Introduction
The Eker rat is a useful animal model with which to
study renal cell carcinoma (RCC) (1). Spontaneous tumors develop in the
kidney due to a germline mutation in the tuberous sclerosis 2
(Tsc2) gene (2,3). Homozygous mutants are embryonic lethal
during midgestation (equivalent to mouse E9.5-E13.5) (4). In contrast, heterozygous mutants
develop bilateral multicentric renal cell carcinomas within one
year after birth (5). The
development of multistage renal carcinogenesis can be monitored at
the histological level (6).
TSC2 (encoding tuberin) is a tumor-suppressor gene
identified as a causative gene of TSC as well as TSC1
(encoding hamartin) (7–9). These products form a complex that
inhibits the mammalian target of rapamycin complex 1 (mTORC1), a
serine/threonine protein kinase essential for the regulation of
cell growth and proliferation (10). Mammalian cells express two
functionally distinct mTOR complexes: mTORC1 and mTORC2. mTORC1
contains mTOR, Raptor and LST8 as primary subunits and is inhibited
by rapamycin (11). In
TSC-associated tumors, the loss of TSC1 or TSC2
induces mTORC1-dependent phosphorylation of p70S6 kinase, ribosomal
protein S6 and 4EBP1 (12). On the
other hand, mTORC2 contains Rictor, LST8 and Sin1 as primary
subunits. This complex functions as a rapamycin-insensitive
regulator of the cytoskeleton and cell survival through Akt
phosphorylation (13).
Few previous studies have explored the
cancer-related cellular mechanisms trigged by the loss of
tumor-suppressor genes using embryonic stem cells (ESCs) such as
Apc-mutated ESCs (14). To
establish a new research model for the analysis of renal
carcinogenesis, we developed ESCs from Eker rat blastocysts
(15). We successfully established
Tsc2+/+, Tsc2+/− and
Tsc2−/− Eker rat ESCs with pluripotency.
Furthermore, in the teratoma formation assay, which is an important
process for pluripotent stem cells (PSCs) to prove differentiation
ability, epithelial tumor-like abnormal ductal structures
resembling Eker rat RCCs were identified in
Tsc2−/− teratomas. Immunohistochemical analysis
revealed the activation of mTORC1 signaling in
Tsc2−/− teratomas, particularly in abnormal
ductal structures, which was suppressed by rapamycin treatment. We
suggested that the appearance of these abnormal structures
indicates a pathogenic mechanism related to renal tumorigenesis in
Eker rats. These teratomas constitute a new research tool for the
analysis of tissue-specific tumorigenesis by suppression of mTORC1
signaling.
Materials and methods
Animal studies
All animal experiments were performed according to
protocols approved by the Animal Care Committee of Juntendo
University of Medicine (Approval no. 250105). All surgeries were
performed under isoflurane anesthesia and measures were taken to
minimize animal suffering.
Cell culture
Rat ESCs were cultured on mitomycin C-treated mouse
embryonic fibroblasts in N2B27-2i medium (1:1 ratio of 2i
medium:N2B27 medium; Axon Medchem, Groningen, The Netherlands)
containing 1,000 U/ml rat leukemia inhibitory factor (LIF; ESGRO
Millipore, Bedford, MA, USA), 3 µM GSK3β inhibitor CHIR99021
(Axon), and 1 µM MEK inhibitor PD0325901 (Axon).
Teratoma formation
A total of 1.5×106
Tsc2+/+, Tsc2+/− or
Tsc2−/− Eker ESCs suspended in Matrigel were
subcutaneously injected into non-obese diabetic/severe combined
immunodeficiency (NOD/SCID) mice. After 5 weeks, 1.5 mg/kg body
weight of rapamycin (Sigma-Aldrich, St. Louis, MO, USA), or vehicle
alone (vehicle control), was intraperitoneally injected every other
day, for a total of three injections. Mice were sacrificed the day
after the last injection.
Immunohistochemistry
Immunohistochemistry was carried out using a
standard method using formalin-fixed, paraffin-embedded tissues.
For chromogenic analysis, EnVision+ System HRP-labeled polymer for
rabbit or mouse Abs (Dako) were primarily used as secondary
antibodies. Alternatively, biotinylated anti-sheep/goat
immunoglobulin from donkey (1:100 dilution) and
streptavidin-biotinylated horseradish peroxidase complex (1:100)
(GE Healthcare Life Sciences, USA) were used for the staining of
goat polyclonal antibodies. For immunofluorescence, the following
secondary Abs were used: Alexa Fluor 568-conjugated donkey
anti-goat Ab (Molecular Probes, Eugene, OR, USA); Alexa Fluor
488-conjugated donkey anti-mouse Ab (Jackson ImmunoResearch
Laboratories, West Grove, PA, USA); Alexa Fluor 488-conjugated
donkey anti-rabbit Ab (Molecular Probes); Alexa Fluor 488 or
568-conjugated goat anti-mouse or anti-rabbit Abs (Molecular
Probes). Nuclei were stained with 6-diamidino-2-phenylindole
(DAPI). Fluorescent images were captured and analyzed using an
Axioplan 2 microscope (Carl Zeiss, Germany).
Following primary mouse monoclonal antibody (mAb),
rabbit polyclonal (pAb), rabbit monoclonal (rAb) and goat
polyclonal (gAb) antibodies were used at indicated dilution rates:
anti-p-S6K (Thr389) rAb (108D; 1:1,000); anti-p-4EBP1 (Thr37/46)
rAb (236B4; 1:100) (both from Cell Signaling Technology, Beverly,
MA, USA); anti-LRP2 (megalin) mAb (CD7D5; 1:300; Novus Biologicals,
Littleton, CO, USA); anti-cubilin gAb (A-20; 1:100; Santa Cruz
Biotechnology, Santa Cruz, CA, USA); anti-Ki67 pAb (1:350; Novus
Biologicals); anti-TFE3 pAb (1:500; Atlas Antibodies AB, Stockholm,
Sweden); anti-TFEB pAb (1:100; Bioss USA Antibodies, Woburn, MA,
USA); anti-β-catenin pAb (1:1,000; Merck Millipore, Darmstadt,
Germany); anti-E-cadherin mAb (1:50; BD Biosciences, San Jose, CA,
USA).
Cell counting was performed using at least 4 fields
(magnification, ×100) of each sample. Ki67 positive-cells were
compared between ductal and abnormal ductal cells of
Tsc2+/+ and Tsc2−/− teratomas,
respectively. For E-cadherin and β-catenin, a cell clearly and
continuously stained at the perimeter was defied as positive.
Statistical analyses were performed with SAS software version R8.1
(SAS Institute Japan, Ltd., Tokyo, Japan). Values of p<0.05 were
considered significant.
Western blotting
For western blotting, teratomas were dissected, snap
frozen and stored until use. Protein samples were obtained by
lysing teratomas in standard sample buffer (50 mM Tris-HCl, pH 6.8,
2% SDS, 10% glycerol) for SDS polyacrylamide gel electrophoresis.
The following primary antibodies were used at the indicated
dilutions: anti-TSC2 pAb (C20; 1:500; Santa Cruz Biotechnology);
anti-p-S6K (Thr389) rmAb (108D; 1:1,000; Cell Signaling
Technology); anti-S6K pAb (C-18; 1:500; Santa Cruz Biotechnology);
anti-p-Akt (Ser473) rmAb (D9E; 1:1,000); anti-Akt pAb (1:1,000);
and β-actin pAb (1:1,000) (all from Cell Signaling Technology).
Results
Tsc2−/− ESCs forms epithelial
tumor-like abnormal ductal structures in teratomas
In the previous study, we found that
Tsc2+/+, Tsc2+/− or
Tsc2−/− Eker ESCs could differentiate to all
three germ layers and epithelial tumor-like abnormal cells forming
ductal structures in Tsc2−/− teratomas by renal
capsule injection in nude mice (15). To confirm this phenomenon in more
mature teratomas, we constructed teratomas by
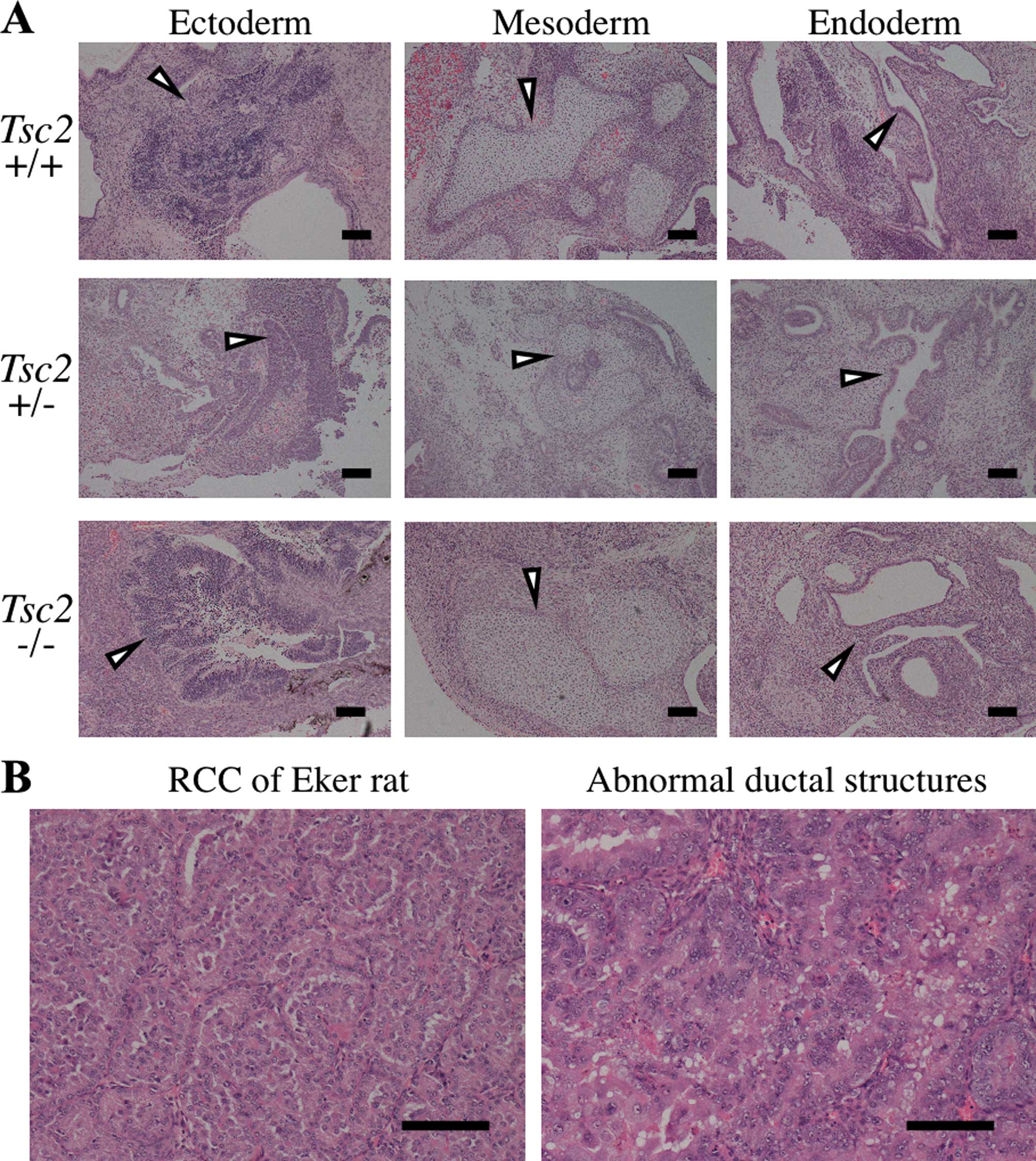
subcutaneous-inject-protocol in NOD/SCID mice. H&E-stained
sections revealed that Tsc2−/− ESCs had the
potential to differentiate into all three germ layers in this
system (Fig. 1A).
Tsc2−/− teratomas also contained
epithelial tumor-like abnormal cells forming ductal structures
(Fig. 1B). They comprised large
cells with clear, finely granular and occasionally vacuolated
cytoplasm. They ruptured the basement membrane and invaded the
surrounding parenchyma. These characteristics are reminiscent of
Eker rat renal carcinoma (16).
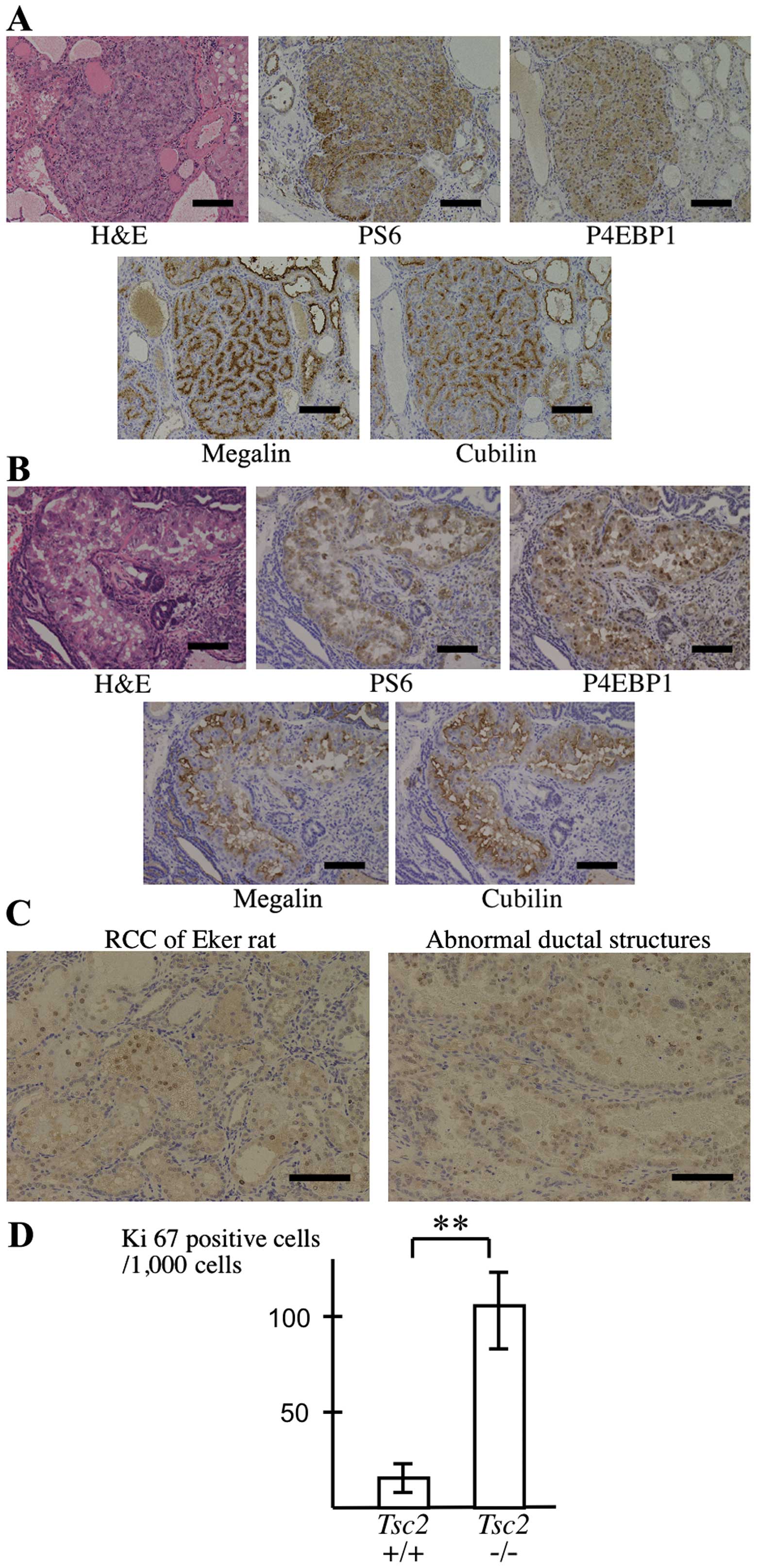
RCCs of Eker rats originate from renal proximal
tubules (17). To explore the cell
types of abnormal ductal structures in Tsc2−/−
teratomas, tissue sections were stained for megalin and cubilin.
Expressed in epithelial cells of renal proximal tubules, these
receptors mediate the endocytosis of numerous ligands. Although
megalin expression is widespread among various epithelial cell
types, cubilin expression is restricted to cells in renal proximal
tubules, small intestine and yolk sac (18). In tissue sections, non-tumorous
renal proximal tubules and Eker rat RCCs stained positive for
megalin and cubilin (Fig. 2A).
Interestingly, abnormal ductal structures of
Tsc2−/− teratomas were also positive for megalin
and cubilin, suggesting epithelial characteristics (Fig. 2B).
Moreover, tissue sections were stained for Ki67 to
check the mitotic activity. Ki67-positive cells were frequently
detected in abnormal ductal structures as observed in Eker rat RCCs
(Fig. 2C). Compared with ductal
parts in Tsc2+/+ teratomas, abnormal ductal
structures in Tsc2−/− teratomas showed a
significant increase in the number of Ki67-positive cells (Fig. 2D). These observations suggest that
cells forming abnormal ductal structures of
Tsc2−/− teratomas possess an epithelial
phenotype. Moreover, these findings suggest that the appearance of
abnormal ductal structures reflects abnormal cellular
differentiation caused by Tsc2 deficiency.
Activation of mTORC1 signaling in
abnormal ductal structures of Tsc2−/− teratomas
The activation state of the mTORC1 pathway in the
Tsc2+/+, Tsc2+/− and
Tsc2−/− teratomas was investigated by
immunostaining of downstream protein targets: phosphorylated S6
(p-S6) and phosphorylated 4EBP1 (p-4EBP1).
Positive staining of p-S6 and p-4EBP1 was broadly
observed throughout Tsc2−/− teratomas and was
considerably more intense in abnormal ductal structures as in Eker
rat RCCs (Fig. 2B). These results
indicated that mTORC1 signaling is activated in abnormal ducts of
Tsc2−/− teratomas. The importance of the mTORC1
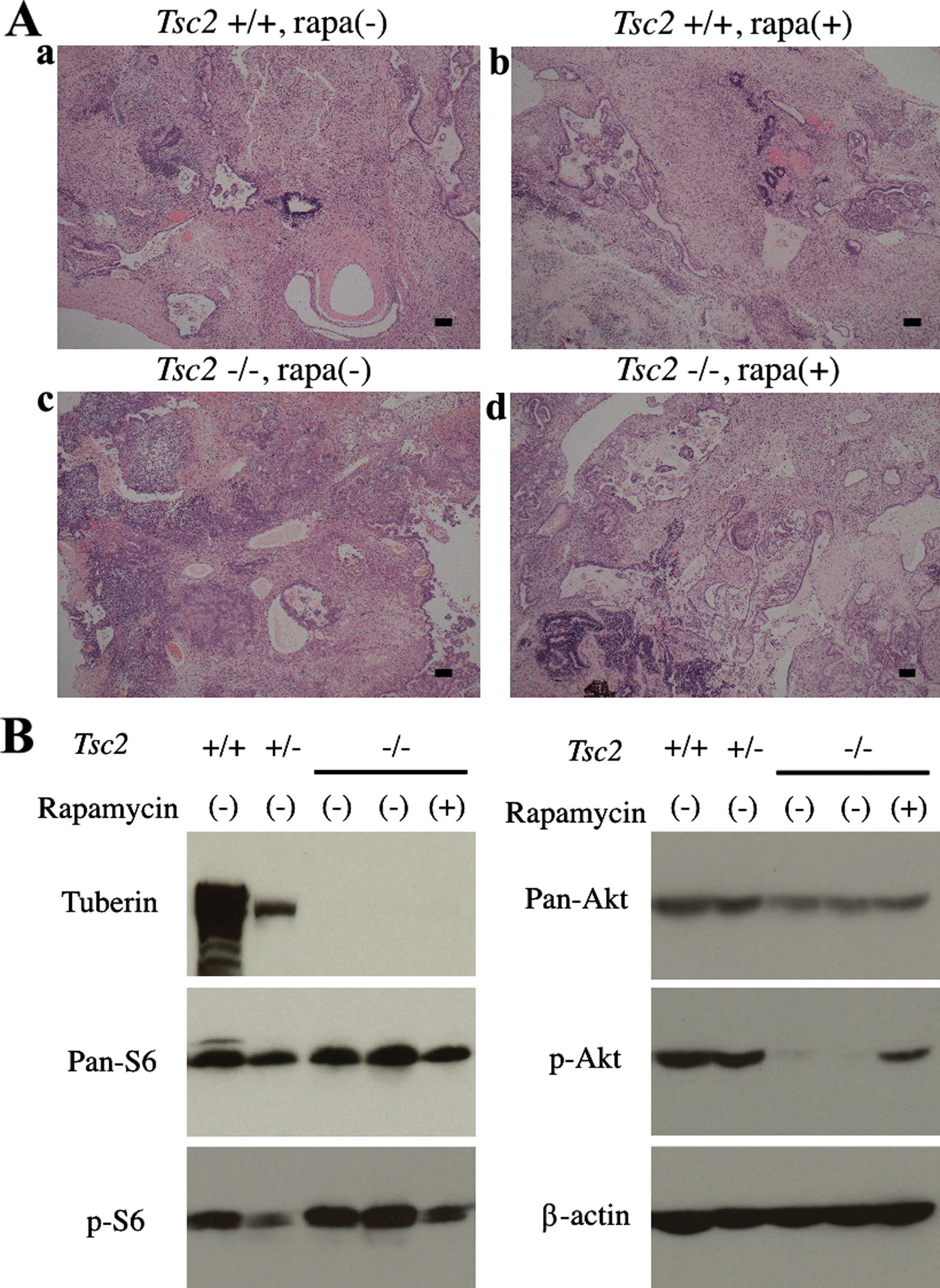
pathway in the formation of abnormal ductal structures was
investigated by treatment with the mTOR inhibitor rapamycin after
solid teratomas were formed. All ESCs maintained differentiation of
three germ layers but the development of abnormal ductal structures
in Tsc2−/− teratomas was suppressed under
rapamycin-treated conditions (Fig.
3A).
Upregulation of S6 phosphorylation in
Tsc2−/− teratomas and its suppression by
rapamycin were confirmed by western blot analysis (Fig. 3B). Interestingly, Akt (S473)
phosphorylation was suppressed in Tsc2−/−
teratomas but was significantly reactivated by rapamycin,
suggesting that the negative feedback on Akt was conferred by an
activated mTORC1 signal. These observations support a relationship
between mTORC1 hyperactivation and the development of abnormal
ductal structures in Tsc2−/− teratomas.
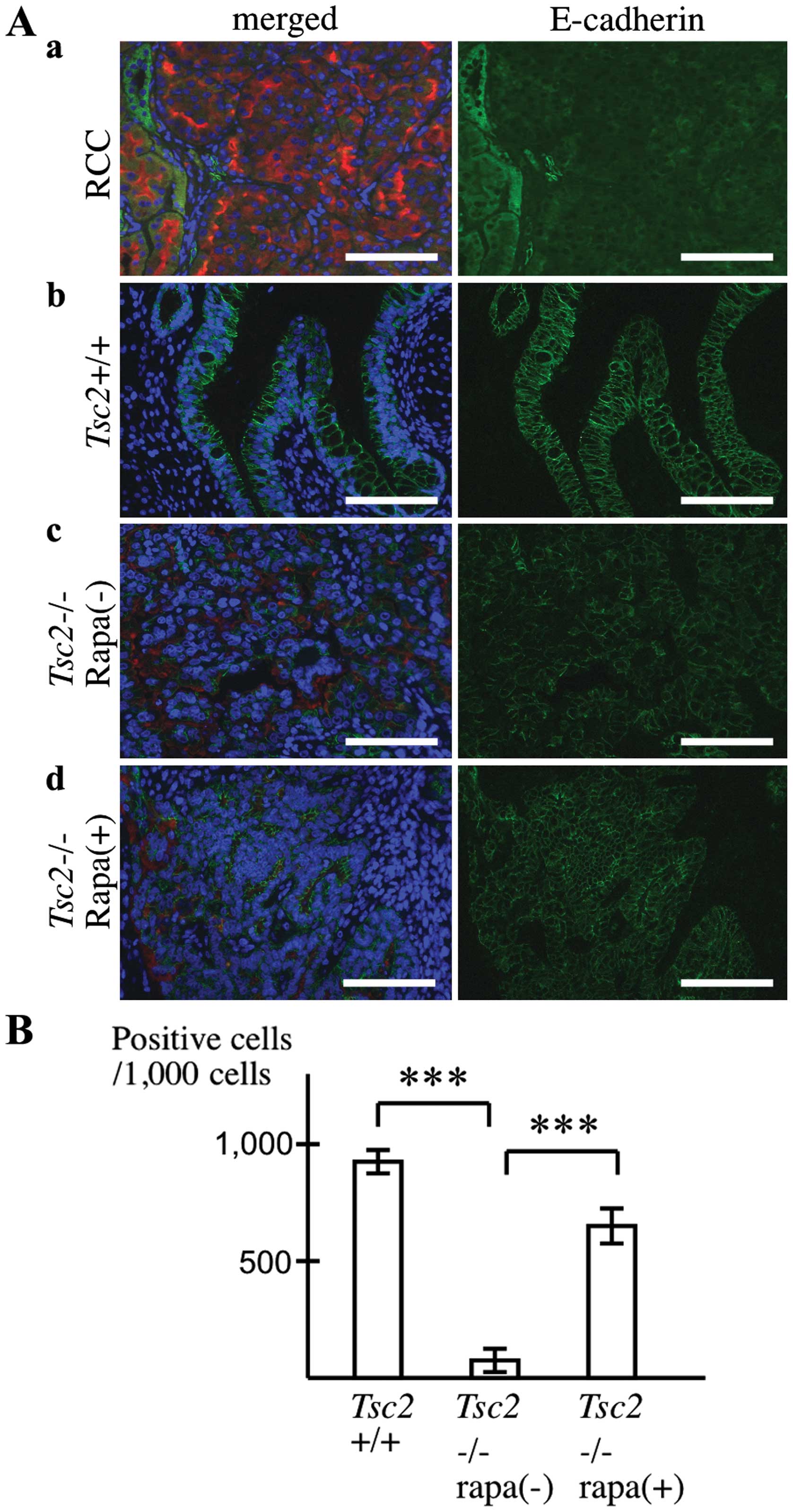
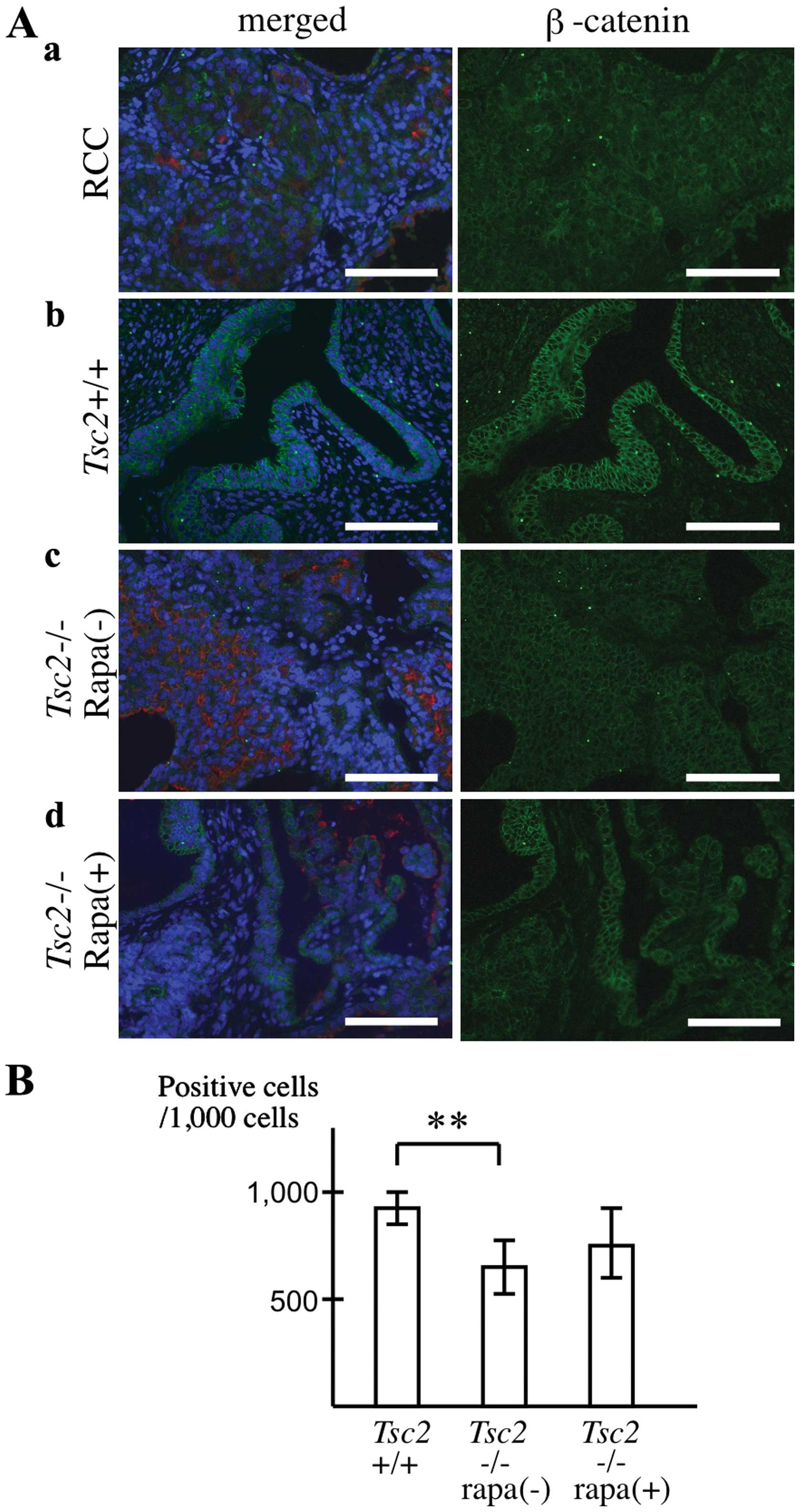
E-cadherin and β-catenin localization are
dysregulated in Tsc2−/− teratomas
Recent evidence suggests that the localization of
E-cadherin is regulated by tuberin via an Akt/mTORC1-dependent
signaling pathway and that Tsc2−/− epithelial cells
display a loss of plasma membrane E-cadherin leading to decreased
cell-cell adhesion (19).
Regulation of β-catenin by hamartin/tuberin complex was also
reported (20,21). Therefore, we examined the expression
of both E-cadherin and β-catenin by dual staining with cubilin.
In Eker rat RCCs, both E-cadherin and β-catenin
primarily localized to the cytoplasm and were weakly detectable in
the plasma membrane (Figs. 4A and
5A). In Tsc2−/−
teratomas, abnormal ductal structures exhibited both E-cadherin and
β-catenin staining at the plasma membrane, but the intensity was
weaker and more heterogeneous compared with that in the normal
ducts (Figs. 4B and 5B). Furthermore, rapamycin treatment
increased the intensity and homogeneity of plasma membrane
E-cadherin and β-catenin in Tsc2−/− teratomas,
although its effect on β-catenin was relatively weak (Figs. 4B and 5B). Taken together, these observations
suggest that the membrane localization of E-cadherin and β-catenin
was dysregulated by Tsc2 deficiency in an mTORC1-dependent
manner.
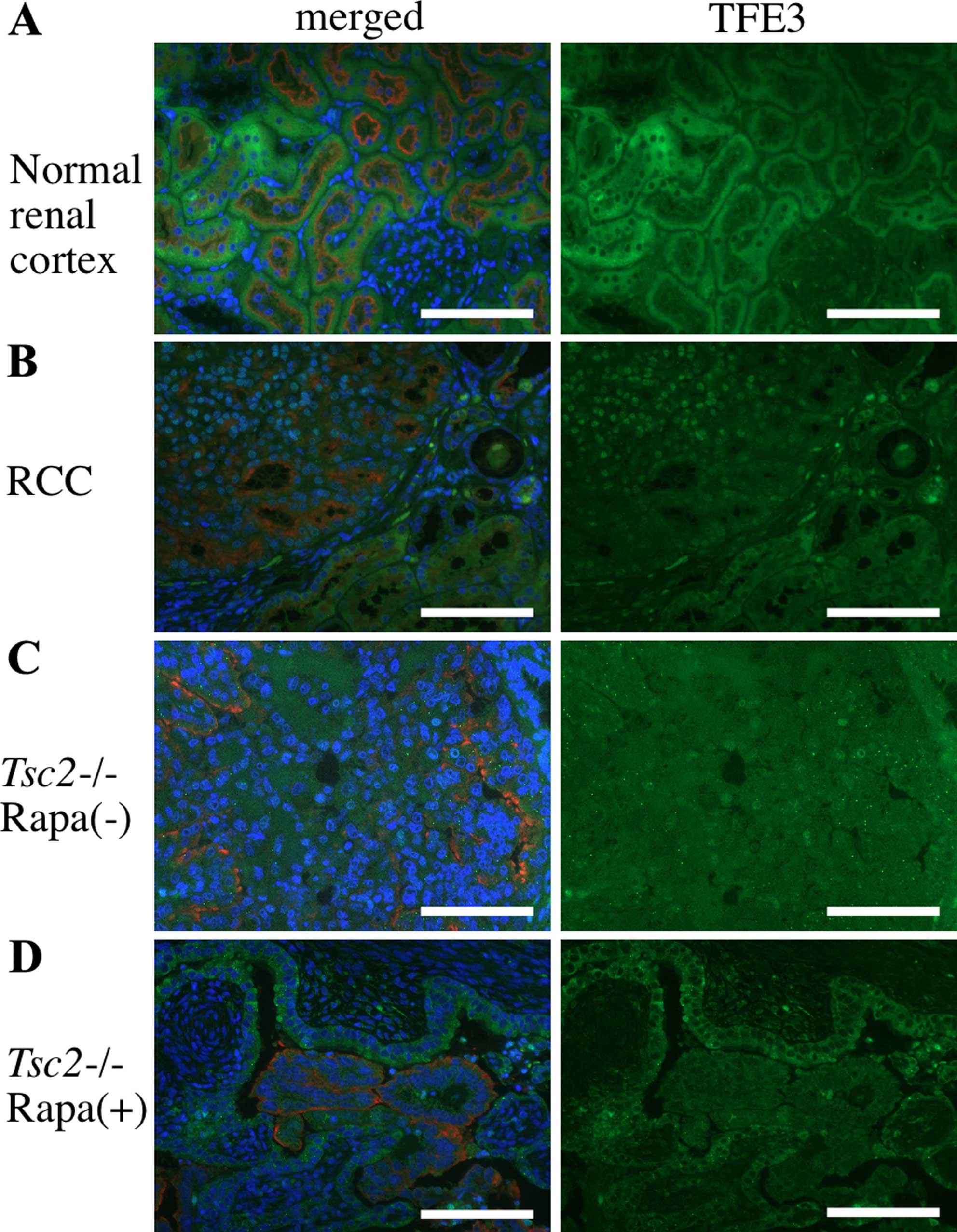
Enhanced nuclear localization of TFE3 in
Tsc2−/− teratomas
A recent study provided evidence regarding the
regulation of transcription factor EB (TFEB) and transcription
factor E3 (TFE3) by mTORC1 (22,23).
Deregulated expression and/or gene rearrangement of these members
of the MiTF/TFE transcription factor family have been implicated in
the development of RCC (24,25).
In fact, TFEB is essential for the expression of genes involved in
autophagy and lysosome biogenesis and is negatively regulated by
mTORC1 (26) On the other hand,
TFE3 was recently identified as a key player in mouse ESCs to
maintain their self-renewal state and prevent differentiation
(22). Interestingly, the knockdown
of Tsc2 increased the nuclear TFE3 concentration through the
mTORC1 pathway. Therefore, the localization of TFEB and TFE3 was
documented in Eker rat kidneys and teratomas by
immunohistochemistry analysis.
In normal kidneys, Tsc2+/+ and
Tsc2+/− teratomas, TFE3 was found in the cytoplasm of
epithelial cells (Fig. 6). On the
other hand, nuclear localization was prominent in Eker rat RCCs,
whereas surrounding normal components maintained a cytoplasmic
localization (Fig. 6). Likewise, in
Tsc2−/− teratomas, abnormal ductal structures and
the stroma clearly revealed high nuclear accumulation of TFE3.
Moreover, rapamycin completely abolished TFE3 accumulation in the
nucleus (Fig. 6). On the other
hand, there was no apparent phenotype regarding TFEB localization
(data not shown).
These results suggest that TFE3 activation by mTORC1
signaling is involved in the development of abnormal ductal
structures in Tsc2−/− teratomas.
Discussion
Various aspects of tissue specificity and
differentiation provide important insights into the mechanisms of
tumorigenesis. Differentiation experiments using pluripotent stem
cells (PSCs) constitute valid methods to explore the mechanism of
tissue-specific tumorigenesis. In general, tumorigenesis is
initiated by the loss of tumor-suppressor gene function according
to Knudson's two-hit model (27).
Since PSCs experience two hits on the tumor-suppressor gene, they
constitute ideal tools to investigate the relationship between
differentiation abnormalities and tumor initiation. In humans, it
is not easy to establish homozygous mutant PSCs for
tumor-suppressor genes. Using reprogramming technology, induced
PSCs were generated from a patient heterozygous for BRCA1 mutation
(5382insC) (28). These mutant
iPSCs exhibited increased protein kinase C θ, but the
differentiation capacity was not different between wild-type and
mutant iPSCs.
In rodents, several studies have documented
homozygous mutant PSCs for tumor-suppressor genes (14,29).
For instance, Kielman et al established mouse ESCs that were
homozygously mutated in the Apc gene (14). In these mutant ESCs, β-catenin was
upregulated. Apc-mutated teratomas revealed severe
differentiation defects in neuroectodermal, dorsal mesodermal and
endodermal lineages. These data suggest that constitutive
activation of the Apc/β-catenin pathway results in differentiation
defects in the possibly underlying tumorigenesis in the colon and
other self-renewing tissues. Kawamata and Ochiya established
Tp53-mutated rat iPSCs and rat cell lines of Tp53
mutant strains (29). Unexpectedly,
female, but not male, homozygous Tp53 mutant rats exhibited
neural tube defects. Concurrently, Tp53-null rat ESCs
resisted differentiation during the embryoid body (EB) formation
assay. Although these studies utilizing PSCs clearly illustrate the
importance of tumor-suppressor gene function in differentiation,
gross abnormalities observed in embryoid bodies or teratomas failed
to capture the detailed tissue specificity.
In the present study, the tissue differentiation of
PSCs with a deficiency in tumor-suppressor gene presented a
different scenario. In Tsc2−/− teratomas, various
tissue types were generated such as neuroepithelial tissue,
squamous epithelium, mesenchyme (undefined immature connective
tissue), smooth muscle, cartilage, bone, adipose, gastrointestinal
epithelium and glandular (unspecified) tissues. Therefore, these
findings suggest that Tsc2−/− ESCs possess the
ability to differentiate into all three germ layers. Nevertheless,
abnormal ductal structures appeared in these differentiated
tissues. In Tsc2−/− teratomas, cells of abnormal
ducts were positive for cubilin and megalin, suggesting aberrant
differentiation of some epithelial components. Although the
identity of the cell types remains unknown, some cell type-specific
effects of Tsc2 deficiency may emerge during the
differentiation of teratomas. We provide evidence that the enhanced
activation of mTORC1 pathways contributes to the development of
Tsc2−/− abnormal ducts. Since abnormal ducts
frequently and reproducibly appeared in Tsc2−/−
teratomas from different cell lines, the accumulation of specific
mutations may not be required for their development. Some
epigenetic mechanisms may support differentiation defects
associated with Tsc2 deficiency in teratomas. Such
mechanisms may be related to the tissue specificity of
tumorigenesis in Eker rats.
E-cadherin plays pivotal roles in epithelial cell
behavior, tissue formation and cancer suppression (30). During embryonic development, the
expression and function of E-cadherin must be normal for the
induction and maintenance of polarized and differentiated epithelia
(31). The lethality of E-cadherin
knockout mice at an early stage of embryogenesis highlights the
significance of E-cadherin in normal development and tissue
function (32,33). The epithelial-to-mesenchymal
transition and loss of E-cadherin expression are closely related
and believed to be involved in tumor initiation as well as
metastasis (34). An imbalance in
β-catenin signaling often results in disease and deregulated growth
related to cancer and metastasis (35). During tumor progression, β-catenin
signaling is inappropriately activated by the loss of E-cadherin or
mutants in various β-catenin signaling components (21). Eker rat RCC and abnormal ductal
structures of Tsc2−/− teratomas revealed
decreased plasma membrane localization of E-cadherin and β-catenin,
which was partially corrected by rapamycin treatment. These data
support findings from previous studies, suggesting that Tsc2
deficiency affects E-cadherin localization through perturbations of
transport mechanisms (19). The
dysregulation of E-cadherin and β-catenin by mTORC1 hyperactivation
may cause a polarity defect during the development of
Tsc2−/− abnormal ducts and tumorigenesis in Eker
rats.
The transcription factor TFE3 has been implicated in
renal carcinogenesis (23).
However, details of TFE3 function have not been elucidated. In a
recent study, TFE3 activation by mTORC1 was determined to be
essential for the maintenance of self-renewal state and the
capacity to withstand differentiation (22). When mTORC1 is activated, TFE3 moves
to the nucleus to promote the transcription of estrogen-related
receptor β genes involved in the maintenance of self-renewal and
pluripotency (22). In our
analysis, TFE3 was detected in the nucleus of both Eker rat RCCs
and Tsc2−/− teratomas, particularly in abnormal
ductal structures.
In conclusion, we established a novel experimental
system to analyze the differentiation and cell-type specific
defects associated with Tsc2 deficiency using ESCs derived
from Eker rats. Future studies should elucidate how mTORC1
hyperactivation and other mechanisms contribute to the development
of abnormal ductal structures in Tsc2−/−
teratomas. Our system will facilitate the understanding of the
pathogenesis caused by Tsc2 deficiency in Eker rats as well
as in human tumor stem cells.
Acknowledgments
The present study was supported in part by the
following grants: Grants-in-Aid for Scientific Research from the
Ministry of Education, Culture, Sports, Science and Technology
(MEXT), Japan, the MEXT-supported Program for the Strategic
Research Foundation at Private Universities, and Science (Japan),
Grants-in-Aid for Scientific Research from the Japan Society for
the Promotion of Science, Japan, and the Grants-in-Aid for
Scientific Research from the Ministry of Health, Labour and Welfare
(Japan). We wish to acknowledge all members of the Department of
Molecular Pathogenesis, Juntendo University Graduate School of
Medicine who participated in this study. The authors would like to
thank Enago for the English language review. Finally, we would like
to thank the Gender Equality Promotion Center, Juntendo University,
for their assistance with our experiments.
References
|
1
|
Eker E and Mossige J: A Dominant gene for
renal adenomas in the rat. Nature. 189:858–859. 1961. View Article : Google Scholar
|
|
2
|
Kobayashi T, Hirayama Y, Kobayashi E, Kubo
Y and Hino O: A germline insertion in the tuberous sclerosis (Tsc2)
gene gives rise to the Eker rat model of dominantly inherited
cancer. Nat Genet. 9:70–74. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yeung RS, Xiao GH, Jin F, Lee WC, Testa JR
and Knudson AG: Predisposition to renal carcinoma in the Eker rat
is determined by germ-line mutation of the tuberous sclerosis 2
(TSC2) gene. Proc Natl Acad Sci USA. 91:11413–11416. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rennebeck G, Kleymenova EV, Anderson R,
Yeung RS, Artzt K and Walker CL: Loss of function of the tuberous
sclerosis 2 tumor suppressor gene results in embryonic lethality
characterized by disrupted neuroepithelial growth and development.
Proc Natl Acad Sci USA. 95:15629–15634. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Everitt JI, Goldsworthy TL, Wolf DC and
Walker CL: Hereditary renal cell carcinoma in the Eker rat: A
rodent familial cancer syndrome. J Urol. 148:1932–1936.
1992.PubMed/NCBI
|
|
6
|
Hino O, Klein-Szanto AJ, Freed JJ, Testa
JR, Brown DQ, Vilensky M, Yeung RS, Hino O, Klein-Szanto AJ, Freed
JJ, Testa JR, Brown DQ, Vilensky M, Yeung RS, Tartof KD and Knudson
AG: Spontaneous and radiation-induced renal tumors in the Eker rat
model of dominantly inherited cancer. Proc Natl Acad Sci USA.
90:327–331. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kandt RS, Haines JL, Smith M, Northrup H,
Gardner RJ, Short MP, Dumars K, Kandt RS, Haines JL, Smith M, et
al: Linkage of an important gene locus for tuberous sclerosis to a
chromosome 16 marker for polycystic kidney disease. Nat Genet.
2:37–41. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
European Chromosome 16 Tuberous Sclerosis
Consortium: Identification and characterization of the tuberous
sclerosis gene on chromosome 16. Cell. 75:1305–1315. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
van Slegtenhorst M, de Hoogt R, Hermans C,
Nellist M, Janssen B, Verhoef S, Lindhout D, van den Ouweland A,
Halley D, Young J, et al: Identification of the tuberous sclerosis
gene TSC1 on chromosome 9q34. Science. 277:805–808. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Orlova KA and Crino PB: The tuberous
sclerosis complex. Ann NY Acad Sci. 1184:87–105. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim DH, Sarbassov DD, Ali SM, King JE,
Latek RR, Erdjument-Bromage H, Tempst P and Sabatini DM: mTOR
interacts with raptor to form a nutrient-sensitive complex that
signals to the cell growth machinery. Cell. 110:163–175. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kenerson HL, Aicher LD, True LD and Yeung
RS: Activated mammalian target of rapamycin pathway in the
pathogenesis of tuberous sclerosis complex renal tumors. Cancer
Res. 62:5645–5650. 2002.PubMed/NCBI
|
|
13
|
Sarbassov DD, Ali SM, Kim DH, Guertin DA,
Latek RR, Erdjument-Bromage H, Tempst P and Sabatini DM: Rictor, a
novel binding partner of mTOR, defines a rapamycin-insensitive and
raptor-independent pathway that regulates the cytoskeleton. Curr
Biol. 14:1296–1302. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kielman MF, Rindapää M, Gaspar C, van
Poppel N, Breukel C, van Leeuwen S, Taketo MM, Roberts S, Smits R
and Fodde R: Apc modulates embryonic stem-cell differentiation by
controlling the dosage of beta-catenin signaling. Nat Genet.
32:594–605. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ito Y, Kawano H, Kanai F, Nakamura E, Tada
N, Takai S, Horie S, Arai H, Kobayashi T and Hino O: Establishment
of Tsc2-deficient rat embryonic stem cells. Int J Oncol.
46:1944–1952. 2015.PubMed/NCBI
|
|
16
|
McDorman KS and Wolf DC: Use of the
spontaneous Tsc2 knockout (Eker) rat model of hereditary renal cell
carcinoma for the study of renal carcinogens. Toxicol Pathol.
30:675–680. 2002. View Article : Google Scholar
|
|
17
|
Wolf DC, Whiteley HE and Everitt JI:
Preneoplastic and neoplastic lesions of rat hereditary renal cell
tumors express markers of proximal and distal nephron. Vet Pathol.
32:379–386. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Verroust PJ and Christensen EI: Megalin
and cubilin - the story of two multipurpose receptors unfolds.
Nephrol Dial Transplant. 17:1867–1871. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Barnes EA, Kenerson HL, Jiang X and Yeung
RS: Tuberin regulates E-cadherin localization: Implications in
epithelial-mesenchymal transition. Am J Pathol. 177:1765–1778.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cadigan KM and Nusse R: Wnt signaling: A
common theme in animal development. Genes Dev. 11:3286–3305. 1997.
View Article : Google Scholar
|
|
21
|
Valenta T, Hausmann G and Basler K: The
many faces and functions of β-catenin. EMBO J. 31:2714–2736. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Betschinger J, Nichols J, Dietmann S,
Corrin PD, Paddison PJ and Smith A: Exit from pluripotency is gated
by intracellular redistribution of the bHLH transcription factor
Tfe3. Cell. 153:335–347. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hong SB, Oh H, Valera VA, Baba M, Schmidt
LS and Linehan WM: Inactivation of the FLCN tumor suppressor gene
induces TFE3 transcriptional activity by increasing its nuclear
localization. PLoS One. 5:e157932010. View Article : Google Scholar
|
|
24
|
Huan C, Sashital D, Hailemariam T, Kelly
ML and Roman CA: Renal carcinoma-associated transcription factors
TFE3 and TFEB are leukemia inhibitory factor-responsive
transcription activators of E-cadherin. Proc Natl Acad Sci USA.
100:6051–6056. 2003.
|
|
25
|
Davis IJ, Hsi BL, Arroyo JD, Vargas SO,
Yeh YA, Motyckova G, Valencia P, Perez-Atayde AR, Argani P, Ladanyi
M, et al: Cloning of an Alpha-TFEB fusion in renal tumors harboring
the t(6;11)(p21;q13) chromosome translocation. Proc Natl Acad Sci
USA. 100:6051–6056. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Martina JA, Chen Y, Gucek M and
Puertollano R: MTORC1 functions as a transcriptional regulator of
autophagy by preventing nuclear transport of TFEB. Autophagy.
8:903–914. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Knudson AG Jr: Mutation and cancer:
Statistical study of retinoblastoma. Proc Natl Acad Sci USA.
68:820–823. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Soyombo AA, Wu Y, Kolski L, Rios JJ,
Rakheja D, Chen A, Kehler J, Hampel H, Coughran A and Ross TS:
Analysis of induced pluripotent stem cells from a BRCA1 mutant
family. Stem Cell Rep. 1:336–349. 2013. View Article : Google Scholar
|
|
29
|
Kawamata M and Ochiya T: Two distinct
knockout approaches highlight a critical role for p53 in rat
development. Sci Rep. 2:9452012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
van Roy F and Berx G: The cell-cell
adhesion molecule E-cadherin. Cell Mol Life Sci. 65:3756–3788.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takeichi M: Cadherin cell adhesion
receptors as a morphogenetic regulator. Science. 251:1451–1455.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Riethmacher D, Brinkmann V and Birchmeier
C: A targeted mutation in the mouse E-cadherin gene results in
defective preimplantation development. Proc Natl Acad Sci USA.
92:855–859. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Larue L, Ohsugi M, Hirchenhain J and
Kemler R: E-cadherin null mutant embryos fail to form a
trophectoderm epithelium. Proc Natl Acad Sci USA. 91:8263–8267.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tiwari N, Gheldof A, Tatari M and
Christofori G: EMT as the ultimate survival mechanism of cancer
cells. Semin Cancer Biol. 22:194–207. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Roose J and Clevers H: TCF transcription
factors: Molecular switches in carcinogenesis. Biochim Biophys
Acta. 1424:M23–M37. 1999.PubMed/NCBI
|