Introduction
Breast cancer is classified into three basic
subtypes including ER-positive subtype, HER2 amplified
subtype and triple-negative breast cancer subtype (1). Approximately 75% breast cancers are
classified into ER-positive subtype and need to be given endocrine
treatment (2). Tamoxifen, a
selective estrogen receptor modulator, has been used as a basic
endocrine drug for the past four decades (3). However, the effect of tamoxifen is
limited by occurrences of de novo and acquired resistance
(4). In previous studies (2,5),
tamoxifen resistance has been attributed to many different reasons
including: loss of ERα, signal pathways crosstalk, deregulation of
coregulators, as well as altered expression of microRNAs. Even so,
there is still a great need to explore the underlying mechanism of
tamoxifen resistance.
The signal pathway of mitogen activated protein
kinases (MAPKs) has been shown to be involved in cancer development
and progression (6). Studies have
also supported that the MAPKs are implicated in tamoxifen
resistance. Activated ERK has been shown to phosphorylate ERα,
leading to ligand-independent activation of ER signal and tamoxifen
resistance (7). High level of
phosphorylated p38 has been observed in tamoxifen treated breast
cancer specimens and activation of p-38 has been proven to induce
tamoxifen resistance through activation of ER (8). Studies have also shown that AP1, a
downstream target of ERK and JNK, promotes transcription in
tamoxifen-treated breast cancer (9). In addition, JNK has been shown to
contribute to tamoxifen pharmacological action through mediating
apoptosis (10).
The MAPK phosphatases (MKPs), which can specifically
bind and dephosphorylate MAPKs, are negative regulators of MAPKs
(11). MKP1 is the first identified
member of MKPs family and thus frequently studied. MKP1 has been
shown to dephosphorylate all three members of MAPKs, particularly
p38 and JNK (12), in inflammatory
response and metabolic homeostasis (13,14).
MKP1 also plays an important role in cancer (15). Li et al (16) reported that MKP1 is a
transcriptional target of tumor suppressor p53, and thus involved
in the negative regulation of cell proliferation. Several studies
have also shown that MKP1 can attenuate chemotherapy-induced
apoptosis in cancers through dephosphorylation of JNK (17–20).
Herein, we show that MKP1 expression was increased
in tamoxifen resistant MCF7 cells. When using MKP1 siRNA knocked
down MKP1 in tamoxifen resistant MCF7 cells, tamoxifen-mediated
cell death increased. On the contrary, overexpression of MKP1 in
MCF7 cells attenuated tamoxifen-mediated cell death. In addition,
change of MKP1 expression was correlated with tamoxifen-induced
activation of JNK in MCF7 and tamoxifen resistant MCF7 cells.
Importantly, using JNK inhibitor SP600125, we showed that the
inhibition effect of MKP1 on tamoxifen-mediated cell death is
through blocking JNK activation.
Material and methods
Reagents and antibodies
Tissue culture media and materials were purchased
from HyClone. Charcoal stripped fetal bovine serum (cs-FBS) and
fetal bovine serum (FBS) were from PAA (A-15–119). 4-Hydroxy
tamoxifen (TAM) was from Sigma and freshly dissolved in ethanol at
stock concentration 10 mmol/l. The TurboFect transfection reagent
was from Thermo Scientific (R0531). Cell Counting Kit-8 (CCK-8) was
from Dojin Laboratories of Kumamoto (CK04-01). Cell apoptosis
detection kit was from Beyotime (C1052). The cDNA synthesis and
real-time PCR reagents were purchased from Takara (D6110A). MKP1
siRNA pool (UGGAGCAUAUCGUGCCGAA and AAGAUAUGCUCGACGCCUU); and
scrambled siRNA (CGTACGCGGAATACTTCGA) were purchased from
Dharmacon. The empty vector pCMV-Tag-2A and mkp1-pCMV-Tag-2A
plasmids were kind gifts from Professor Lianfeng Zhang. The
SP600125 (sc-200635), triptolide (sc-200122) and dexamethasone were
from Santa Cruz Biotechnology. The following antibodies were used
in the present study: anti-MKP-1 (sc-370; Santa Cruz); anti-MAPKs
(cst-9926; Cell Signaling Technology), anti-p-MAPKs (cst-9938),
anti p-c-jun (cst-9164) and anti-PARP (cst-9532); anti-β-actin
(A-5316; Sigma). Caspase 3/7 activity assay kit was from
Beyotime.
Cell lines and treatment
Human breast cancer MCF7 cell line was purchased
from the Cell Bank of Chinese Academy of Science. Tamoxifen
resistant MCF7 cell line LCC2 (21)
was a kind gift from Professor Xinyi Chen. MCF7 cells were
maintained in Dulbecco's modified Eagle's medium (DMEM) containing
10% FBS supplemented with 2 µM L-glutamine, 50 U/ml
penicillin and 50 µg/ml streptomycin. LCC2 cells were
maintained in phenol red free DMEM containing 10% cs-FBS
supplemented with 2 µM L-glutamine, 5,050 U/ml penicillin
and 50 µg/ml streptomycin. High-dose tamoxifen resistant
MCF7 cells (MCF7-TR) were derived from LCC2 cells based on the
following strategy: LCC2 cells were treated with gradually
increased concentration of TAM (1–5 µM) for two months, and
then were kept in medium containing 5 µM TAM for 6 months.
All of the cells were incubated at 37°C in 5% CO2. Prior
to experiments, all of the cells were cultured in phenol red free
DMEM containing 5% cs-FBS for one week.
MTT analysis
The cell viability was determined by CCK-8 assay
according to the manufacturer's instruction. Cells were incubated
with 10 µl CCK-8 for 3 h at 37°C. The absorbance was
measured at 450 nm. The viability of cells was calculated as:
(OD450 nm of sample/OD450 nm of control) × 100%. All experiments
were performed in triplicate.
Flow cytometric analysis of cell
apoptosis
Equal numbers of cells (2×106) were
seeded into 60 mm dishes and incubated at 37°C in 5%
CO2. Overnight, cells were treated with different doses
of TAM for 48 h. Both floating and adherent cells were collected by
trypsin/EDTA-free, washed in cold PBS and centrifuged at 1,000 rpm
for 5 min. The supernatant was discarded. Binding buffer (500
µl) was added to the deposit which was resuspended, then 5
µl Annexin V-FITC and 5 µl propidium iodide were
added. After incubated at RT for 15 min, cells were analyzed by
FACScan flow cytometer. For each sample, 10,000 events were
analyzed.
cDNA synthesis and quantitative RT-PCR
analysis
Cell monolayers were lysed and total RNA was
isolated using TRIzol reagent according to the manufacturer's
protocol. Total RNA (100 ng) was reverse transcribed using Takara
PrimeScript First Strand cDNA Synthesis kit. Real-time PCR was
performed using the Takara SYBR Premix Ex Taq kit. Primers
used to amplify MKP1 and 18S were as follows: MKP1F,
5′-gggacgcgcggtgaag-3′ and MKP1R, 5′-gatcttgt-gcggttttttgtgg-3′;
18SF, 5′-tctcaaagattcgccatgc-3′ and 18SR,
5′-tcaccaacggagaccttg-3′.
MKP1 silencing with small interfering
RNA
MCF7-TR cells were transiently transfected with MKP1
siRNA and scrambled siRNA into 6-well plates with TurboFect reagent
according to the manufacturer's instruction. Forty-eight hours
after transfection, cells were used for the experiments. Cells
transfected with scrambled siRNA were used as control.
Establishment of MKP1 overexpressing MCF7
cells
MCF7 cells were transfected with either the empty
vector pCMV-Tag-2A plasmid or mkp1-pCMV-Tag-2A plasmid using
TurboFect reagent according to the instruction of TurboFect
reagent. Transfected cells were selected with 400 µg/ml G418
(Sigma) for 4 weeks. Single clones were picked up for establishing
stable clones. MKP-1 expression was determined by western
blotting.
Western blot analysis
Western blot analysis in cultured cells was carried
out as previously reported (22).
Briefly, cells were washed in PBS and scraped, and whole-cell
lysates were prepared and centrifuged at 12,000 rpm for 20 min at
4°C. Equal amounts of total protein (40 µg) were loaded on
10% SDS-PAGE gels and then transferred onto a polyvinylidene
fluoride membrane with a Semi-Dry Transfer Cell. Membranes were
blocked in TBS-Tween (0.1%)/5% milk and were then hybridized with
primary antibody overnight at 4°C. Membranes were incubated with
the secondary antibody for 1 h at RT. Resulting bands were
visualized using enhanced chemiluminescence reagents (Millipore).
Images were scanned and quantified by ChemiDoc™ MP System
(Bio-Rad).
Determination of caspase 3/7
activity
Caspase 3/7 activity was detected using the Caspase
3/7 Activity Assay kit following the protocol provided by the
manufacturer. Cells were harvested and then incubated with
Ac-DEVD-Pna for 2 h at 37°C. The absorbance was measured at 405 nm.
The caspase 3/7 activity of the cells was calculated as: (OD405 nm
of sample/OD405 nm of control) × 100%. All experiments were
performed in triplicate.
Statistical analysis
Statistical analysis was carried out with
Statistical Package for the Social Sciences version 19.0 (SPSS,
Inc.). Statistical significance (p<0.05) between experimental
and control groups was calculated by LSD-t test. All data are
expressed as the means ± SD for the number of experiments.
Results
MKP1 expression was upregulated in
tamoxifen resistant breast cancer MCF7 cells
The LCC2 cells were derived from MCF7 cells and have
been used as a tamoxifen resistant cell model for several years
(21). In the present study, we
established a high-dose tamoxifen resistant MCF7 cell line
(MCF7-TR) from LCC2 cells by treating LCC2 with 5 µM TAM for
>6 months. In order to verify resistant character of the MCF7-TR
cells, we treated parental MCF7, LCC2 and MCF7-TR cells with
estrogen (E2), tamoxifen (TAM) and control medium for 7 days. The
cell proliferation was detected by MTT test and cell growth curves
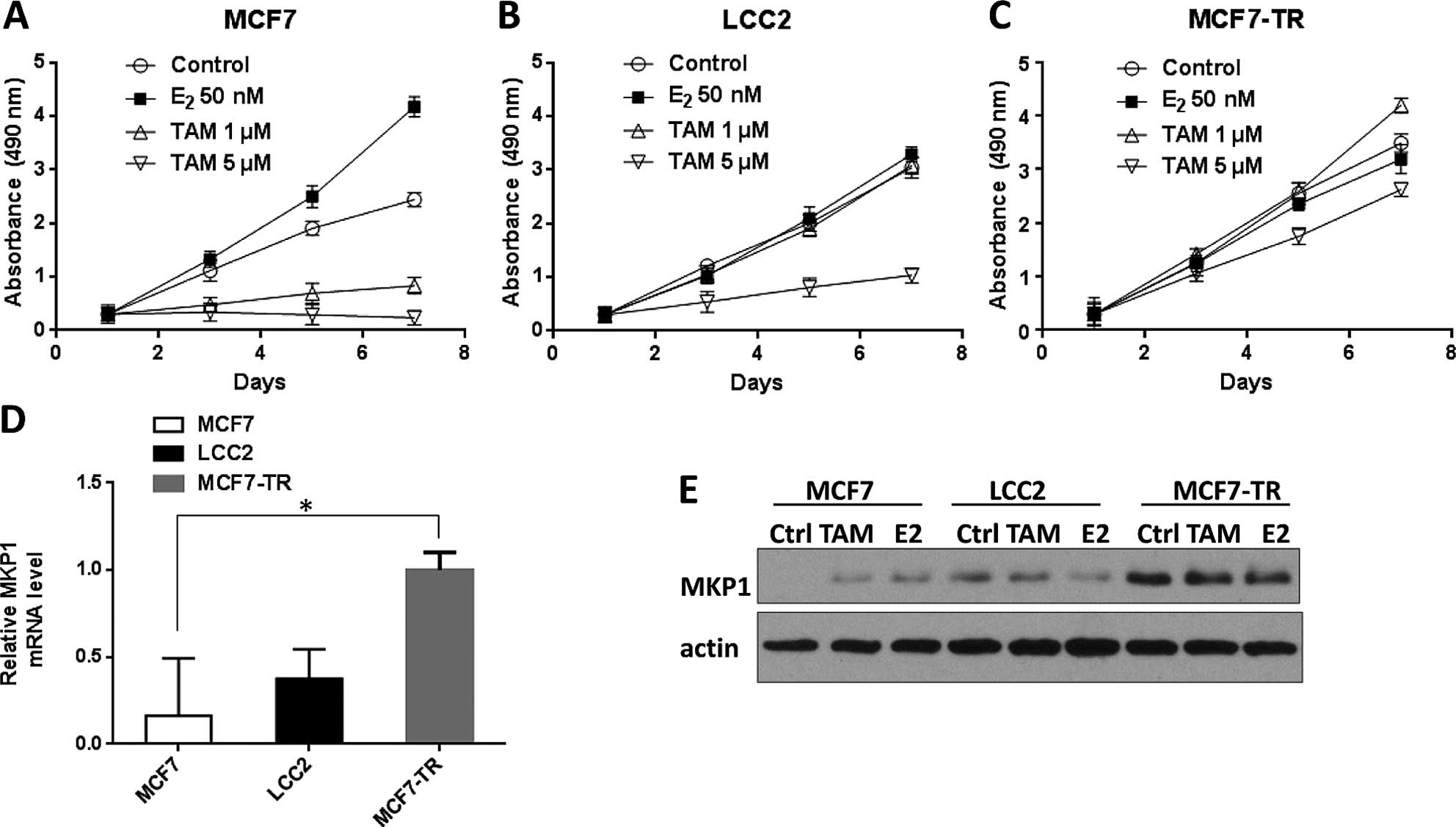
were depicted. As shown in Fig.
1A–C, MCF7-TR cells were resistant to high-dose (5 µM)
tamoxifen, whereas MCF7 and LCC2 cells were not. For exploring the
possible role of MKP1 in tamoxifen resistance, we examined mRNA
level of MKP1 in all three cell lines using quantitative RT-PCR.
The results indicated that MKP1 mRNA level was increased in MCF7-TR
cells compared with MCF7 and LCC2 cells (Fig. 1D). Consistent with MKP1 mRNA, MKP1
protein expression was also increased in MCF7-TR cells (Fig. 1E). Taken together, all the above
results suggest that MKP-1 expression is upregulated in high-dose
tamoxifen resistant MCF7 cells.
MKP1 inhibits tamoxifen-mediated
apoptosis in MCF7-TR and MCF7 cells
As MKP1 expression increased in MCF7-TR cells
compared with MCF7 and LCC2 cells, we asked whether MKP1 is
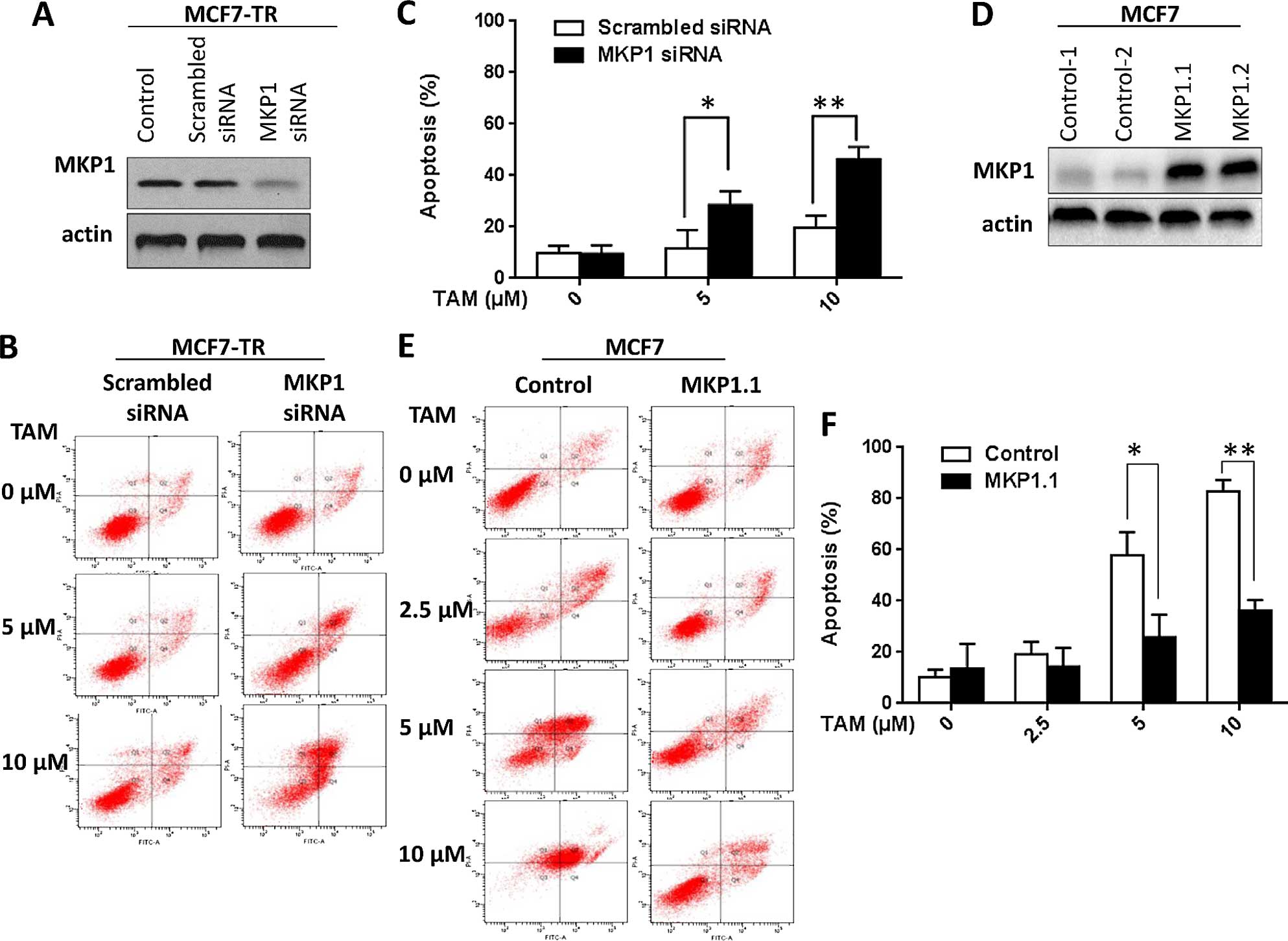
correlated with tamoxifen resistance. To this end, we transiently
transfected scrambled siRNA and MKP1 siRNA to MCF7-TR cells. The
knockdown efficiency was determined by western blotting and the
result is shown in Fig. 2A. We then
asked whether MKP1 plays a role in tamoxifen-induced apoptosis
(23). To this end, both scrambled
siRNA and MKP1 siRNA transfected MCF7-TR cells were treated with
gradually increased micromolar concentration of tamoxifen (0–10
µM) for 48 h. Apoptosis was determined by flow cytometry. As
shown in the Fig. 2B and C,
tamoxifen-induced apoptosis was sharply increased in MKP1 siRNA
transfected MCF7-TR cells compared to scrambled siRNA transfected
MCF7-TR cells, indicating that knockdown of MKP1 enhanced tamoxifen
sensitivity of MCF7-TR cells. On the other hand, we established
MKP1 stably overexpressing MCF7 cell clones by transfection with
mkp1-pCMV-Tag-2A plasmid. The cell clones were verified by western
blotting (Fig. 2D). We also
detected the apoptosis induced by tamoxifen (0–10 µM) in
MKP1 overexpressed MCF7 cells (MCF7/MKP1.1) and empty vector
plasmid transfected MCF7 cells (MCF7/control). As shown in Fig. 2E and F, results of flow cytometry
indicated that overexpression of MKP1 effectively inhibits
tamoxifen-induced apoptosis in MCF7 cells. Taken together, the
above results suggest that MKP1 can protect MCF7 and MCF7-TR cells
from tamoxifen-induced apoptosis.
MKP1 blocks TAM-mediated activation of
MAPKs
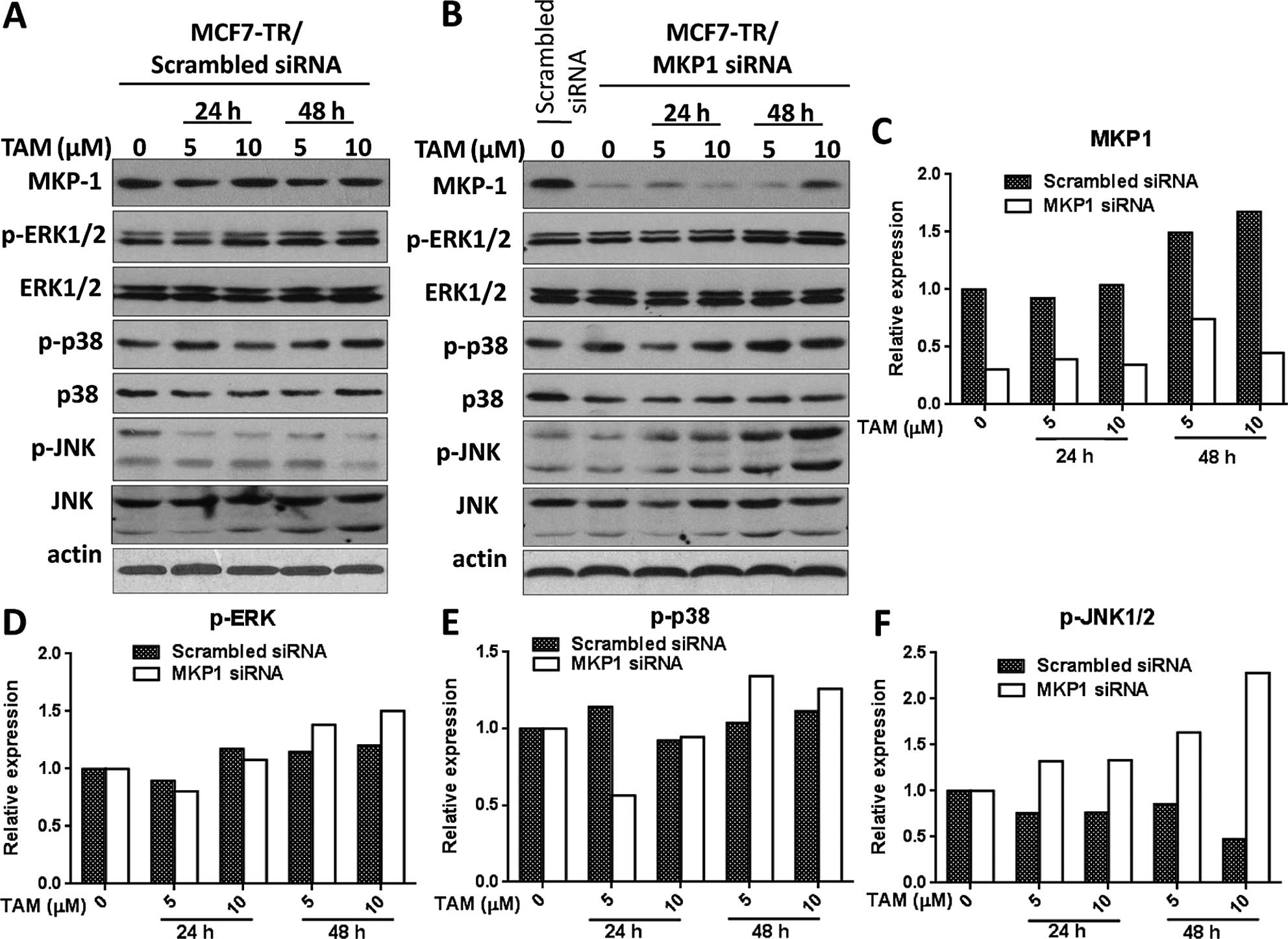
Since MKP1 is a phosphatase which specifically
targets activated MAPKs (11), we
hypothesized that knockdown of MKP1 influences the activation of
MAPKs in tamoxifen-treated MCF7-TR cells. The phosphorylated MAPKs
and total MAPKs were analyzed by western blotting in scrambled
siRNA and MKP1 siRNA transfected MCF7-TR cells which were treated
with tamoxifen for 24 and 48 h (Fig. 3A
and B). As shown in the relative quantitation histograms of
western blotting bands (Fig. 3D and
E), the increased level of phosphorylated ERK1/2 and p-38 were
comparable between scrambled siRNA transfected MCF7-TR cells and
MKP1 siRNA transfected MCF7-TR cells after tamoxifen treatment.
Interestingly, the expression of phosphorylated JNK in MKP1 siRNA
transfected MCF7-TR cells was sharply increased compared to
expression of phosphorylated JNK in scrambled siRNA transfected
MCF7-TR cells after tamoxifen treatment (Fig. 3F), indicating that knockdown of MKP1
discharges the inhibition on tamoxifen-induced activation of JNK in
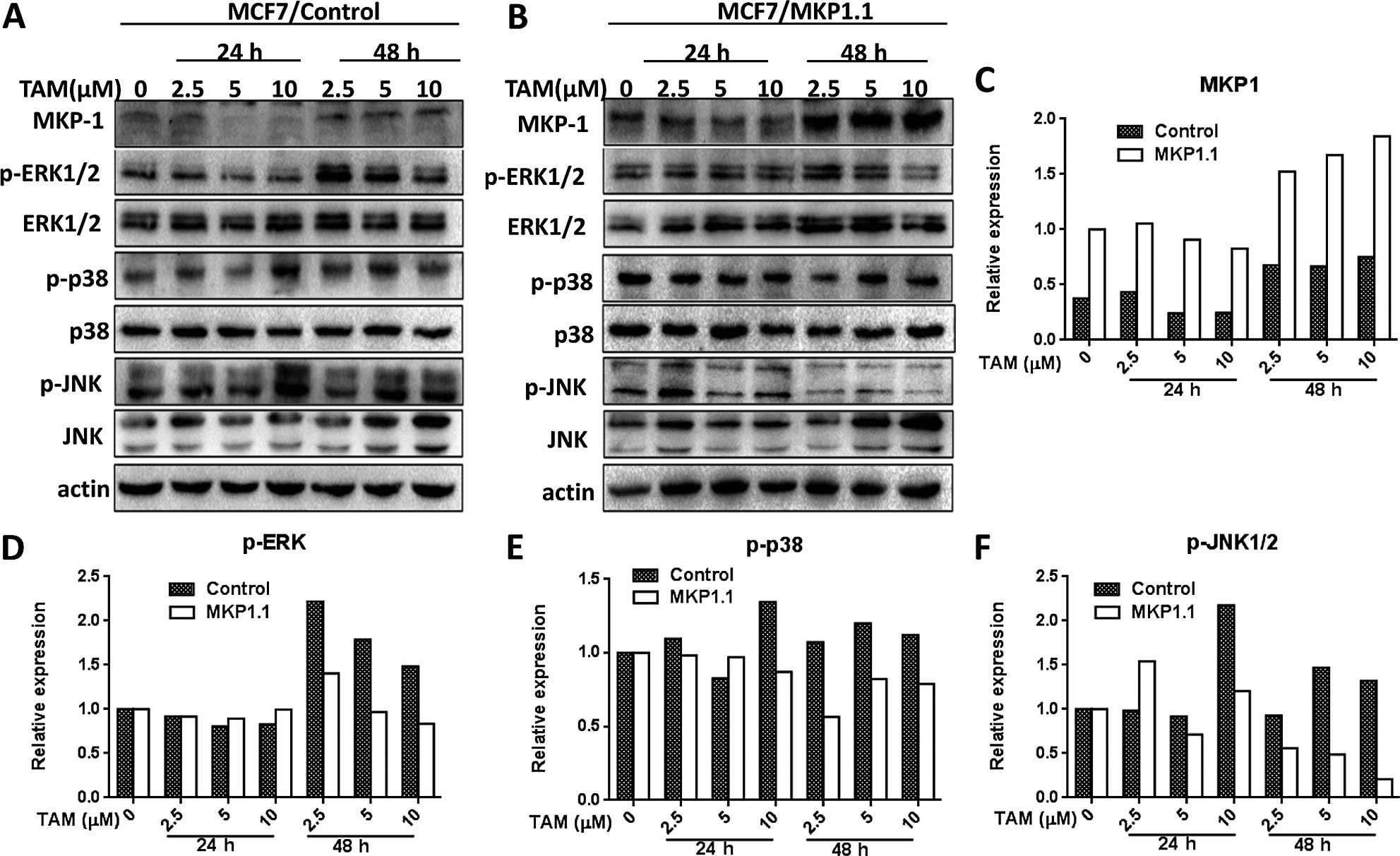
MCF7-TR cells. Tamoxifen has been shown to activate several
signaling pathways which were upstream of MAPKs and then induce
activation of MAPKs in MCF7 cells (23,24).
We next wanted to know whether overexpression of MKP1 affects the
tamoxifen-induced activation of MAPKs in MCF7 cells. The
phosphorylated MAPKs and total MAPKs were determined by western
blotting in MCF7/control and MCF7/MKP1.1 cells after tamoxifen
treatment for 24 and 48 h (Fig. 4A and
B). As shown in the relative quantitation histograms of western
blotting bands (Fig. 4D–F),
tamoxifen-induced phosphorylation of MAPKs was decreased in
MCF7/MKP1.1 cells compared to MCF7/control cells (Fig. 4D–F), indicating that overexpression
of MKP1 in MCF7 attenuates the tamoxifen-induced activation of
MAPKs. Taken together, these results suggest that MKP1 can
negatively regulate tamoxifen-induced activation of MAPKs,
especially JNK, in MCF7-TR and MCF7 cells.
MKP1 inhibits tamoxifen-induced apoptosis
through blocking JNK signaling
It has been shown that activation of JNK contributes
to tamoxifen-mediated apoptosis (10,25,26).
Our results showed that MKP1 inhibited tamoxifen-induced apoptosis
and JNK activation in MCF7-TR and MCF7 cells. Hence, we asked
whether MKP1 mediates inhibition of tamoxifen-induced apoptosis is
through blocking JNK activation. To this end, we used JNK inhibitor
SP600125 (SP) to inhibit the tamoxifen-induced JNK activation in
MKP1 siRNA transfected MCF7-TR and MCF7 cells. All cells were
treated with the following protocol: incubated with 20 µM
SP600125 for 3 h and then treated with tamoxifen for 48 h or only
treated with tamoxifen for 48 h. After that, cells were harvested
and expression of MKP1, p-JNK, JNK, p-c-jun and cleaved PARP were
determined by western blotting (Fig. 5A
and B). As shown in Fig. 5A,
SP600125 effectively inhibited tamoxifen-mediated JNK activation
and cleaved PARP in MKP1 siRNA transfected MCF7-TR cells.
Tamoxifen-induced apoptosis in MKP1 siRNA transfected MCF7-TR cells
was sharply reduced (Fig. 5C). On
the other hand, SP600125 also inhibited tamoxifen-mediated JNK
activation in MCF7 cells and protected MCF7 cells from
tamoxifen-induced apoptosis (Fig. 5B
and C). We also analyzed the caspase 3/7 activity of cells
after treatment with tamoxifen alone or tamoxifen in combination
with SP600125. As shown in Fig. 5D,
SP600125 treatment attenuated TAM-induced caspase 3/7 activity in
MKP1 siRNA-transfected MCF7-TR cells. Taken together, these results
suggest that JNK signaling is involved in TAM-induced apoptosis and
MKP1 inhibition of TAM-induced apoptosis is through blocking JNK
activation.
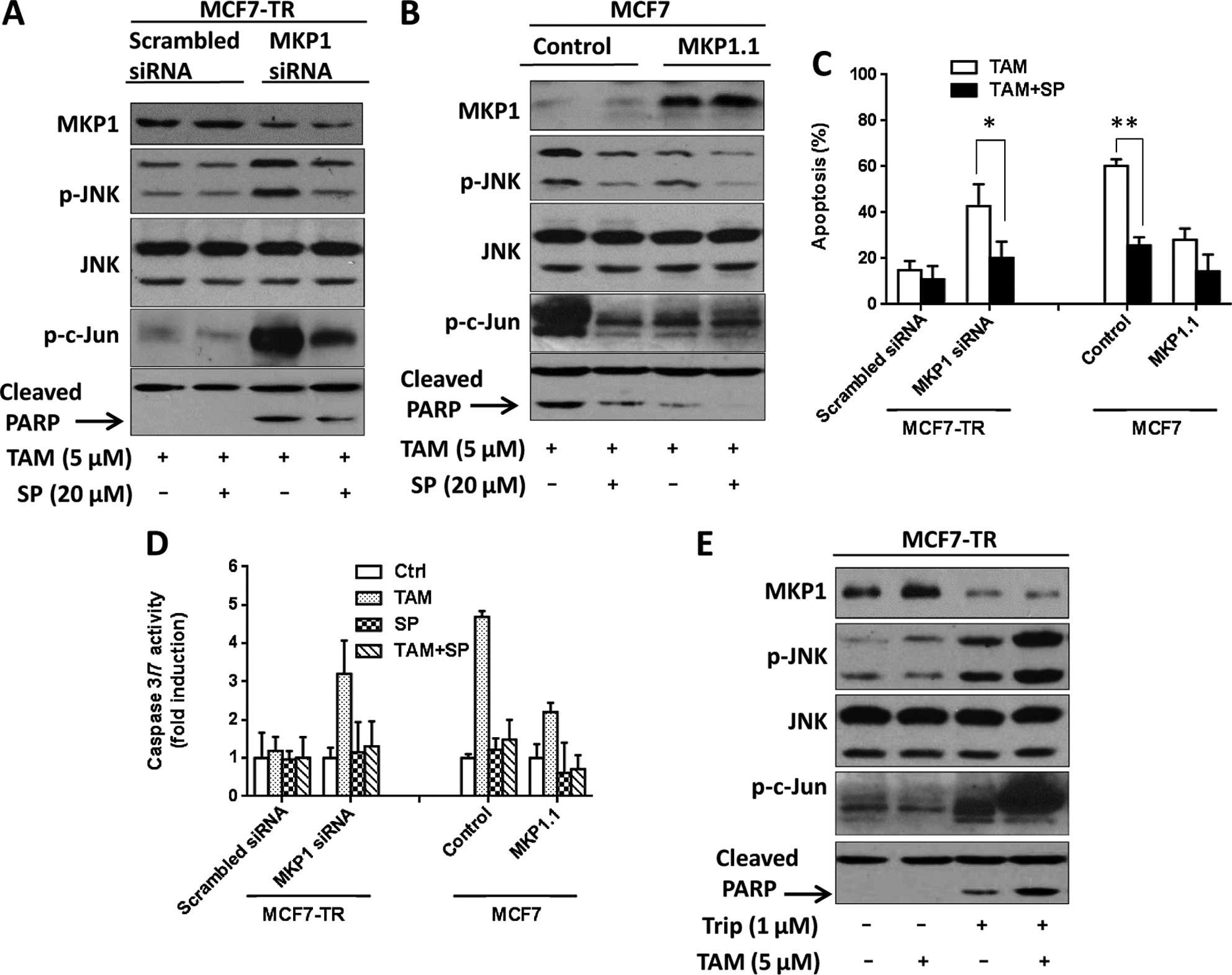 | Figure 5(A) Expression of MKP1, p-JNK, JNK,
p-c-jun and cleaved PARP were determined by western blotting in
scrambled siRNA and MKP1 siRNA transfected MCF7-TR cells, which
were treated with either 5 µM TAM for 48 h or 20 µM
SP600125 (SP) for 3 h and then 5 µM TAM for 48 h. (B)
Expression of MKP1, p-JNK, JNK, p-c-jun and cleaved PARP were
determined by western blotting in MCF7/control and MCF7/MKP1.1
cells, which were treated with either 5 µM TAM for 48 h or
20 µM SP600125 (SP) for 3 h and then 5 µM TAM for 48
h. (C) Histograms represent the apoptosis rate of cells in A and B.
Significant difference *P<0.05;
**P<0.01 (mean ± SD, n=3). (D) Histograms represent
caspase 3/7 activity of cells in A and B. (E) MCF7-TR cells were
treated with vehicle (−), 5 µM TAM, 1 µM triptolide
or both of compounds for 48 h. Western blot analyses were performed
using MKP1, p-JNK, JNK, p-c-jun and PARP antibodies |
Triptolide has been shown to inhibit MKP1 expression
(27,28). In the present study, we combined
triptolide with tamoxifen to treat MCF7-TR cells. As shown in
Fig. 5E, triptolide reduced MKP1
expression in MCF7-TR cells. Moreover, combined application of
triptolide and tamoxifen sharply enhanced tamoxifen-induced
activation of JNK and cleaved PARP compared with tamoxifen
treatment alone, indicating that MKP1 inhibitor may be a new
therapeutic strategy for tamoxifen resistance.
Discussion
Tamoxifen has been shown to induce both cytostatic
(proliferation arrest) and cytotoxic effect (cell death or
apoptosis) through ERα-dependent and -independent pathways
(23,24). In MCF7 cells, the antitumor effects
of tamoxifen through ERα and non-ERα signal pathway were dependent
on concentration of tamoxifen (nanomolar or micromolar) (23,29).
In other words, when used at nanomolar concentration, such as 100
nM, tamoxifen mainly induced cell growth arrest (cytostatic
effects) through inhibition of ER signaling, whereas used at
micromolar concentration, such as 5 µM, tamoxifen mainly
induced cell death (cytotoxic effect) through ER-independent signal
pathways. In the present study, we established a high-dose TAM
resistant MCF7 cell line (MCF7-TR) from the verified tamoxifen
resistant LCC2 cells (21). MCF7-TR
cells are not only resistant to nanomolar (low-dose)
tamoxifen-induced cell growth arrest, but also resistant to
micromolar (high-dose) tamoxifen-induced cell death. We detected
the MKP1 expression in MCF7-TR cells and MCF7 cells, and found that
both of MKP1 mRNA and protein expression increased in MCF7-TR
cells. Several studies have shown that MKP1 increases in poorly
differentiated and late stage of breast cancer but not in early
stage of breast cancer (30,31).
Our data indicated that MKP1 expression is higher in tamoxifen
resistant MCF7-TR cells compared with parental MCF7 cells, which is
consistent with the previous studies.
MKP1 has been shown to induce drug resistance
through inhibiting apoptosis (18–20,32).
In the present study, knockdown of MKP1 in MCF7-TR cells increased
cell death induced by micromolar concentration of tamoxifen. On the
other hand, overexpression of MKP1 in MCF7 cells protected cells
from cytotoxic effect mediated by micromolar concentration of
tamoxifen. These results showed that MKP1 expression in MCF7-TR and
MCF7 cells protects cell from tamoxifen-induced cytotoxic effect,
indicating that MKP1 may be involved in tamoxifen resistance of
MCF7 cells.
It has been reported that tamoxifen induces
activation of MAPKs in breast cancer (33,34).
In the present study, detection of phosphorylated MAPKs in MKP1
knockdown MCF7-TR and MCF7 cells after tamoxifen treatment showed
similar results (as shown in Figs.
3B and 4A). Since MKP1
specifically and negatively regulated MAPKs, we explored whether
MKP1 inhibits tamoxifen-induced activation of JNK in MCF7-TR and
MCF7 cells. As shown in Figs. 3A
and 4B, the MKP1 highly-expressed
MCF7-TR and MCF7 cells exhibited declining phosphorylation of
MAPKs, especially phosphorylation of JNK, after tamoxifen
treatment, indicating that MKP1 expressed in MCF7-TR and MCF7 cells
inhibits the activation of MAPKs.
It has been reported that activation of JNK
contributes to tamoxifen cytotoxic effect through the process of
mediating apoptosis (10,25,26,29),
we thus hypothesized that MKP1 may protect cells by inactivating
JNK. We used both JNK specific inhibitor SP600125 and tamoxifen to
treat cells. The results showed that inhibition of JNK activation
sharply reduced tamoxifen-induced apoptosis and cleaved PARP
expression in MKP1 knockdown MCF7-TR cells and MKP1 low-expressed
MCF7 cells, suggesting that JNK signal pathway is indeed involved
in the tamoxifen-induced apoptosis, and MKP1 inhibits
tamoxifen-induced apoptosis through blocking JNK activation.
Furthermore, we used MKP1 inhibitor triptolide in combination with
tamoxifen to treat MCF7-TR cells, and found that triptolide
promoted tamoxifen-induced activation of JNK and apoptosis in
MCF7-TR cells.
In conclusion, we showed that MKP1 expression
increased in tamoxifen resistant MCF7-TR cells compared with
parental MCF7 cells. We also showed that high expression of MKP1 in
MCF7-TR and MCF7 cells can protect cells from tamoxifen-induced
apoptosis, and this is dependent on the role MKP1 played in
regulation of JNK activation. Moreover, our result suggested that
targeting MKP1 may be a potential therapeutic strategy for
tamoxifen resistance.
Abbreviations:
|
MKP1
|
mitogen-activated protein kinase
phosphatase 1
|
|
MAPKs
|
mitogen-activated protein kinases
|
|
ER
|
estrogen receptor
|
|
JNK
|
JUN NH2-terminal kinases
|
|
TAM
|
4-hydroxy tamoxifen
|
Acknowledgments
This study was supported by grant from the Science
and Technology Research and Development Foundation of Shaanxi
Province (no. 2011K13-01-08 and no. 2014KRM99-02). We thank
Professor Xinyi Chen (Beijing University of Chinese Medicine) for
providing tamoxifen resistant MCF7 cells and Professor Lianfeng
Zhang (Peking Union Medical Collage) for providing MKP-1 cDNA
plasmid.
References
|
1
|
Cancer Genome Atlas N: Cancer Genome Atlas
Network: Comprehensive molecular portraits of human breast tumours.
Nature. 490:61–70. 2012. View Article : Google Scholar
|
|
2
|
Osborne CK and Schiff R: Mechanisms of
endocrine resistance in breast cancer. Annu Rev Med. 62:233–247.
2011. View Article : Google Scholar
|
|
3
|
Osborne CK and Osborne CK: Tamoxifen in
the treatment of breast cancer. N Engl J Med. 339:1609–1618. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Riggins RB, Schrecengost RS, Guerrero MS
and Bouton AH: Pathways to tamoxifen resistance. Cancer Lett.
256:1–24. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
García-Becerra R, Santos N, Díaz L and
Camacho J: Mechanisms of resistance to endocrine therapy in breast
cancer: Focus on signaling pathways, miRNAs and genetically based
resistance. Int J Mol Sci. 14:108–145. 2012. View Article : Google Scholar
|
|
6
|
Kim EK and Choi EJ: Pathological roles of
MAPK signaling pathways in human diseases. Biochim Biophys Acta.
1802:396–405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Britton DJ, Hutcheson IR, Knowlden JM,
Barrow D, Giles M, McClelland RA, Gee JM and Nicholson RI:
Bidirectional cross talk between ERalpha and EGFR signalling
pathways regulates tamoxifen-resistant growth. Breast Cancer Res
Treat. 96:131–146. 2006. View Article : Google Scholar
|
|
8
|
Gutierrez MC, Detre S, Johnston S, Mohsin
SK, Shou J, Allred DC, Schiff R, Osborne CK and Dowsett M:
Molecular changes in tamoxifen-resistant breast cancer:
Relationship between estrogen receptor, HER-2, and p38
mitogen-activated protein kinase. J Clin Oncol. 23:2469–2476. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dumont JA, Bitonti AJ, Wallace CD, Baumann
RJ, Cashman EA and Cross-Doersen DE: Progression of MCF-7 breast
cancer cells to antiestrogen-resistant phenotype is accompanied by
elevated levels of AP-1 DNA-binding activity. Cell Growth Differ.
7:351–359. 1996.PubMed/NCBI
|
|
10
|
Mandlekar S, Yu R, Tan TH and Kong AN:
Activation of caspase-3 and c-Jun NH2-terminal kinase-1 signaling
pathways in tamoxifen-induced apoptosis of human breast cancer
cells. Cancer Res. 60:5995–6000. 2000.PubMed/NCBI
|
|
11
|
Theodosiou A and Ashworth A: MAP kinase
phosphatases. Genome Biol. 3:S30092002. View Article : Google Scholar
|
|
12
|
Camps M, Nichols A and Arkinstall S: Dual
specificity phosphatases: A gene family for control of MAP kinase
function. FASEB J. 14:6–16. 2000.PubMed/NCBI
|
|
13
|
Wu JJ, Roth RJ, Anderson EJ, Hong EG, Lee
MK, Choi CS, Neufer PD, Shulman GI, Kim JK and Bennett AM: Mice
lacking MAP kinase phosphatase-1 have enhanced MAP kinase activity
and resistance to diet-induced obesity. Cell Metab. 4:61–73. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao Q, Wang X, Nelin LD, Yao Y, Matta R,
Manson ME, Baliga RS, Meng X, Smith CV, Bauer JA, et al: MAP kinase
phosphatase 1 controls innate immune responses and suppresses
endotoxic shock. J Exp Med. 203:131–140. 2006. View Article : Google Scholar
|
|
15
|
Wancket LM, Frazier WJ and Liu Y:
Mitogen-activated protein kinase phosphatase (MKP)-1 in immunology,
physiology, and disease. Life Sci. 90:237–248. 2012. View Article : Google Scholar :
|
|
16
|
Li M, Zhou JY, Ge Y, Matherly LH and Wu
GS: The phosphatase MKP1 is a transcriptional target of p53
involved in cell cycle regulation. J Biol Chem. 278:41059–41068.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sánchez-Pérez I, Martínez-Gomariz M,
Williams D, Keyse SM and Perona R: CL100/MKP-1 modulates JNK
activation and apoptosis in response to cisplatin. Oncogene.
19:5142–5152. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Z, Xu J, Zhou JY, Liu Y and Wu GS:
Mitogen-activated protein kinase phosphatase-1 is required for
cisplatin resistance. Cancer Res. 66:8870–8877. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Small GW, Shi YY, Higgins LS and Orlowski
RZ: Mitogen-activated protein kinase phosphatase-1 is a mediator of
breast cancer chemoresistance. Cancer Res. 67:4459–4466. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Z, Zhou JY, Kanakapalli D, Buck S, Wu
GS and Ravindranath Y: High level of mitogen-activated protein
kinase phosphatase-1 expression is associated with cisplatin
resistance in osteosarcoma. Pediatr Blood Cancer. 51:754–759. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brünner N, Frandsen TL, Holst-Hansen C,
Bei M, Thompson EW, Wakeling AE, Lippman ME and Clarke R:
MCF7/LCC2: A 4-hydroxytamoxifen resistant human breast cancer
variant that retains sensitivity to the steroidal antiestrogen ICI
182,780. Cancer Res. 53:3229–3232. 1993.PubMed/NCBI
|
|
22
|
Takeuchi K, Shin-ya T, Nishio K and Ito F:
Mitogen-activated protein kinase phosphatase-1 modulated JNK
activation is critical for apoptosis induced by inhibitor of
epidermal growth factor receptor-tyrosine kinase. FEBS J.
276:1255–1265. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mandlekar S and Kong AN: Mechanisms of
tamoxifen-induced apoptosis. Apoptosis. 6:469–477. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bogush T, Dudko E, Bogush E, Polotsky B,
Tjulandin S and Davydov M: Tamoxifen non-estrogen receptor mediated
molecular targets. Oncol Rev. 6:e152012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Madeo A, Vinciguerra M, Lappano R, Galgani
M, Gasperi-Campani A, Maggiolini M and Musti AM: c-Jun activation
is required for 4-hydroxytamoxifen-induced cell death in breast
cancer cells. Oncogene. 29:978–991. 2010. View Article : Google Scholar
|
|
26
|
Wang N, Li Z, Tian F, Feng Y, Huang J, Li
C and Xie F: PKCα inhibited apoptosis by decreasing the activity of
JNK in MCF-7/ADR cells. Exp Toxicol Pathol. 64:459–464. 2012.
View Article : Google Scholar
|
|
27
|
Chen P, Li J, Barnes J, Kokkonen GC, Lee
JC and Liu Y: Restraint of proinflammatory cytokine biosynthesis by
mitogen-activated protein kinase phosphatase-1 in
lipopolysaccharide-stimulated macrophages. J Immunol.
169:6408–6416. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao Q, Shepherd EG, Manson ME, Nelin LD,
Sorokin A and Liu Y: The role of mitogen-activated protein kinase
phosphatase-1 in the response of alveolar macrophages to
lipopolysaccharide: Attenuation of proinflammatory cytokine
biosynthesis via feedback control of p38. J Biol Chem.
280:8101–8108. 2005. View Article : Google Scholar
|
|
29
|
Obrero M, Yu DV and Shapiro DJ: Estrogen
receptor-dependent and estrogen receptor-independent pathways for
tamoxifen and 4-hydroxytamoxifen-induced programmed cell death. J
Biol Chem. 277:45695–45703. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang HY, Cheng Z and Malbon CC:
Overexpression of mitogen-activated protein kinase phosphatases
MKP1, MKP2 in human breast cancer. Cancer Lett. 191:229–237. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rojo F, González-Navarrete I, Bragado R,
Dalmases A, Menéndez S, Cortes-Sempere M, Suárez C, Oliva C,
Servitja S, Rodriguez-Fanjul V, et al: Mitogen-activated protein
kinase phosphatase-1 in human breast cancer independently predicts
prognosis and is repressed by doxorubicin. Clin Cancer Res.
15:3530–3539. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang J, Zhou JY and Wu GS: ERK-dependent
MKP-1-mediated cisplatin resistance in human ovarian cancer cells.
Cancer Res. 67:11933–11941. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rabenoelina F, Semlali A, Duchesne MJ,
Freiss G, Pons M and Badia E: Effect of prolonged hydroxytamoxifen
treatment of MCF-7 cells on mitogen activated kinase cascade. Int J
Cancer. 98:698–706. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Creighton CJ, Hilger AM, Murthy S, Rae JM,
Chinnaiyan AM and El-Ashry D: Activation of mitogen-activated
protein kinase in estrogen receptor alpha-positive breast cancer
cells in vitro induces an in vivo molecular phenotype of estrogen
receptor alpha-negative human breast tumors. Cancer Res.
66:3903–3911. 2006. View Article : Google Scholar : PubMed/NCBI
|