Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignancies worldwide, particularly in Asia. The current
treatments for HCC patients include surgical resection, local
ethanol injection, transarterial chemoembolization (TACE), liver
transplant, radiation therapy, chemotherapy and target therapy
(1–3). However, more than two-thirds of HCC
patients are not indicated for surgical resection due to large
tumor size, poor hepatic function or metastasis (4). Moreover, HCC seems to be resistant to
most chemotherapies and radiation therapy (2). Therefore, it is urgent to improve the
drug sensitivity of HCC and to develop new strategies to treat HCC
patients.
Most cancer cells prefer to use glycolysis rather
than utilize oxidative phosphorylation (OXPHOS) for glucose
metabolism even in oxygen-rich conditions, this is termed aerobic
glycolysis or the 'Warburg effect' (5). Deregulated cellular energetics was
recently included as one new cancer hallmark (6). This is due to the fast progress in
understanding various molecular mechanisms of the Warburg effect in
cancer cells. These mechanisms include oncogenic activation,
inhibition of tumor-suppressor genes or mitochondrial dysfunction
due to nuclear/mitochondrial DNA mutations (7–9). These
metabolic features facilitate the survival, proliferation and
metastasis of cancer cells (10).
Therefore, targeting energy metabolism in cancer cells has become
an important focus of cancer therapy (11).
Mammalian target of rapamycin (mTOR) is a
serine/threonine kinase which modulates numerous cellular functions
including cell growth, migration and protein translation (12,13).
Highly activated mTOR signaling due to mutations of receptor
tyrosine kinase (RTK), amplification of AKT or loss of PTEN has
been observed in several types of cancer including HCC (14,15).
The p70S6K and eIF4E-binding proteins (4E-BPs), which
can be phosphorylated by the mTOR complex, promote protein
synthesis for cell growth and survival (12,16).
AMP-activated protein kinase (AMPK) is an energy sensor and is
involved in the regulation of mTOR signaling. During energy stress,
AMPK is activated by its upstream liver kinase B1 (LKB1) and
further suppresses mTOR signaling for cellular adaptation in a
stress condition (17,18). LKB1 is thought to be a tumor
suppressor, and genetic loss of LKB1 is a frequent event in several
types of cancer, including HCC (19–22).
The loss of LKB1 expression contributes to the aberrant activation
of mTOR signaling in cancer cells (23). Hence, identification of ways to
reduce mTOR signaling is a therapeutic strategy against cancer.
Biguanide drugs, particularly metformin, are used to
treat type 2 diabetic patients (24). Studies show that the biguanide drugs
reduce the risk of HCC in type 2 diabetes mellitus patients (T2DM)
and thus suggest that biguanide drugs can be used as adjuvant
reagents for the treatment of HCC patients (25–29).
The biguanides were found to inhibit Complex I of the mitochondrial
respiratory chain (29), and to
repress mTOR signaling through AMPK-dependent (30) and -independent pathways (31) in cellular experiments. However, it
is unclear whether alteration of the cellular energy metabolism
affects the sensitivity of HCC cells to biguanide drugs.
In HepG2 HCC cells, mitochondrial inhibitors have
been shown to activate AMPK and repress mTOR signaling, which
downregulates HIF-1α protein expression (32). In the present study, we found that
various HCC cell lines (Mahlavu, SK-HEP-1 and HA22T/VGH) exhibited
resistance to mitochondrial inhibitors and biguanide drugs in the
examined HCC cell lines. The role of energy metabolism in
regulating the sensitivity of HCC cells to mitochondrial inhibitors
and biguanides was further evaluated.
Materials and methods
Reagents and antibodies
Biguanide drugs including metformin hydrochloride
(cat. no. PHR1084, purity >99.9%) and phenformin hydrochloride
(cat. no. P7045, purity >97%), and mitochondrial inhibitors
including oligomycin (cat. no. O4876, purity >90%) and rotenone
(cat. no. R8875, purity >95%) were purchased from Sigma-Aldrich
(St. Louis, MO, USA). Cell culture medium was purchased from Gibco
(Grand Island, NY, USA). Fetal bovine serum (FBS), L-glutamine and
non-essential amino acids were obtained from Biological Industries
(Kibbutz Beit Haemek, Israel). The antibodies against 4E-BP-1,
phospho-4E-BP-1 (Thr70), ACC, phospho-ACC (Ser79), AMPKα,
phospho-AMPKα (Thr172), cyclin D1, LKB1, phospho-LKB1 (Ser428),
Mcl-1, p70S6 kinase, phospho-p70S6 kinase (Thr389), raptor and
phosphor-raptor (Ser792) were purchased from Cell Signaling
Technology (Beverly, MA, USA). Aprotinin, EGTA, FCCP,
Na3VO4, PMSF, D-glucose, D-galactose and the
antibody against α-tubulin were purchased from Sigma-Aldrich.
AICAR-riboside (cat. no. 123040, purity >99.6%) was purchased
from Merck Millipore (West Point, PA, USA).
Cell cultures
Human hepatoma cells (HepG2, Mahlavu, SK-HEP-1 and
HA22T/VGH) were cultured in Dulbecco's modified Eagle's medium
(DMEM) supplemented with 10% FBS, 2 mmol/l L-glutamine, 10 mmol/l
non-essential amino acids, 100 U/ml of penicillin and 0.1 mg/ml of
streptomycin at 37°C in a humidified 5% CO2
incubator.
For changing cellular energy metabolism from
glycolysis to mitochondrial OXPHOS, the hepatoma cells were grown
in medium in which glucose was replaced with galactose according to
a previous study (33). Briefly,
the hepatoma cells were cultured in D-glucose-free DMEM
supplemented with 25 mM D-galactose, 10% FBS, 2 mmol/l L-glutamine,
10 mmol/l non-essential amino acids, 100 μg/ml pyruvate, 100
U/ml of penicillin, and 0.1 mg/ml of streptomycin at 37°C in a
humidified 5% CO2 incubator.
Cell viability analysis
Cell viability was determined using sulforhodamine B
(SRB) assay. The cells (5×103) were seeded on 96-well
plates overnight before each experiment. After treatment with
mitochondrial inhibitors or biguanide drugs for 24 h, the cells
were fixed with 10% ice-cold trichloroacetic acid (TCA)
(Sigma-Aldrich) at 4°C for 1 h, rinsed four times with distilled
water and air dried. The cells were then stained with 0.057% SRB
(Sigma-Aldrich) in 1% acetic acid for 30 min at room temperature.
After rinsing four times with 1% acetic acid and air dried, 50
μl of 10 mM Tris-base (pH 10.5) was added into each well for
30 min. The colorimetric level was read by a microplate reader
(Tecan) at 510 nm.
Western blot analysis
Whole cell extracts were prepared using
radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 50 mM
Tris-HCl, 0.1% SDS, 0.5% sodium deoxycholate, 0.1% Triton X-100)
plus 10 μg/ml aprotinin, 2 mM EGTA, 2 mM
Na3VO4 and 1 mM PMSF. The protein
concentrations were determined using the Bradford assay
(Sigma-Aldrich) and samples were diluted in 5X Laemmli buffer [300
mM Tris-HCl pH 6.8, 10% SDS (w/v), 5%, 2-mercaptoethanol, 25%
glycerol (v/v), 0.1% bromphenol blue (w/v)] and boiled for 5 min.
Proteins (40 μg) were separated by 8–15% SDS-PAGE and
transferred onto polyvinylidene fluoride (PVDF) membranes (Pall
Life Sciences). Non-specific binding sites on the PVDF membranes
were blocked with 5% non-fat milk in TBST (20 mM Tris-HCl, pH 7.6,
137 mM NaCl, 1% Tween-20). Membranes were then hybridized with
primary antibodies overnight at 4°C, followed by incubation with
horseradish peroxidase (HRP)-conjugated secondary antibodies. The
membranes were then developed using Immobilon Western
Chemiluminescence HRP Substrates (Millipore). Images were captured
by a Luminescence/Fluorescence Imaging System (GE Healthcare), and
signal intensities were quantified using Multi Guage image analysis
software (Fujifilm).
OCR and ECAR analyses
The oxygen consumption rate (OCR) and extracellular
acidification rate (ECAR) of the examined cells were determined by
a Seahorse Extracellular Flux XF-24 analyzer (Seahorse Bioscience,
North Billerica, MA, USA) according to the manufacturer's
instructions. Cells (3×104) were seeded in a 24-well
custom-made plate for the XF-24 analyzer. The culture medium was
replaced with sodium carbonate-free DMEM (pH 7.4). Prior to the
assay, the cell plate and sensor cartridge were kept with 1 ml
Seahorse Bioscience XF-24 Calibrant/well in an incubator
maintaining 37°C without CO2 overnight. The basal,
proton-leaked, maximal and non-OXPHOS OCRs were sequentially
measured before and after the injection of 75 μl of
oligomycin (2 μg/ml), FCCP (2 μM) or antimycin A (2
μM), respectively. The program of the Seahorse XF-24
analyzer was set according to the manufacturer's instructions. The
mitochondrial OCR was calculated by subtracting the residual rate
after injection of antimycin A. The OCR and ECAR were expressed in
pmol/min and mpH/min, respectively, and normalized to the examined
cell number.
Determination of intracellular ATP
content
Cells (2×105) were seeded on 6-well
plates overnight before each experiment. After treatment with
vehicle (DMSO) or the mitochondrial inhibitors for 3 h, the cells
were collected and the intracellular ATP content was measured by
the ATP bioluminescence assay kit (Roche Applied Science) according
to the manufacturer's instructions.
Determination of lactate production
Cells (2×105) were seeded on 6-well
plates overnight, and the culture medium was then replaced by fresh
culture medium and cells were incubated for an additional 6 h.
Lactate levels in the culture medium were measured by the Lactate
Assay kit (Trinity Biotech) according to the manufacturer's
instructions.
Statistical analysis
Data are presented as the mean ± SEM. Statistical
differences between the control and treated groups were determined
using Student's t-test, and results were considered to be a
statistically significant at P<0.05.
Results
Differential effects of the mitochondrial
inhibitors and biguanide drugs on AMPK-mTOR signaling and cell
survival among the HCC cell lines
To examine the effect of the mitochondrial
inhibitors and biguanide drugs on mTOR signaling in different HCC
cell lines, four HCC cell lines (HepG2, Mahlavu, SK-HEP-1 and
HA22T/VGH cells) were treated with rotenone (a Complex I inhibitor)
or oligomycin (a Complex V inhibitor). We found that the repression
of mTOR signaling (indicated by the phosphorylation levels of
p70S6K and 4E-BP-1) by the mitochondrial inhibitors was
only observed in the HepG2 cells (Fig.
1A, lanes 1–3) but not in the Mahlavu, SK-HEP-1 and HA22T/VGH
cells (Fig. 1A, lanes 4–12).
Similarly, the activation of AMPK by the mitochondrial inhibitors
was only observed in the HepG2 cells (Fig. 1B, lanes 1–3), but not in the other
three HCC cell lines (Fig. 1B,
lanes 4–12). Consistently, the activation of AMPK and inhibition of
mTOR signaling by metformin and phenformin were only observed in
the HepG2 cells but not in the other three HCC cell lines (Fig. 1C and D).
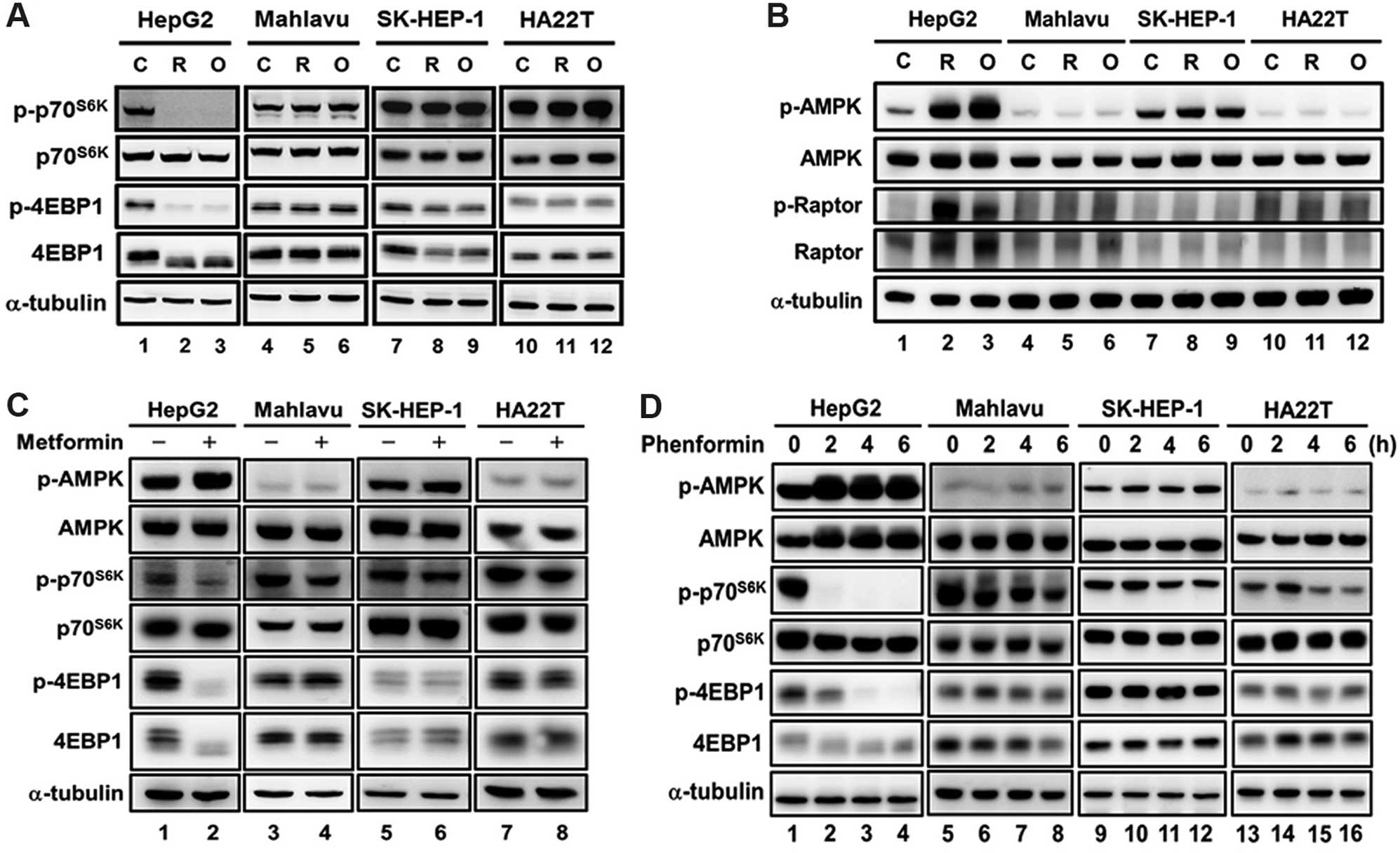 | Figure 1Effect of mitochondrial inhibitors
and biguanides on the AMPK-mTOR signaling in HCC cells. HCC cell
lines (HepG2, Mahlavu, SK-HEP-1 and HA22T/VGH) were treated with (A
and B) mitochondrial inhibitors [0.2 μM rotenone (R) and 1
μg/ml oligomycin (O)] for 3 h or (C and D) biguanide drugs
(5 mM metformin for 9 h and 0.1 mM phenformin for 0, 2, 4 and 6 h),
and the mTOR signaling was analyzed using western blotting with
antibodies specific for p-p70S6K, p70S6K,
p-4E-BP-1, 4E-BP-1, p-AMPK, AMPK, p-Raptor and Raptor. α-tubulin
was used as an internal control for protein loading. Each western
blot is a representative result obtained from three independent
experiments. AMPK, AMP-activated protein kinase. |
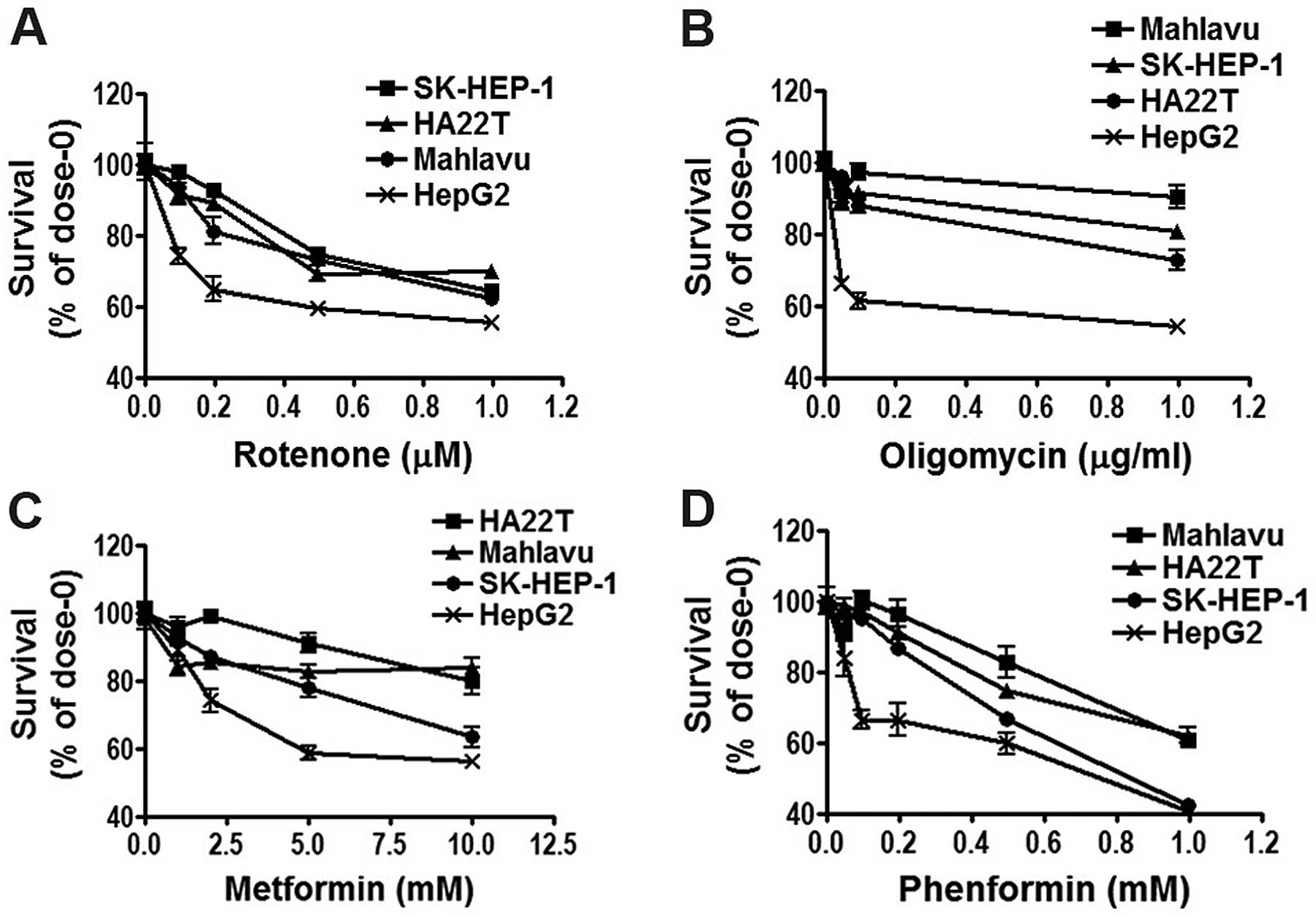
Due to the importance of mTOR signaling in the
growth and survival of cancer cells, we next examined the effect of
the mitochondria inhibitors and biguanide drugs on cell survival in
the four HCC cell lines. The results revealed that the Mahlavu,
SK-HEP-1 and HA22T/VGH cell lines were more resistance to the
mitochondrial inhibitors (rotenone and oligomycin) (Fig. 2A and B) and biguanide drugs
(metformin and phenformin) (Fig. 2C and
D). These results indicated that Mahlavu, SK-HEP-1 and
HA22T/VGH cells were more resistant to mitochondrial inhibitors and
biguanide drugs as compared with the HepG2 cells.
HCC cells with higher glycolysis activity
are more resistant to mitochondrial inhibitors and biguanide
drugs
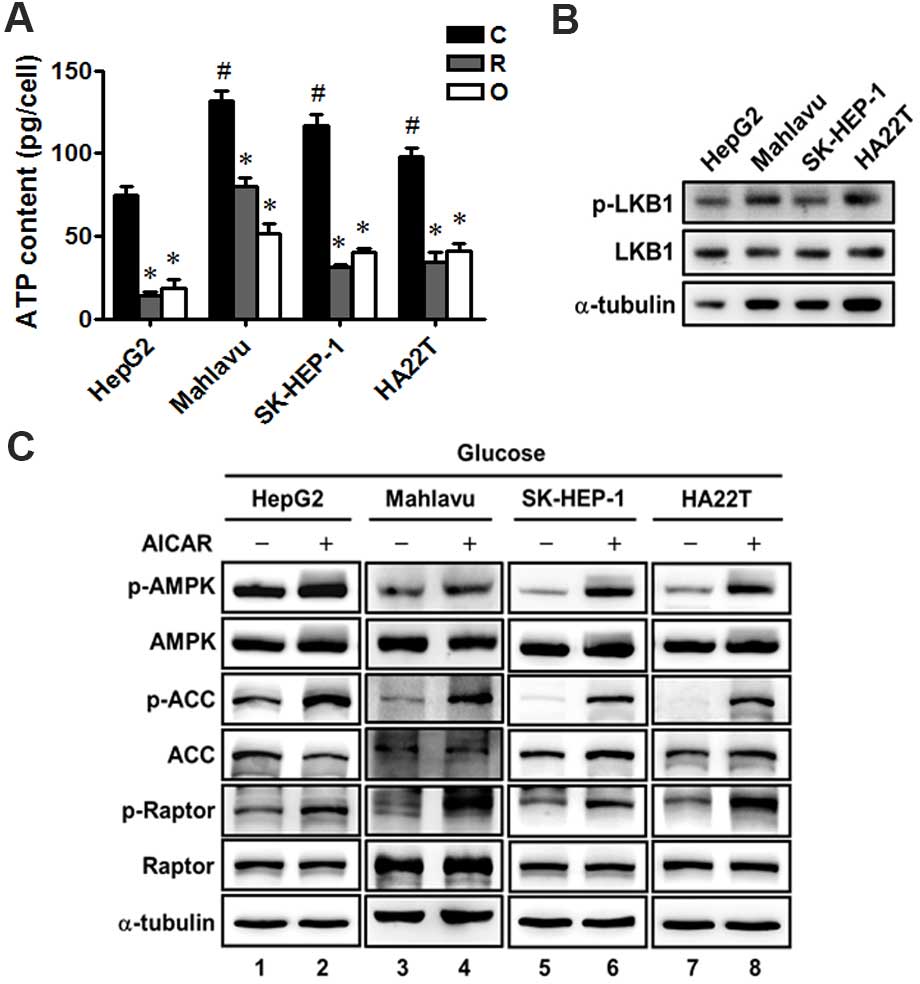
We further determined the intracellular ATP levels
and found that the ATP level in the HepG2 cells was lower than
levels in the Mahlavu, SK-HEP-1 and HA22T/VGH cells (Fig. 3A), and the mitochondrial inhibitors
markedly decreased the intracellular ATP to a similar extent in the
four HCC cell lines (Fig. 3A).
Moreover, all the four HCC cells were found to express LKB1 protein
and similar levels of phosphorylated LKB1, indicating that LKB1 was
not deficient in the four HCC cell lines (Fig. 3B). In addition, we treated the four
HCC cell lines with an AMPK activator AICAR, and found that AICAR
significantly increased the phosphorylation of AMPK and the AMPK
downstream proteins, such as acetyl-CoA carboxylase (ACC) and
Raptor, in the four HCC cell lines (Fig. 3C). These results revealed that there
were no significant differences in the ATP changes in response to
mitochondrial inhibitors, LKB1 protein expression and AMPK function
among the four HCC cells. Therefore, these factors did not play a
major role in the resistance of the Mahlavu, SK-HEP-1 and HA22T/VGH
cells to mitochondrial inhibitors and biguanide.
We next determined the energy metabolism in the HCC
cell lines using the Seahorse Extracellular Flux XF-24 analyzer. We
found that mitochondrial OCR including the basal, proton-leaked,
ATP-link and maximal OCRs in the HepG2 cells were higher than those
in the Mahlavu, SK-HEP-1 and HA22T/VGH cells (Fig. 4A). Moreover, the ratio of OCR/ECAR
(Fig. 4B) in HepG2 cells was higher
than that in the Mahlavu, SK-HEP-1 and HA22T/VGH cells. In
addition, the lactate production rate in HepG2 cells was lower than
that in the Mahlavu, SK-HEP-1 and HA22T/VGH cells (Fig. 4C). These results indicated that
HepG2 cells have higher OXPHOS activity; while Mahlavu, SK-HEP-1
and HA22T/VGH cells have higher glycolysis activity. These results
suggest that HCC cells with high glycolysis activity are more
resistant to mitochondrial inhibitors and biguanide drugs.
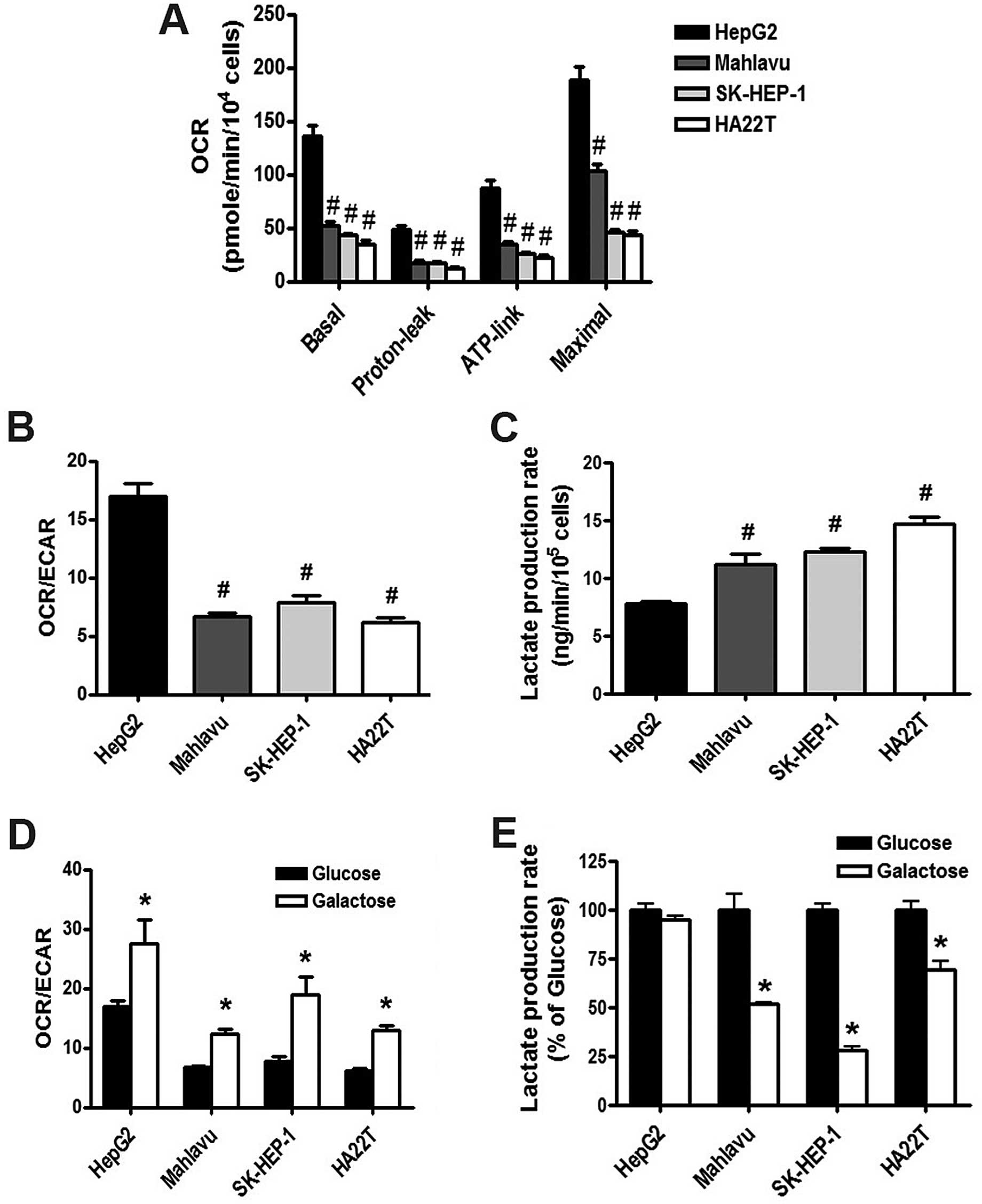 | Figure 4Energy metabolism characteristics of
the HCC cells; replacement of glucose with galactose changes the
energy metabolism from glycolysis to mitochondrial respiration. HCC
cell lines (HepG2, Mahlavu, SK-HEP-1 and HA22T/VGH) were cultured
in DMEM with 25 mM glucose or galactose, and the parameters of
energy metabolism including (A) basal OCR, proton leak, ATP-linked
OCR, maximal OCR and (B and D) OCR/ECAR in the HepG2, Mahlavu,
SK-HEP-1 and HA22T/VGH cell lines were analyzed using Seahorse
Extracellular Flux XF analyzer. (C and E) The lactate production
was detected using a lactate analysis kit (#P<0.05 as
compared with HepG2 cells; *P<0.05 as compared with
the glucose group). HCC, hepatocellular carcinoma, OCR, oxygen
consumption rate; ECAR, extracellular acidification rate. |
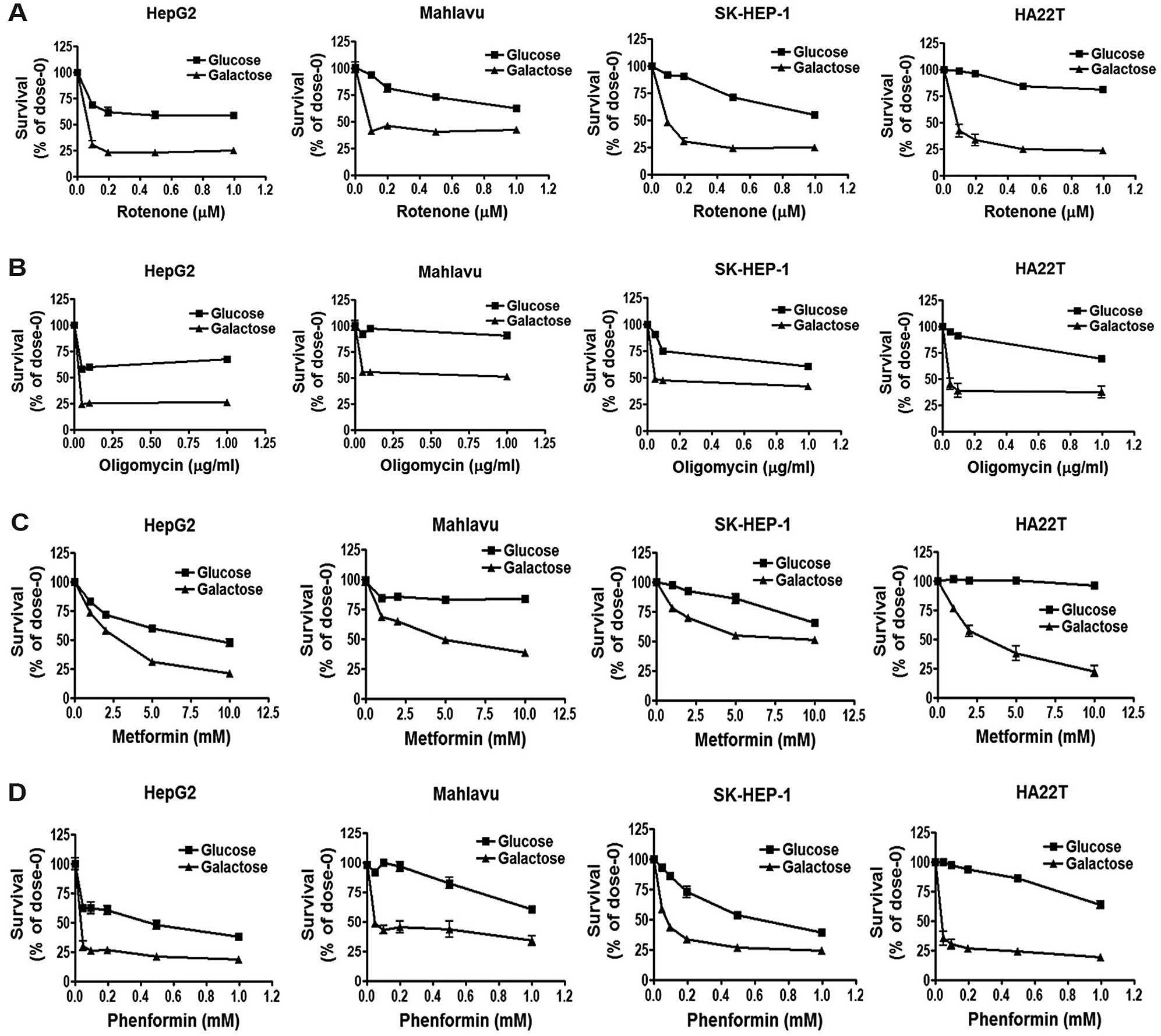
Increased OCR/ECAR by galactose medium
enhances the cell sensitivity to mitochondrial inhibitors and
biguanide drugs
To examine whether energy metabolism determines the
response to mitochondrial inhibitors, we replaced glucose with
galactose in the culture medium. The results revealed that the
galactose medium increased the OCR/ECAR ratio of the HCC cells
(Fig. 4D), and decreased the
lactate production rate (Fig. 4E)
as compared with parameters in the HCC cells grown in the glucose
medium. Parental HepG2 cells have higher OXPHOS activity; as a
result, the lactate production rate did not show a significant
change between the glucose and galactose medium in the HepG2 cells
(Fig. 4E). These results indicate
that the galactose medium altered the energy metabolism to enhance
mitochondrial respiration in the HCC cells.
We further evaluated whether the change in energy
metabolism alters the effect of mitochondrial inhibitors and
biguanides on AMPK-mTOR signaling and cell survival. We found that
the extent of AMPK activation (Fig. 5A,
C and D), repression of mTOR signaling (Fig. 5B–D) and the cytotoxicity (Fig. 6) were significantly increased in the
HCC cells when they were grown in the galactose medium containing
mitochondrial inhibitors and biguanides. These results together
suggest that the increase in the OCR/ECAR ratio enhances the cell
sensitivity to mitochondrial inhibitors and biguanide drugs.
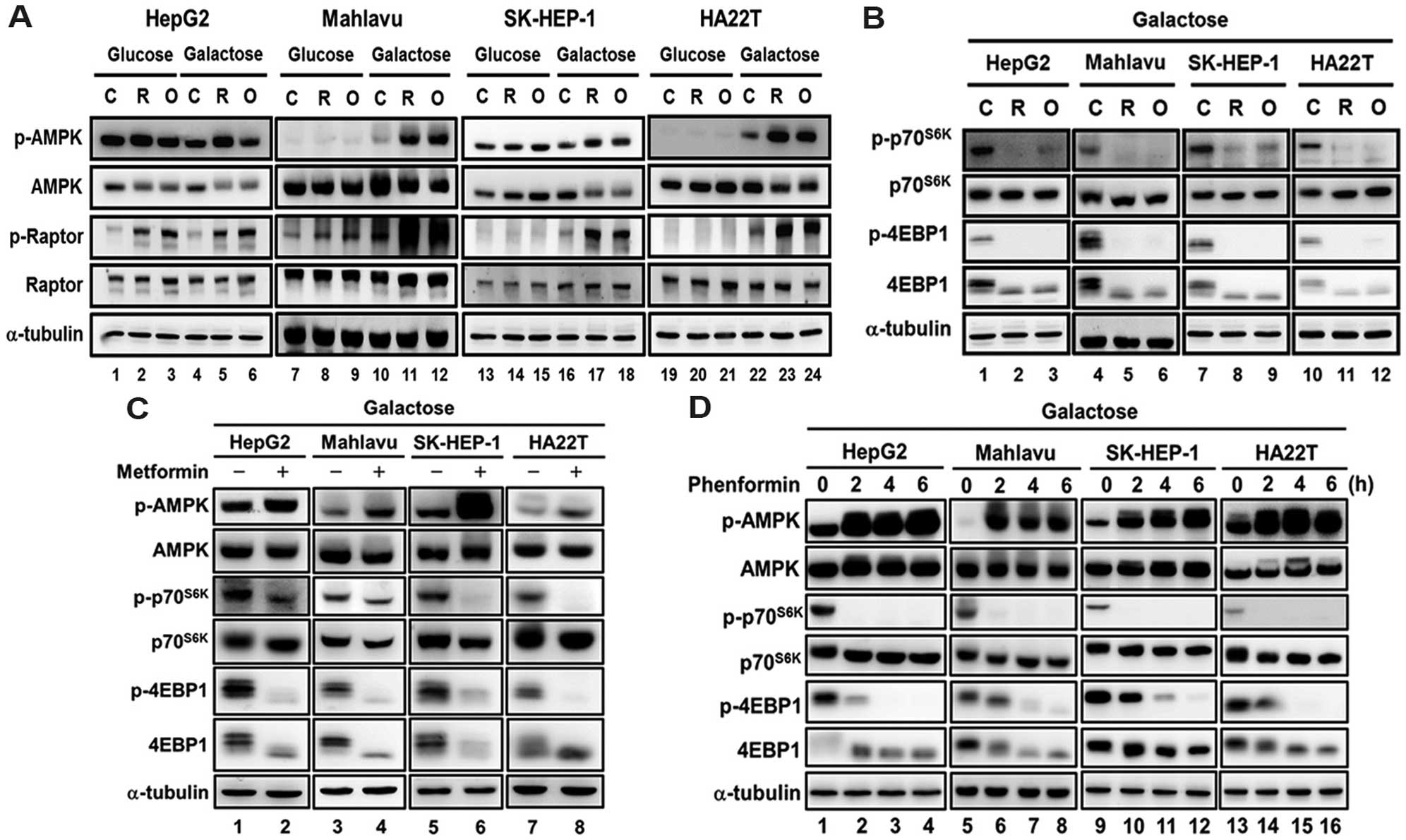 | Figure 5Replacing glucose with galactose
increases the response to mitochondrial inhibitors and biguanides
in regards to AMPK-mTOR signaling in HCC cells. HCC cell lines
(HepG2, Mahlavu, SK-HEP-1 and HA22T/VGH) were cultured in DMEM with
25 mM glucose or galactose, and then treated with (A and B)
mitochondrial inhibitors (0.2 μM rotenone (R) and 1
μg/ml oligomycin (O)] for 3 h or (C and D) biguanide drugs
(5 mM metformin for 9 h, and 0.1 mM phenformin for 0, 2, 4 and 6
h), and the AMPK and mTOR signaling was analyzed using western
blotting with antibodies specific for p-p70S6K,
p70S6K, p-4E-BP-1, 4E-BP-1, p-AMPK, AMPK, p-Raptor and
Raptor. α-tubulin was used as an internal control for protein
loading. Each western blot shown is a representative result
obtained from three independent experiments. AMPK, AMP-activated
protein kinase; HCC, hepatocellular carcinoma. |
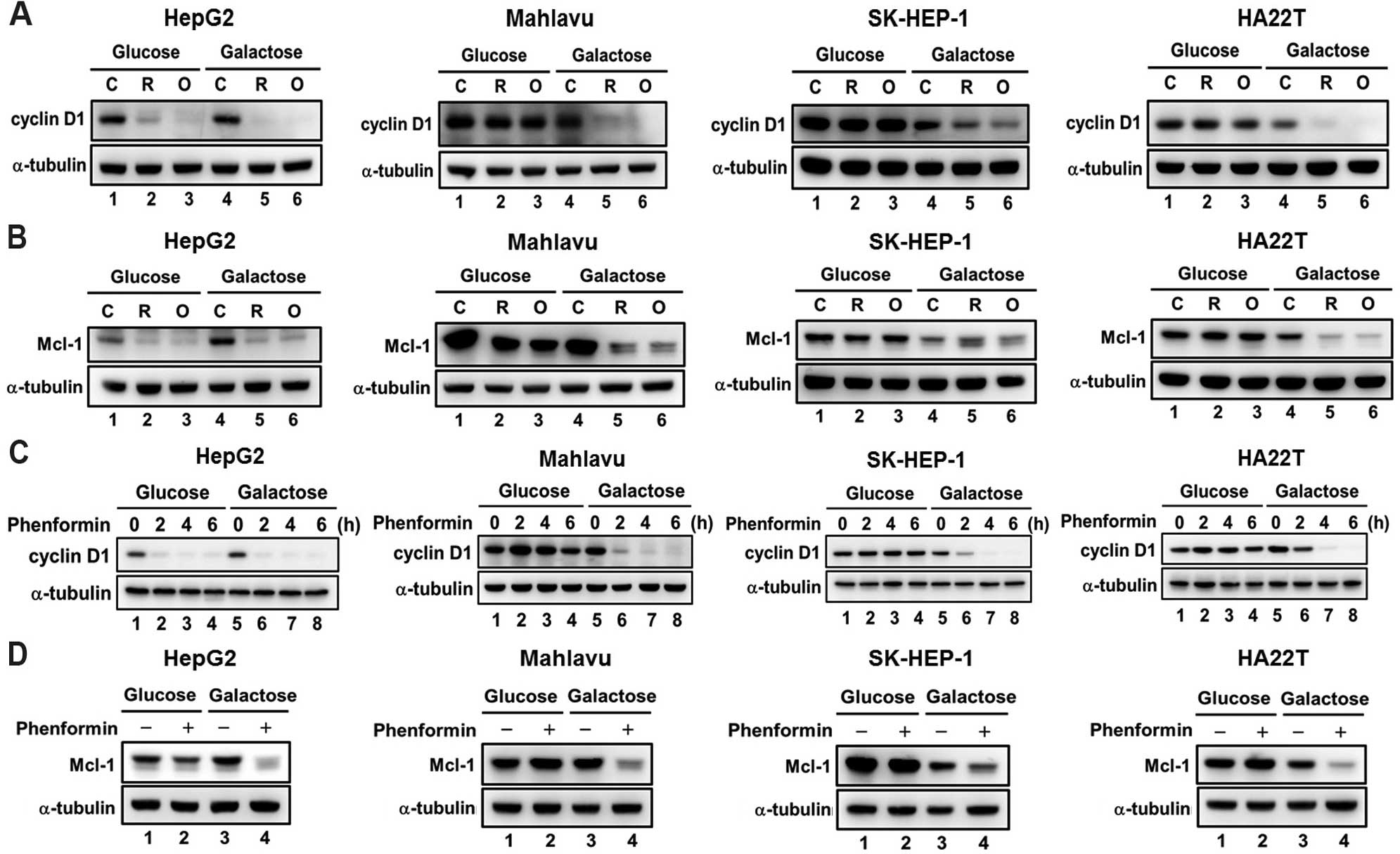
Decreases in cyclin D1 and Mcl-1 are
associated with the cytotoxicity in response to mitochondrial
inhibitors and biguanide drugs
Due to the importance of cyclin D1 and Mcl-1 for
cell cycle progression and cell survival in cancer cells, we
further investigated the effect of mitochondrial inhibitors and
phenformin on the levels of cyclin D1 and Mcl-1 in the HCC cells
grown in glucose or galactose medium. We found that downregulation
of cyclin D1 and Mcl-1 expression by mitochondrial inhibitors
(Fig. 7A and B, lanes 1–3) and
phenformin (Fig. 7C, lane 1–4;
Fig. 7D, lane 1 and 2) were
observed only in HepG2 cells but not in the Mahlavu, SK-HEP-1 and
HA22T/VGH cell lines cultured in glucose medium. In the galactose
medium, mitochondrial inhibitors and phenformin significantly
decreased the protein expression of cyclin D1 and Mcl-1 in the four
HCC cell lines (Fig. 7A and B,
lanes 4–6; Fig. 7C, lanes 5–8;
Fig. 7D, lanes 3 and 4). These
results suggest that the downregulation of cyclin D1 and Mcl-1 are
associated with the cytotoxicity induced by mitochondrial
inhibitors and biguanide drugs.
Discussion
In the present study, we demonstrated that changing
energy metabolism from glycolysis to mitochondrial respiration
enhances the sensitivity of HCC cell lines to mitochondrial
inhibitors and biguanide drugs. HepG2 cells had a higher
mitochondrial respiration rate (OCR), lower glycolysis (ECAR) and
lower lactate production rate, and were found to be more sensitive
to mitochondrial inhibitors, suggesting that the energy metabolism
of the cells is more dependent on mitochondrial OXPHOS. In
contrast, the Mahlavu, SK-HEP-1 and HA22T/VGH cell lines had higher
glycolysis, lower mitochondrial respiration and were more resistant
to mitochondrial inhibitors, indicating that their metabolism was
more dependent on glycolysis. Thus, high glycolysis activity may
render HCC cells more resistant to biguanide drugs. In addition, we
found that a change in energy metabolism from glycolysis to OXPHOS
sensitized the HCC cells to mitochondrial inhibitors and biguanide
drugs. These results suggest that energy metabolism plays an
important role in regulating the sensitivity of HCC cells to
biguanide drugs.
The biguanide drugs were reported to inhibit
mitochondrial respiratory chain Complex I (29) and activate AMPK (33). Accumulating evidence indicates that
the biguanide drugs reduce the risk of breast cancer (34,35),
HCC (25–27) and pancreatic cancer (36,37),
and thus these drugs have been proposed as adjuvant reagents for
cancer therapy (28,29). Recent studies have also revealed
that OXPHOS plays an essential role in tumor initiation and
metastasis (38,39). These findings suggest that the
OXPHOS can be therapeutically targeted in cancers. Our present
results revealed that HCC cells (for example, HepG2 cells)
exhibiting higher OXPHOS activity were more sensitive to biguanide
drugs, which further suggest that the biguanide drugs may be
potential agents to inhibit cancer metastasis and progression.
Cancer cells usually exhibit various energy
metabolism characteristics and have different responses to
therapeutic agents. To evaluate whether cellular energy metabolism
regulates the sensitivity to biguanide drugs of HCC cells, we used
mitochondrial inhibitors to reduce the intracellular ATP content in
the HCC cell lines (Fig. 3A), and
found that HepG2 cells were more sensitive than the Mahlavu,
SK-HEP-1 and HA22T/VGH cells. Moreover, the activation of AMPK was
detected only in HepG2 cells rather than the other HCC cell lines
(Mahlavu, SK-HEP-1 and HA22T/VGH cells) (Fig. 1B). The lower response of AMPK to the
mitochondrial inhibitors in the Mahlavu, SK-HEP-1 and HA22T/VGH
cells was associated with their higher glycolysis and lower
mitochondrial respiration rate. Moreover, we altered the cellular
energy metabolism from glycolysis to OXPHOS in the Mahlavu,
SK-HEP-1 and HA22T/VGH cells, and found that AMPK was activated by
these treatments (Fig. 5A, C and
D). These results indicated that the cell sensitivity and the
activation of AMPK in response to mitochondrial inhibitors or
biguanide drugs are associated with cellular energy metabolism.
Further investigation is warranted to ascertain whether the change
in energy metabolism from glycolysis to OXPHOS alters the
sensitivity in other types of cancer such as breast and pancreatic
cancers.
In the present study, galactose was used for
altering the cellular metabolism from glycolysis to mitochondrial
OXPHOS and this enhanced cell sensitivity to the biguanides. This
combination of galactose with biguanide drugs may provide a
beneficial strategy for TACE in HCC patients. Recent progress in
understanding the mechanisms for exhibiting the Warburg effect in
cancer cells has identified several proteins, including lactate
dehydrogenase A (LDHA), pyruvate kinase M2 isoform (PKM2) and
pyruvate dehydrogenase kinases (PDKs) (40–42).
Many proliferating cells or cancer cells express a high level of
LDHA resulting in glycolytic flux toward lactate production even in
the presence of oxygen, and inhibition of LDHA was found to induce
the oxygen consumption rate and inhibit tumor growth (41,43).
In addition, it has been reported that PKM2 is highly expressed in
several types of cancer including breast, cervical, colon,
gastrointestinal, hepatoma and lung cancer (44), and knockdown of PKM2 in several
types of cancer was found to lead to increased OCR, decreased
lactate production and inhibition of tumor growth (45). Moreover, PDKs are upregulated in
cancer cells due to high expression of HIF-1α, and activated PDKs
inhibit pyruvate dehydrogenases, which convert pyruvate into
acetyl-CoA for tricarboxylic acid cycle, and thereby switch the
energy metabolism from OXPHOS to aerobic glycolysis (40). Therefore, further investigation is
warranted to ascertain whether inhibition of these enzymes (LDHA,
PKM2 or PDKs) alters the energy metabolism in HCC cells and
sensitizes HCC cells to mitochondrial inhibitors or biguanide
drugs.
In conclusion, we found that HCC cells which exhibit
higher glycolysis and lower mitochondrial respiration are more
resistant to mitochondrial inhibitors and biguanide drugs. Our
findings also provide evidence to suggest that altering the energy
metabolism from aerobic glycolysis to OXPHOS enhances the cell
sensitivity to biguanides. These findings suggest that a change in
energy metabolism from glycolysis to OXPHOS enhances the effect of
biguanide drugs in HCC therapy.
Acknowledgments
We thank Ms. Shu-Hui Li for the excellent technical
assistance. This study was partly supported by a grant from the
Center of Excellence for Cancer Research at Taipei Veterans General
Hospital, the Ministry of Health and Welfare, Executive Yuan
(MOHW103-TD-B-111-02; MOHW104-TDU-B-211-124-001), a grant from the
Ministry of Education, Aim for the Top University Plan, and the
grant MOST101-2320-B-010-068-MY3 from the Ministry of Science and
Technology, Taiwan.
Abbreviations:
|
4E-BPs
|
eIF4E-binding proteins
|
|
AMPK
|
AMP-activated protein kinase
|
|
FCCP
|
carbonyl cyanide 4-(trifluoromethoxy)
phenylhydrazone
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
ECAR
|
extracellular acidification rate
|
|
HA22T
|
HA22T/VGH
|
|
HCC
|
hepatocellular carcinoma
|
|
HIF-1α
|
hypoxia inducible factor-1α
|
|
LDHA
|
lactate dehydrogenase A
|
|
LKB1
|
liver kinase B1
|
|
mTOR
|
mammalian target of rapamycin
|
|
OCR
|
oxygen consumption rate
|
|
OXPHOS
|
oxidative phosphorylation
|
|
PDK
|
pyruvate dehydrogenase kinase
|
|
PI
|
propidium iodide
|
|
PKM2
|
pyruvate kinase M2
|
|
RTK
|
receptor tyrosine kinase
|
|
SRB
|
sulforhodamine B
|
|
T2DM
|
type 2 diabetes mellitus
|
|
TACE
|
transarterial chemoembolization
|
References
|
1
|
Belghiti J and Kianmanesh R: Surgical
treatment of hepatocellular carcinoma. HPB Oxf. 7:42–49. 2005.
View Article : Google Scholar
|
|
2
|
Marin JJ, Castaño B, Martinez-Becerra P,
Rosales R and Monte MJ: Chemotherapy in the treatment of primary
liver tumours. Cancer Ther. 6:711–728. 2008.
|
|
3
|
Wang Y and Shen Y: Unresectable
hepatocellular carcinoma treated with transarterial
chemoembolization: Clinical data from a single teaching hospital.
Int J Clin Exp Med. 6:367–371. 2013.PubMed/NCBI
|
|
4
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen Z, Lu W, Garcia-Prieto C and Huang P:
The Warburg effect and its cancer therapeutic implications. J
Bioenerg Biomembr. 39:267–274. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hsu CC, Lee HC and Wei YH: Mitochondrial
DNA alterations and mitochondrial dysfunction in the progression of
hepatocellular carcinoma. World J Gastroenterol. 19:8880–8886.
2013. View Article : Google Scholar :
|
|
9
|
Lee HC and Wei YH: Mitochondrial DNA
instability and metabolic shift in human cancers. Int J Mol Sci.
10:674–701. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
DeBerardinis RJ, Lum JJ, Hatzivassiliou G
and Thompson CB: The biology of cancer: Metabolic reprogramming
fuels cell growth and proliferation. Cell Metab. 7:11–20. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tennant DA, Durán RV and Gottlieb E:
Targeting metabolic transformation for cancer therapy. Nat Rev
Cancer. 10:267–277. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma XM and Blenis J: Molecular mechanisms
of mTOR-mediated translational control. Nat Rev Mol Cell Biol.
10:307–318. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shimobayashi M and Hall MN: Making new
contacts: The mTOR network in metabolism and signalling crosstalk.
Nat Rev Mol Cell Biol. 15:155–162. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Engelman JA, Luo J and Cantley LC: The
evolution of phosphatidylinositol 3-kinases as regulators of growth
and metabolism. Nat Rev Genet. 7:606–619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yuan TL and Cantley LC: PI3K pathway
alterations in cancer: Variations on a theme. Oncogene.
27:5497–5510. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gwinn DM, Shackelford DB, Egan DF,
Mihaylova MM, Mery A, Vasquez DS, Turk BE and Shaw RJ: AMPK
phosphorylation of raptor mediates a metabolic checkpoint. Mol
Cell. 30:214–226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hardie DG, Ross FA and Hawley SA: AMPK: A
nutrient and energy sensor that maintains energy homeostasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fernandez P, Carretero J, Medina PP,
Jimenez AI, Rodriguez-Perales S, Paz MF, Cigudosa JC, Esteller M,
Lombardia L, Morente M, et al: Distinctive gene expression of human
lung adenocarcinomas carrying LKB1 mutations. Oncogene.
23:5084–5091. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang YH, Chen ZK, Huang KT, Li P, He B,
Guo X, Zhong JQ, Zhang QY, Shi HQ, Song QT, et al: Decreased
expression of LKB1 correlates with poor prognosis in hepatocellular
carcinoma patients undergoing hepatectomy. Asian Pac J Cancer Prev.
14:1985–1988. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sahin F, Maitra A, Argani P, Sato N,
Maehara N, Montgomery E, Goggins M, Hruban RH and Su GH: Loss of
Stk11/Lkb1 expression in pancreatic and biliary neoplasms. Mod
Pathol. 16:686–691. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shen Z, Wen XF, Lan F, Shen ZZ and Shao
ZM: The tumor suppressor gene LKB1 is associated with prognosis in
human breast carcinoma. Clin Cancer Res. 8:2085–2090.
2002.PubMed/NCBI
|
|
23
|
Hezel AF and Bardeesy N: LKB1; linking
cell structure and tumor suppression. Oncogene. 27:6908–6919. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bailey CJ and Turner RC: Metformin. N Engl
J Med. 334:574–579. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen HP, Shieh JJ, Chang CC, Chen TT, Lin
JT, Wu MS, Lin JH and Wu CY: Metformin decreases hepatocellular
carcinoma risk in a dose-dependent manner: Population-based and in
vitro studies. Gut. 62:606–615. 2013. View Article : Google Scholar
|
|
26
|
Hassan MM, Curley SA, Li D, Kaseb A,
Davila M, Abdalla EK, Javle M, Moghazy DM, Lozano RD, Abbruzzese
JL, et al: Association of diabetes duration and diabetes treatment
with the risk of hepatocellular carcinoma. Cancer. 116:1938–1946.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Libby G, Donnelly LA, Donnan PT, Alessi
DR, Morris AD and Evans JM: New users of metformin are at low risk
of incident cancer: A cohort study among people with type 2
diabetes. Diabetes Care. 32:1620–1625. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Petrushev B, Tomuleasa C, Soritau O, Aldea
M, Pop T, Susman S, Kacso G, Berindan I, Irimie A and Cristea V:
Metformin plus PIAF combination chemotherapy for hepatocellular
carcinoma. Exp Oncol. 34:17–24. 2012.PubMed/NCBI
|
|
29
|
Pollak M: Potential applications for
biguanides in oncology. J Clin Invest. 123:3693–3700. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dowling RJ, Zakikhani M, Fantus IG, Pollak
M and Sonenberg N: Metformin inhibits mammalian target of
rapamycin-dependent translation initiation in breast cancer cells.
Cancer Res. 67:10804–10812. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kalender A, Selvaraj A, Kim SY, Gulati P,
Brûlé S, Viollet B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, et
al: Metformin, independent of AMPK, inhibits mTORC1 in a rag
GTPase-dependent manner. Cell Metab. 11:390–401. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hsu CC, Wang CH, Wu LC, Hsia CY, Chi CW,
Yin PH, Chang CJ, Sung MT, Wei YH, Lu SH, et al: Mitochondrial
dysfunction represses HIF-1α protein synthesis through AMPK
activation in human hepatoma HepG2 cells. Biochim Biophys Acta.
1830:4743–4751. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stephenne X, Foretz M, Taleux N, van der
Zon GC, Sokal E, Hue L, Viollet B and Guigas B: Metformin activates
AMP-activated protein kinase in primary human hepatocytes by
decreasing cellular energy status. Diabetologia. 54:3101–3110.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bodmer M, Meier C, Krähenbühl S, Jick SS
and Meier CR: Long-term metformin use is associated with decreased
risk of breast cancer. Diabetes Care. 33:1304–1308. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chlebowski RT, McTiernan A,
Wactawski-Wende J, Manson JE, Aragaki AK, Rohan T, Ipp E, Kaklamani
VG, Vitolins M, Wallace R, et al: Diabetes, metformin, and breast
cancer in postmenopausal women. J Clin Oncol. 30:2844–2852. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bodmer M, Becker C, Meier C, Jick SS and
Meier CR: Use of antidiabetic agents and the risk of pancreatic
cancer: A case-control analysis. Am J Gastroenterol. 107:620–626.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li D, Yeung SC, Hassan MM, Konopleva M and
Abbruzzese JL: Antidiabetic therapies affect risk of pancreatic
cancer. Gastroenterology. 137:482–488. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
LeBleu VS, O'Connell JT, Gonzalez Herrera
KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A,
Domingos Chinen LT, Rocha RM, et al: PGC-1α mediates mitochondrial
biogenesis and oxidative phosphorylation in cancer cells to promote
metastasis. Nat Cell Biol. 16:992–1003. 1001–1015. 2014. View Article : Google Scholar
|
|
39
|
Tan AS, Baty JW, Dong LF, Bezawork-Geleta
A, Endaya B, Goodwin J, Bajzikova M, Kovarova J, Peterka M, Yan B,
et al: Mitochondrial genome acquisition restores respiratory
function and tumorigenic potential of cancer cells without
mitochondrial DNA. Cell Metab. 21:81–94. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cairns RA, Harris IS and Mak TW:
Regulation of cancer cell metabolism. Nat Rev Cancer. 11:85–95.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lunt SY and Vander Heiden MG: Aerobic
glycolysis: Meeting the metabolic requirements of cell
proliferation. Annu Rev Cell Dev Biol. 27:441–464. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Vander Heiden MG, Christofk HR, Schuman E,
Subtelny AO, Sharfi H, Harlow EE, Xian J and Cantley LC:
Identification of small molecule inhibitors of pyruvate kinase M2.
Biochem Pharmacol. 79:1118–1124. 2010. View Article : Google Scholar :
|
|
43
|
Le A, Cooper CR, Gouw AM, Dinavahi R,
Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL and Dang
CV: Inhibition of lactate dehydrogenase A induces oxidative stress
and inhibits tumor progression. Proc Natl Acad Sci USA.
107:2037–2042. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wong N, De Melo J and Tang D: PKM2, a
central point of regulation in cancer metabolism. Int J Cell Biol.
2013:2425132013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Christofk HR, Vander Heiden MG, Harris MH,
Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL and
Cantley LC: The M2 splice isoform of pyruvate kinase is important
for cancer metabolism and tumour growth. Nature. 452:230–233. 2008.
View Article : Google Scholar : PubMed/NCBI
|