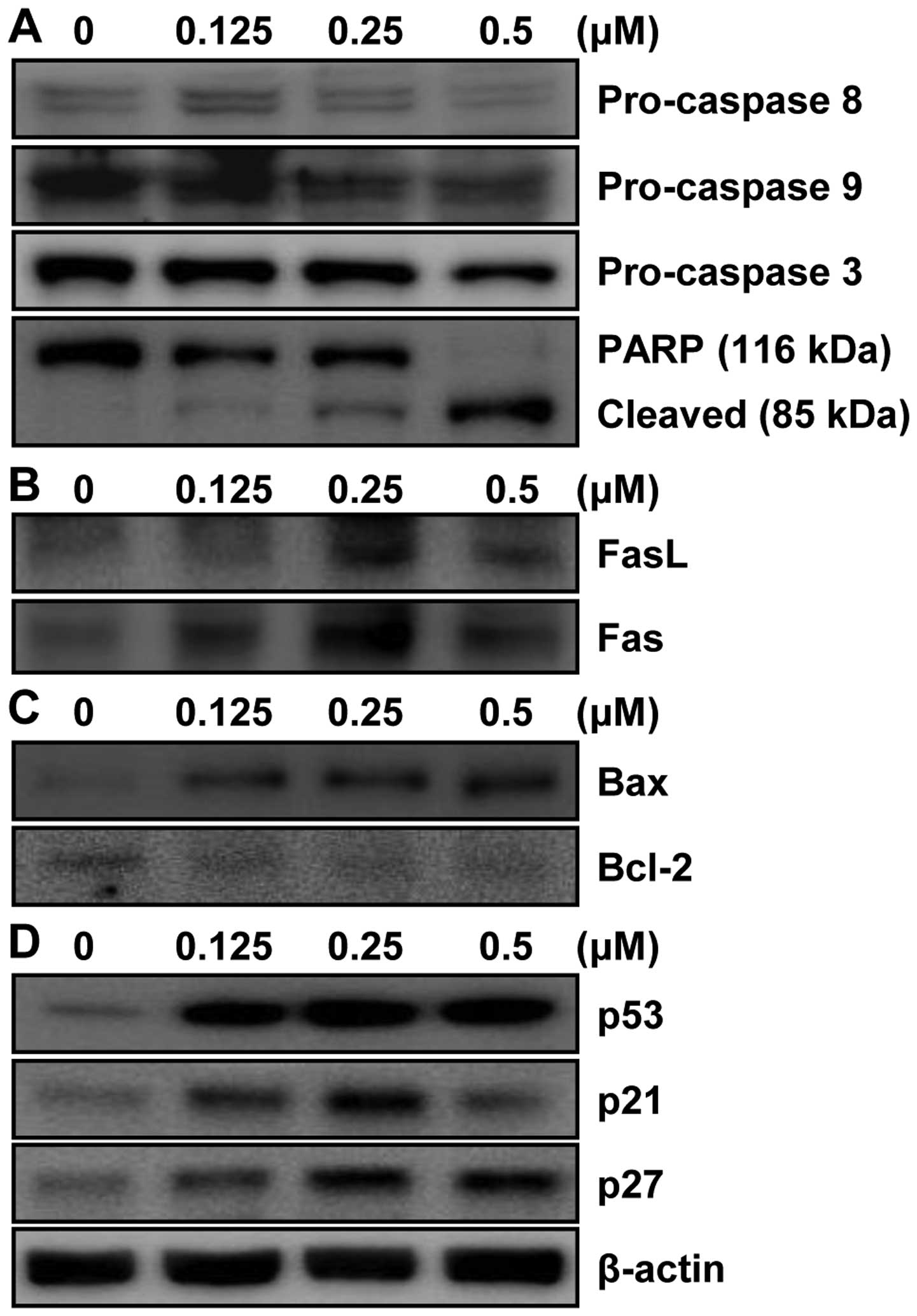
MHY-449 triggers the apoptosis of AGS
cells via extrinsic and intrinsic pathways
We analyzed the modulation of apoptotic markers in
MHY-449-induced apoptotic AGS cells, to investigate the mechanisms
involved. Based on results from Fig.
2, concentration ranges of 0–0.5 µM of MHY-449 were used
to examine its effect on apoptosis-related protein expression. As
shown in Fig. 4A, MHY-449 decreased
the expression of pro-caspase-3, -8, and -9 in a
concentration-dependent manner. Furthermore, MHY-449-treated AGS
cells showed proteolytic degradation of PARP, a molecular marker of
apoptosis (Fig. 4A).
MHY-449 treatment decreased pro-caspase-8, and
therefore, we postulated that its proapoptotic effect may partially
be mediated via the extrinsic pathway. We found that MHY-449
treatment upregulated Fas, a death receptor, and its ligand FasL in
a concentration-dependent manner (Fig.
4B). We also investigated whether the intrinsic pathway
contributed to MHY-449-induced apoptosis. The results showed that
levels of the intrinsic apoptotic Bax and the anti-apoptotic Bcl-2
proteins were upregulated and downregulated, respectively, in AGS
cells (Fig. 4C). Taken together,
these observations suggested that MHY-449 induces intrinsic and
extrinsic apoptotic pathways in AGS cells.
MHY-449 upregulates the expression of
tumor-suppressor proteins in AGS cells
To explore the anticancer mechanisms of MHY-449, we
examined its effects on tumor-suppressor genes. MHY-449 increased
the expression of p53 and one of its downstream target genes
p21WAF1/CIP1 in a concentration-dependent manner
(Fig. 4D). We investigated whether
MHY-449 also modulates p27KIP1, which is
reportedly involved in the regulation of cancer cell apoptosis
(6). As shown in Fig. 4D, MHY-449 induced the expression of
p27 in a concentration-dependent manner.
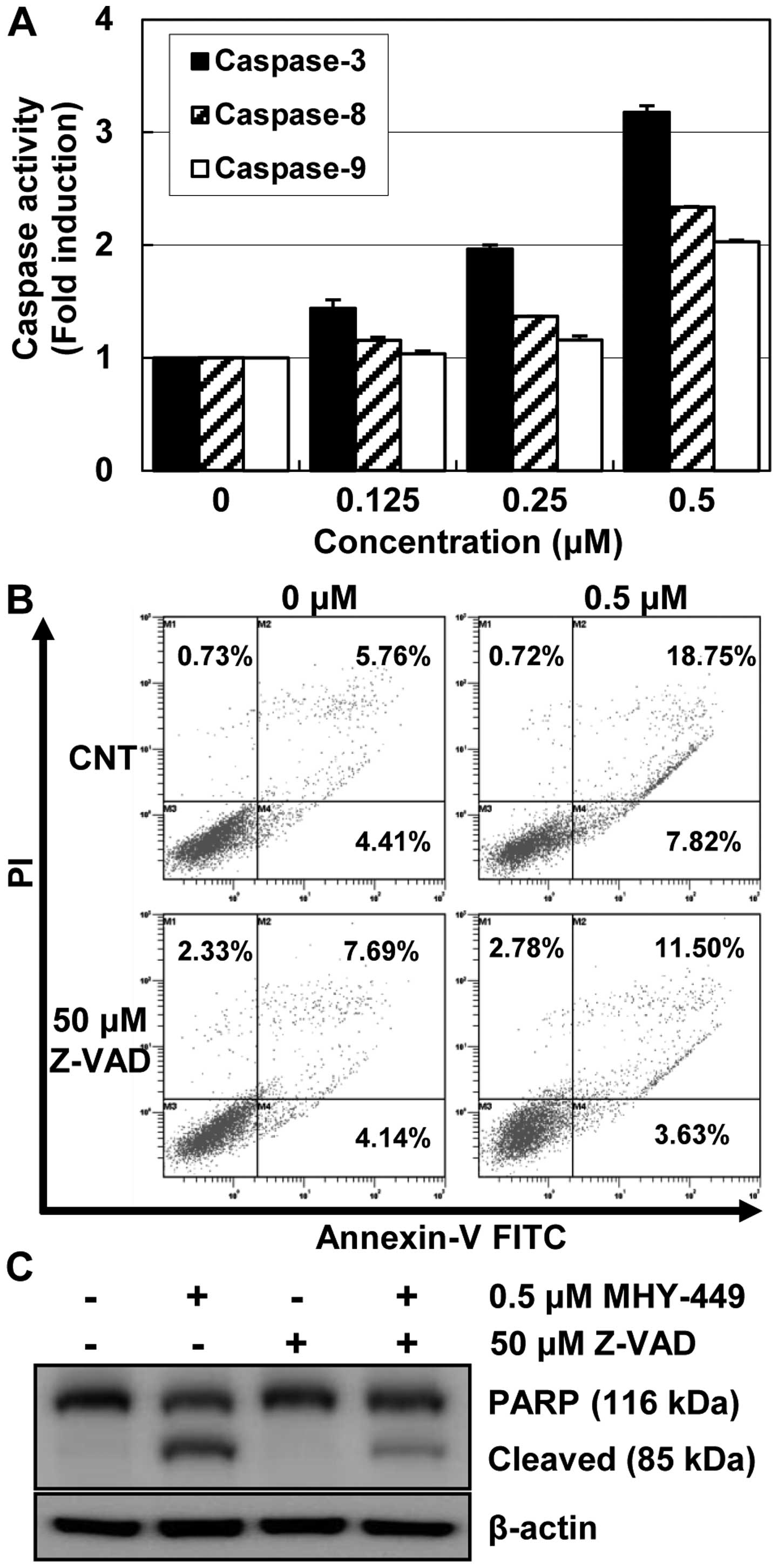
MHY-449 promotes apoptosis via the
activation of caspases
Pro-caspases are the precursors of caspases, which
are executors of the apoptotic process (7). However, a decrease in pro-caspase
levels may not accurately reflect the activation of caspases.
Therefore, we evaluated the effects of MHY-449 on caspase
activation by using specific substrates. AGS cells treated with
MHY-449 showed a concentration-dependent increase in the activities
of caspases (Fig. 5A). In addition,
the results clearly indicated an almost 3-fold increase in
caspase-3 activation, following 24 h treatment with MHY-449. This
process was activated by upstream signaling molecules such as
caspase-8 and -9.
To confirm the involvement of caspase activation in
MHY-449-induced apoptosis, AGS cells were cultured in the presence
and absence of the broad-spectrum caspase inhibitor Z-VAD-FMK and
analyzed using Annexin V-FITC/PI double staining. As shown in
Fig. 5B, the pretreatment of cells
with Z-VAD-FMK inhibited the MHY-449-induced apoptosis albeit not
completely. This result was further confirmed by measuring PARP
cleavage, which is a substrate of the effector caspase-3, under
identical experimental conditions. Consistent with the cell death
measured by flow cytometry, western blot analysis of PARP showed
that pretreatment with Z-VAD-FMK markedly inhibited MHY-449-induced
cleavage of PARP (Fig. 5C). This
result strongly suggested that caspase activation plays an
important role in caspase-induced apoptosis in AGS cells.
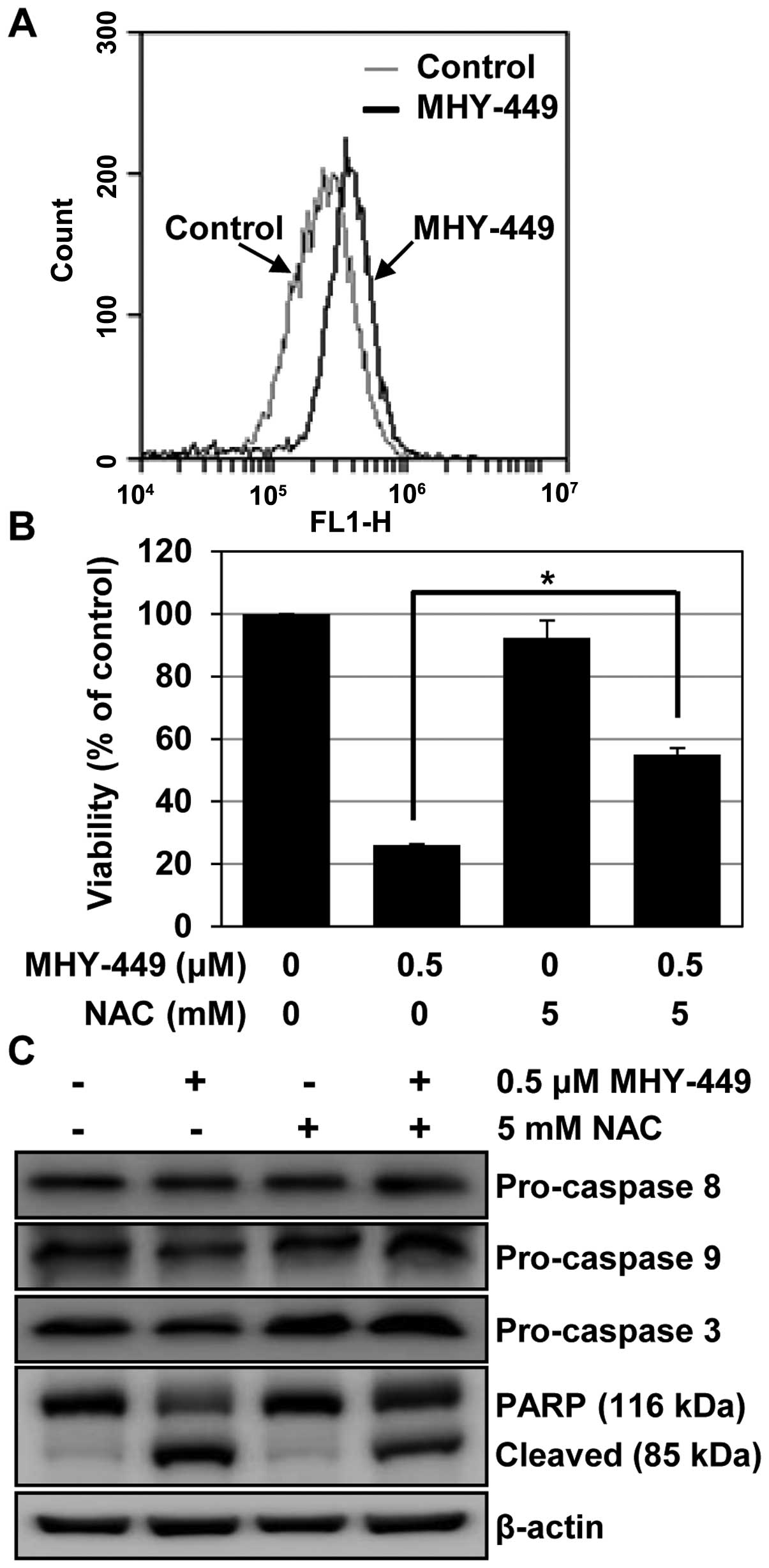
MHY-449 induces apoptosis in AGS cells
via ROS generation
The double-edged sword property of ROS reported to
play a critical role in determining death or survival cell fate
(8,9). Thus, inducing ROS is considered as a
strategy to treat cancer (10).
Therefore, we examined the generation of ROS by MHY-449, to
determine whether this was the mechanism of apoptosis induction.
The intracellular ROS level was quantified using DCF-DA, a
fluorescent probe. We found that MHY-449 induced ROS, and the
maximum intracellular level was observed 8 h after exposure to 0.5
µM (Fig. 6A). To identify
the cause-effect relationship between ROS generation and apoptosis,
we examined the effects of NAC, a widely used thiol-containing ROS
scavenger, on MHY-449-treated cells. As shown in Fig. 6B, the pretreatment of cells with NAC
significantly abrogated cell death in MHY-449-treated cells
(p<0.05), but affected cell viability minimally in untreated
cells. In agreement with these observations, we found that
sequestration of ROS by NAC effectively suppressed the decline in
pro-caspase levels and prevented MHY-449-induced PARP cleavage in
AGS cells (Fig. 6C). Collectively,
these results suggested that an increase in ROS generation is
required for MHY-449-induced apoptosis in AGS cells.
Discussion
A previous study indicated that MHY-449 exhibits
anticancer properties in human prostate and breast cancer cells
(3). These anticancer effects were
mediated by cytotoxicity and induction of cell cycle arrest
(3). Our previous studies have also
shown that MHY-449 induced the apoptosis of colon and prostate
cancer cells by caspase-mediated or extracellular signal-regulated
kinases (ERK)-dependent signaling pathways or both (4,5).
However, the anticancer potential of MHY-449 in other types of
cancer and its detailed mechanism of action has not been fully
elucidated. As mentioned previously, MHY-449 is a novel molecule,
based on psorospermin with a xanthone template and acronycine
derivatives (3).
The molecular mechanism of action of the
benzoacronycine derivative S23906-1 is the most characterized of
several synthetic derivatives. S23906-1 is reported to alkylate
guanines in the minor groove of the DNA helix and subsequently
destabilize base pairing (11,12).
This destabilization causes helix opening, which appears to be
associated with its cytotoxic activity (11,12).
In addition, despite its promising in vivo activity, the
mechanism of the anticancer cell cytotoxicity of this acronycine
derivative largely remains unknown (13). It was shown recently that S23906-1
induces cyclin E, inhibits DNA synthesis (14), alters mitochondrial functions,
stimulates ROS production, and activates caspases (15). The structural similarity between
MHY-449 and S23906-1 led to the hypothesis that their underlying
anticancer mechanisms may also be similar and mediated via
DNA-intercalation and alkylation, cell cycle perturbation or
apoptosis. Of note, our recent findings showed the anticancer
activity of MHY-449 (4). In
addition, we showed that MHY-449-induced apoptosis was mediated
through downregulation of the Akt/Forkhead-Box Class O (FoxO) 1 and
activation of ERK (4). Those
results provided a possibility that the anticancer property of
MHY-449 is not dependent on DNA alkylating or stability. Therefore,
it is postulated that other mechanisms may mediate the apoptotic
effect.
The present study provides insight into the mode of
action of MHY-449 and finds several important points: i) MHY-449
significantly inhibits GC cell growth; ii) MHY-449 induces
apoptosis; iii) caspase activation appears to be one of the major
mechanisms of MHY-449-triggered apoptosis; iv) MHY-449 modulates
the expression of genes involved in the extrinsic and intrinsic
apoptotic pathways; and v) MHY-449-induced apoptosis is
ROS-dependent, as shown by the ability of MHY-449 to generate ROS,
and that NAC-mediated quenching of ROS abrogated the
MHY-449-induced apoptosis.
We found that MHY-449 inhibited GC cell growth in a
concentration-dependent manner. This result is in agreement with
those of recent studies showing that MHY-449 suppresses cell
proliferation in breast (3),
prostate (3,4) and colonic cancer cells (5). The mechanism by which MHY-449 inhibits
cell growth is not completely understood. However, it has been
shown that MHY-449 upregulates Fas/FasL or Bax or both, and
downregulates Bcl-2 expression, which leads to apoptosis (4,5).
Apoptosis can be induced via activation of the intrinsic or
extrinsic pathways. The intrinsic pathway involves mitochondrial
disruption by pro-apoptotic Bcl-2 family members and by Bax
oligomerization, which consequently releases cytochrome c
(16). We found that the expression
of Bcl-2 decreased whereas that of Bax increased in MHY-449-treated
AGS cells. In addition, we observed that MHY-449 induces the
expression of Fas and FasL, which are the major mediators of
extrinsic apoptosis signaling (17). Therefore, MHY-449 activated major
extrinsic and intrinsic pathways, leading to apoptosis.
The tumor-suppressor p53 is known to induce cell
cycle arrest, apoptosis, or senescence as a protective measure
against cancer (18). As a
transcription factor, p53 regulates multiple target genes, such as
p21WAF1/CIP1, a cyclin-dependent kinase (CDK)
inhibitor (18). This inhibitor
binds and inhibits the cyclin/CDK complexes, thereby effectively
blocking cell cycle progression (18). In addition to its CDK inhibitory
function, p21WAF1/C1P1 also negatively or
positively modulates the apoptosis-inducing function of p53
(19). In our study, MHY-449
increased the expression of p53 and p21WAF1/CIP1
in GC cells in a concentration-dependent manner, suggesting that
MHY-449 has potential cytotoxicity against GC. Previous studies
reported that the upregulation of p27KIP1, an
endogenous CDK inhibitor, inhibits the growth of various cancer
cell models (6,20). We also found that MHY-449 induced
p27KIP1 protein levels. In agreement with this
finding, our previous study showed that MHY-449 caused G2/M phase
arrest in HCT116 colon cancer cells, which was associated with the
upregulation of p21WAF1/CIP1 and
p27KIP1 (5). The
effect of MHY-449 on cell cycle progression in AGS cells remains to
be explored. However, a possible explanation is that the inhibition
of cell growth by MHY-449 may be attributable to the induction of
p21WAF1/CIP1, p27KIP1, or p53
or their combination, in response to MHY-449.
We also found that MHY-449 activates caspases, which
is a pivotal process in apoptosis. This corroborates our previous
studies that MHY-449 activates caspase-3, -8 and -9, which leads to
cleavage of PARP in HCT116 colon and PC-3 prostate cancer cells
(4,5). Apoptosis can occur through
caspase-dependent and -independent mechanisms (21,22).
Therefore, we used a pan-caspase inhibitor Z-VAD-FMK to define the
role of caspase activation in MHY-449-induced cell death. Of note,
50 µM Z-VAD-FMK effectively prevented but did not completely
rescue cells from MHY-449-induced apoptosis. These results suggest
the possibility that mechanisms other than apoptosis may be
responsible for the MHY-449-induced cell death. However, the flow
cytometry data showed clearly that the MHY-449-mediated
caspase-independent cell death was not caused by necrosis. More
studies are required to determine the molecules involved and their
underlying mechanisms of mediating MHY-449-induced
caspase-independent cell death. Therefore, it would also be worth
to investigate the ability of MHY-449 to induce cell death via
pathways other than apoptosis such as autophagy. These potential
alternative pathways may contribute to caspase-independent cell
death.
To the best of our knowledge, the present study is
the first to investigate the ROS-mediated anticancer effects of the
novel dihydrobenzofuro[4,5-b][1,8]naphthyridin-6-one derivative.
Several anticancer agents such as cisplatin, doxorubicin, mitomycin
C, and etoposide reportedly exert their effects at least in part,
via induction of ROS (23). ROS,
which were predominantly produced in the mitochondria, may play a
role as active mediators in the regulation of programmed cell death
when produced in excess. ROS achieve this by inducing mitochondrial
membrane depolarization and release of mitochondrial factors, which
activate caspases and cause nuclear condensation (24). Pretreatment with the antioxidant NAC
significantly abrogated the MHY-449-induced decrease in pro-caspase
levels as well as subsequent cell death, thereby, suggesting that
ROS was involved in the upstream events of caspase activation by
MHY-449 in AGS cells. ROS also modulate apoptosis by controlling
apoptosis-related proteins. For instance, ROS exert a direct role
in the induction of Fas or FasL or both (20,21).
They also regulate the expression of Bcl-2 proteins and eventually
trigger apoptosis (22,23). Although the effect of NAC on the
modulation of those proapoptotic or anti-apoptotic proteins by
MHY-449 have yet to be investigated, it is very likely that the
apoptogenic effect of MHY-449 occurs through generation of ROS and
the ROS-associated apoptotic pathway.
In conclusion, MHY-449 is a potent inducer of
apoptosis in AGS cells. MHY-449 causes apoptosis in AGS cells
primarily through ROS generation and partly through activation of
the caspase cascade. Based on these results, further preclinical
studies using appropriate animal models are required to explore the
potential of MHY-449 as an anticancer agent.
Acknowledgments
The present study was supported by the National
Research Foundation of Korea (NRF) grant, funded by the Korea
Government (MSIP, no. 2009-0083538). We would like to thank the
Aging Tissue Bank for providing research information.
References
|
1
|
Orditura M, Galizia G, Sforza V,
Gambardella V, Fabozzi A, Laterza MM, Andreozzi F, Ventriglia J,
Savastano B, Mabilia A, et al: Treatment of gastric cancer. World J
Gastroenterol. 20:1635–1649. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sun J, Song Y, Wang Z, Chen X, Gao P, Xu
Y, Zhou B and Xu H: Clinical significance of palliative gastrectomy
on the survival of patients with incurable advanced gastric cancer:
A systematic review and meta-analysis. BMC Cancer. 13:5772013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kang JA, Yang Z, Lee JY, De U, Kim TH,
Park JY, Lee HJ, Park YJ, Chun P, Kim HS, et al: Design, synthesis
and anticancer activity of novel
dihydrobenzofuro[4,5-b][1,8]naphthyridin-6-one derivatives. Bioorg
Med Chem Lett. 21:5730–5734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee SH, Kang YJ, Sung B, Kim DH, Lim HS,
Kim HR, Kim SJ, Yoon JH, Moon HR, Chung HY, et al: MHY-449, a novel
dihydrobenzofuro[4,5-b][1,8] naphthyridin-6-one derivative, induces
apoptotic cell death through modulation of Akt/FoxO1 and ERK
signaling in PC3 human prostate cancer cells. Int J Oncol.
44:905–911. 2014.PubMed/NCBI
|
|
5
|
Hwang HJ, Kang YJ, Hossain MA, Kim DH,
Jang JY, Lee SH, Yoon JH, Moon HR, Kim HS, Chung HY, et al: Novel
dihydrobenzofuro[4,5-b][1,8]naphthyridin-6-one derivative, MHY-449,
induces apoptosis and cell cycle arrest in HCT116 human colon
cancer cells. Int J Oncol. 41:2057–2064. 2012.PubMed/NCBI
|
|
6
|
Hiromura K, Pippin JW, Fero ML, Roberts JM
and Shankland SJ: Modulation of apoptosis by the cyclin-dependent
kinase inhibitor p27(Kip1). J Clin Invest. 103:597–604. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kaufmann SH, Lee SH, Meng XW, Loegering
DA, Kottke TJ, Henzing AJ, Ruchaud S, Samejima K and Earnshaw WC:
Apoptosis-associated caspase activation assays. Methods.
44:262–272. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang J and Yi J: Cancer cell killing via
ROS: To increase or decrease, that is the question. Cancer Biol
Ther. 7:1875–1884. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu WS: The signaling mechanism of ROS in
tumor progression. Cancer Metastasis Rev. 25:695–705. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Watson J: Oxidants, antioxidants and the
current incurability of metastatic cancers. Open Biol.
3:1201442013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
David-Cordonnier MH, Laine W, Lansiaux A,
Kouach M, Briand G, Pierré A, Hickman JA and Bailly C: Alkylation
of guanine in DNA by S23906-1, a novel potent antitumor compound
derived from the plant alkaloid acronycine. Biochemistry.
41:9911–9920. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thi Mai HD, Gaslonde T, Michel S,
Tillequin F, Koch M, Bongui JB, Elomri A, Seguin E, Pfeiffer B,
Renard P, et al: Structure-activity relationships and mechanism of
action of antitumor benzo[b]pyrano[3,2-h]acridin-7-one acronycine
analogues. J Med Chem. 46:3072–3082. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guilbaud N, Kraus-Berthier L, Meyer-Losic
F, Malivet V, Chacun C, Jan M, Tillequin F, Michel S, Koch M,
Pfeiffer B, et al: Marked antitumor activity of a new potent
acronycine derivative in orthotopic models of human solid tumors.
Clin Cancer Res. 7:2573–2580. 2001.PubMed/NCBI
|
|
14
|
Léonce S, Pérez V, Lambel S, Peyroulan D,
Tillequin F, Michel S, Koch M, Pfeiffer B, Atassi G, Hickman JA, et
al: Induction of cyclin E and inhibition of DNA synthesis by the
novel acronycine derivative S23906-1 precede the irreversible
arrest of tumor cells in S phase leading to apoptosis. Mol
Pharmacol. 60:1383–1391. 2001.PubMed/NCBI
|
|
15
|
Kluza J, Lansiaux A, Wattez N, Hildebrand
MP, Léonce S, Pierré A, Hickman JA and Bailly C: Induction of
apoptosis in HL-60 leukemia and B16 melanoma cells by the
acronycine derivative S23906-1. Biochem Pharmacol. 63:1443–1452.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Suen DF, Norris KL and Youle RJ:
Mitochondrial dynamics and apoptosis. Genes Dev. 22:1577–1590.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Waring P and Mullbacher A: Cell death
induced by the Fas/Fas ligand pathway and its role in pathology.
Immunol Cell Biol. 77:312–317. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Craig C, Wersto R, Kim M, Ohri E, Li Z,
Katayose D, Lee SJ, Trepel J, Cowan K and Seth P: A recombinant
adenovirus expressing p27Kip1 induces cell cycle arrest and loss of
cyclin-Cdk activity in human breast cancer cells. Oncogene.
14:2283–2289. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tait SW and Green DR: Caspase-independent
cell death: Leaving the set without the final cut. Oncogene.
27:6452–6461. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kolenko VM, Uzzo RG, Bukowski R and Finke
JH: Caspase-dependent and -independent death pathways in cancer
therapy. Apoptosis. 5:17–20. 2000. View Article : Google Scholar
|
|
23
|
Fang J, Nakamura H and Iyer AK:
Tumor-targeted induction of oxystress for cancer therapy. J Drug
Target. 15:475–486. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zamzami N, Marchetti P, Castedo M,
Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B and
Kroemer G: Sequential reduction of mitochondrial transmembrane
potential and generation of reactive oxygen species in early
programmed cell death. J Exp Med. 182:367–377. 1995. View Article : Google Scholar : PubMed/NCBI
|