Introduction
Epithelial ovarian cancer, being the fourth leading
cause of female cancer-related death in the developed world, is one
of the most common and lethal gynecologic malignancies (1). The majority of ovarian cancer patients
are diagnosed at advanced stages and undergo maximal cytoreductive
surgery followed by a program of chemotherapy with a platinum agent
and paclitaxel (2). While response
to initial chemotherapy is remarkable in most cases, a significant
proportion of ovarian cancers are originally resistant to platinum-
and taxane-based chemotherapy, and even those ovarian cancers which
respond well to the initial chemotherapy eventually recur with a
high probability, most likely with increased chemoresistance
(1,2). Such intrinsic and acquired
chemoresistance makes the management of ovarian cancer difficult
and is therefore a contributing factor to the dismal prognosis of
ovarian cancer. Apparently, elucidation of the underlying mechanism
and development of novel measures to overcome the chemoresistance
of ovarian cancer are direly needed to improve its prognosis. To
date, extensive studies have identified a plethora of genes and
molecular pathways implicated in the chemoresistance of ovarian
cancer (3,4); however, such information has not yet
been fully translated into specific, effective measures to overcome
the chemoresistance of ovarian cancer. Here in the present study,
we investigated the molecular pathways responsible for the
resistance of chemoresistant ovarian cancer cells. Our data suggest
that JNK pathway activation plays a significant role in the
chemoresistance of ovarian cancer cells and that JNK inhibition
prior to the application of paclitaxel or cisplatin effectively
sensitizes chemoresistant ovarian cancer cells to these
chemotherapeutic agents.
Materials and methods
Reagents and antibodies
SP600125 was purchased from Calbiochem (La Jolla,
CA, USA) and dissolved in dimethyl-sulfoxide (DMSO) to prepare a 50
mM stock solution. Cisplatin and paclitaxel were purchased from
Sigma-Aldrich (St. Louis, MO, USA) and Wako Pure Chemical
Industries, Ltd. (Osaka, Japan), respectively, and were dissolved
in DMSO to prepare 100 mM and 1 mM stock solutions, respectively.
Anti-c-Jun (#9165), anti-phospho-c-Jun (#9261) and anti-phospho-JNK
(#9251) antibodies were purchased from Cell Signaling Technology,
Inc. (Beverly, MA, USA). Anti-JNK1 (sc-474) and anti-JNK2 (sc-7345)
antibodies were purchased from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA), whereas anti-β-actin (A1978) was from
Sigma-Aldrich.
Cell culture
Human ovarian cancer cell lines, TOV21-G and SKOV-3,
were purchased from the American Type Culture Collection (ATCC,
Manassas, VA, USA). A2780 was a kind gift from Dr T. Tsuruo at the
Institute of Molecular and Cellular Biosciences, University of
Tokyo, Japan and Drs R.F. Ozols and T.C. Hamilton at the National
Institutes of Health, USA (5).
A2780CP was kindly provided by the Department of Obstetrics and
Gynecology, Osaka University, Japan. SKOV-3ip1 was a kind gift from
Dr M.C. Hung at MD Anderson Cancer Center, University of Texas, USA
(6). RMG-1 was kindly provided by
Dr S. Nozawa and Dr D. Aoki at Keio University, Japan (7). TOV-21G, SKOV-3 and SKOV-3ip1 cell
lines were maintained in M199:105 medium, a 1:1 mixture of M199 and
MCDB105 media supplemented with 10% (for SKOV-3 and SKOV-3ip1) or
15% (for TOV-21G) fetal bovine serum (FBS; Sigma) (8,9).
RMG-1, A2780 and A2780CP cell lines were maintained in DMEM/F12
medium supplemented with 10% FBS (10,11).
Normal human IMR90 fetal lung fibroblasts, NIH3T3 mouse fibroblasts
and Rat1 rat fibroblasts were obtained from ATCC and maintained in
DMEM supplemented with 10% FBS. The culture medium was also
supplemented with 100 U/ml penicillin and 100 µg/ml
streptomycin. The culture medium was changed every 3 days. The
authenticity of A2780CP, RMG-1, and SKOV-3 cells was verified by
genotyping of short tandem repeat (STR) loci (BEX Co., Ltd., Tokyo,
Japan) followed by comparison with the ATCC STR database for human
cell lines. All IMR90 experiments were performed using low passage
number (<8) cells.
Cell viability assays
Viable and dead cells were identified by their
ability and inability to exclude vital dyes, respectively (12). In brief, cells were stained with
0.2% trypan blue, and the numbers of viable and dead cells were
determined using a hemocytometer. Cell viability (%) was defined as
100 × [number of viable cells/(number of viable + dead cells)],
whereas the percentage of dead cells was defined as 100 × [number
of dead cells/(number of viable + dead cells)]. To determine the
IC50 values of cisplatin and paclitaxel for the ovarian
cancer cell lines used in the present study, we treated the cells
with varying concentrations of cisplatin or paclitaxel for 3 days
and then determined their viability. The IC50 values
were calculated using the following formula (13):
IC50=10[log(A/B)×(50−C)]/[(D−C)+Log(B)] where
A and B are the corresponding concentrations of the test drug
directly above and below 50% inhibition, respectively, and C and D
are the percentage of inhibition directly below and above 50%
inhibition, respectively.
Immunoblot analysis
Immunoblot analysis was conducted as described
previously (14). In brief, cells
were washed with ice-cold PBS and lysed in RIPA buffer [10 mM
Tris-HCl (pH 7.4), 0.1% SDS, 0.1% sodium deoxycholate, 1% NP-40,
150 mM NaCl, 1 mM EDTA, 1.5 mM Na3VO4, 10 mM
NaF, 10 mM sodium pyrophosphate, 10 mM sodium β-glycerophosphate
and 1% protease inhibitor cocktail set III (Sigma-Aldrich)]. After
centrifugation for 10 min at 14,000 × g at 4°C, the supernatants
were recovered as the cell lysates, and the protein concentration
of the cell lysates was determined using a BCA protein assay kit
(Pierce Biotechnology, Inc., Rockford, IL, USA). Cell lysates
containing equal amounts of protein were separated by SDS-PAGE and
transferred to a polyvinylidene difluoride membrane. The membrane
was probed with a primary antibody and then with an appropriate
HRP-conjugated secondary antibody according to the protocol
recommended by the manufacturer of each antibody. Immunoreactive
bands were visualized using Immobilon Western Chemiluminescent HRP
Substrate (Millipore, Billerica, MA, USA).
Colony formation assay
Colony formation assay was performed as described
previously (15,16). In brief, cells were seeded at a low,
colony-forming density (1×103 cells/60-mm dish) and
cultured for ~2 weeks. The cells were then fixed with formaldehyde
(4% v/v), followed by staining with crystal violet (0.1% w/v).
Colonies [consisting of ≥50 cells derived from a single cell
(progenies)] were counted using a microscope.
Statistical analysis
Results are expressed as the means ± standard
deviations (SD), and differences were compared using the two-tailed
Student's t-test. P<0.05 was considered to indicate a
statistically significant difference and is indicated with
asterisks in the figures.
Results
Increased JNK pathway activity in human
ovarian cancer cell lines resistant to cisplatin and
paclitaxel
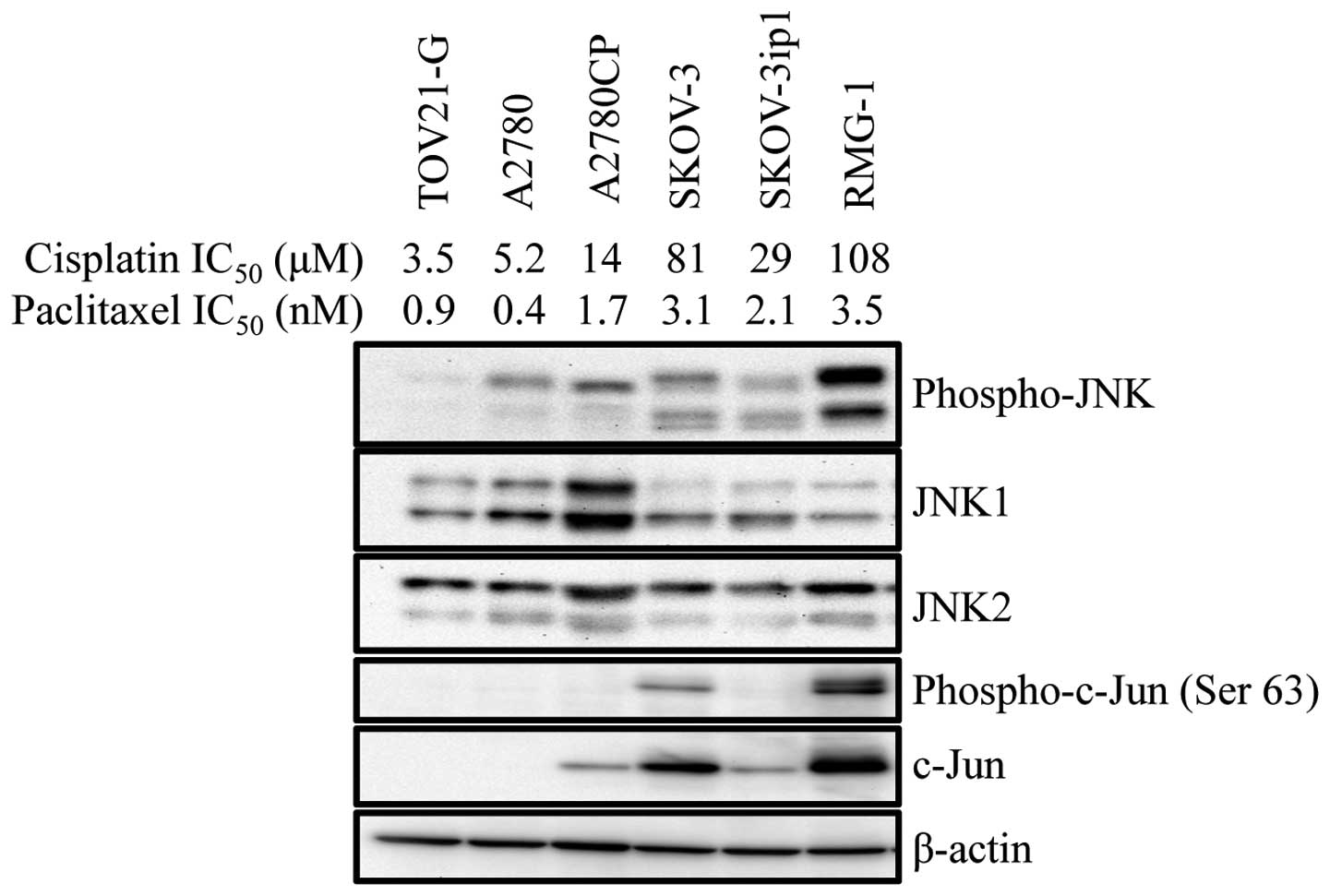
We previously reported that the expression of a
dominant-negative c-Jun mutant sensitizes human ovarian cancer cell
lines (A2780 and Caov-3) to cisplatin (17), which prompted us to hypothesize that
the JNK pathway may have a role in the development of
chemoresistance in ovarian cancer cells. As an initial approach to
test this idea, we first examined the basal JNK activity in human
ovarian cancer cell lines with various sensitivity/resistance to
cisplatin. The results indicated that cisplatin-resistant cell
lines tended to have higher basal JNK activity. Intriguingly, we
also noticed at the same time that there was some parallelism
between the sensitivity/resistance to cisplatin and paclitaxel. For
instance, A2780CP, a cisplatin-resistant subline of A2780, was more
resistant than the original A2780 cell line not only to cisplatin
but also to paclitaxel (Fig. 1).
Thus, the findings suggested that the basal JNK activity may be
associated with cisplatin- and paclitaxel-resistance of human
ovarian cancer cell lines.
Enhanced anticancer effects of cisplatin
in ovarian cancer cells in the presence of a pharmacological JNK
inhibitor SP600125
We next examined whether JNK is involved in the
cisplatin resistance of the ovarian cancer cell lines. Since our
previous study using a genetic approach showed that constitutive
expression of a dominant-negative c-Jun mutant sensitized A2780 and
Caov-3 cells to cisplatin (17), we
wished to investigate in this particular study whether direct
inhibition of JNK through a pharmacological approach could overcome
the cisplatin-resistance of ovarian cancer cells, and that, of
ovarian cancer cell lines other than A2780 and Caov-3. We therefore
used SP600125, the most widely used pharmacological inhibitor of
JNK (18), and tested its effect in
three cisplatin-resistant ovarian cancer cell lines, A2780CP,
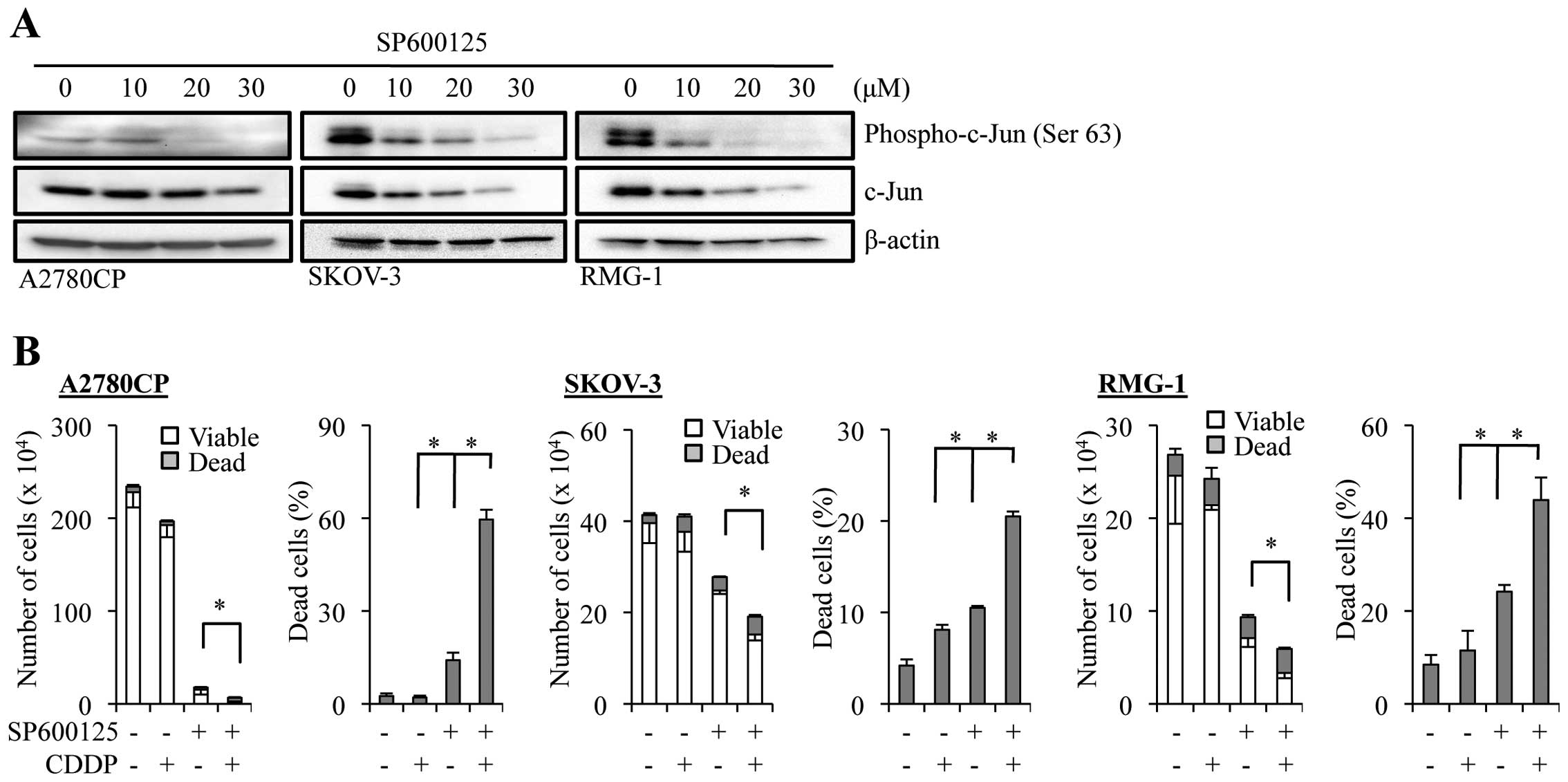
SKOV-3, and RMG-1. Since SP600125 effectively inhibited the JNK
activity at 20 µM in all three cell lines as indicated by
the reduced phosphorylation of c-Jun at the JNK phosphorylation
site (Fig. 2A), we treated the
cells with SP600125 at 20 µM, alone or in combination with
cisplatin (50 µM), and examined the effect of the drug
treatment on their growth and viability (Fig. 2B). The results indicated that
treatment with SP600125 alone caused a substantial inhibition of
cell growth as well as a modest increase in the proportion of dead
cells in the three cell lines. On the other hand, treatment of the
cells with cisplatin alone affected their growth and viability only
marginally, as expected from their inherent resistance to
cisplatin. However, when the cells were treated with cisplatin in
the presence of SP600125, the combination treatment caused a marked
decrease in the number of viable cells accompanied by the
corresponding increase in the proportion of dead cells, compared to
treatment with either SP600125 or cisplatin alone. These findings
extended our previous findings and suggested that the role of JNK
in cisplatin resistance may be shared by different human ovarian
cancer cell lines beyond A2780 and Caov-3, and that JNK could be a
druggable target to overcome cisplatin resistance of ovarian cancer
cells.
Diminished anticancer effects of
paclitaxel in ovarian cancer cells in the presence of SP600125
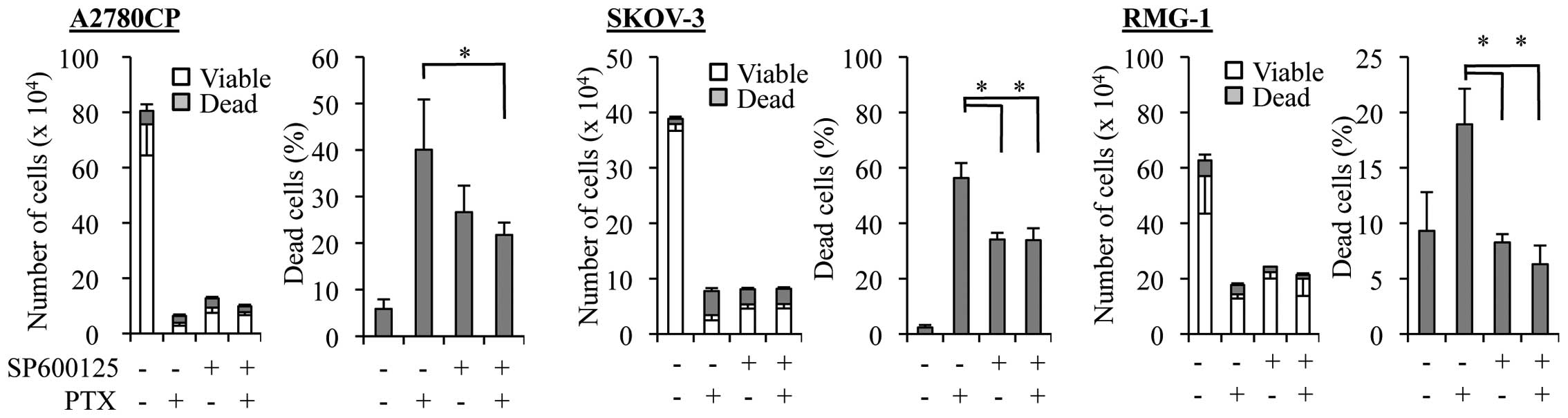
Given our earlier observation in this study that the
ovarian cancer cell lines with increased basal JNK activity were
resistant not only to cisplatin but also to paclitaxel (Fig. 1), we next asked whether SP600125
treatment could enhance the anticancer effects of paclitaxel
similarly to those of cisplatin. However, we found that the growth
inhibitory effect of paclitaxel was not at all enhanced in the
presence of SP600125. On the contrary, SP600125 even compromised
the paclitaxel effect on ovarian cancer cells. Indeed, when the
paclitaxel-resistant cell lines were treated with paclitaxel at a
sufficiently high concentration (5 nM) to cause growth inhibition
in the presence and absence of SP600125, the number of viable cells
increased whereas that of dead cells decreased in the presence of
SP600125 as compared with its absence (Fig. 3).
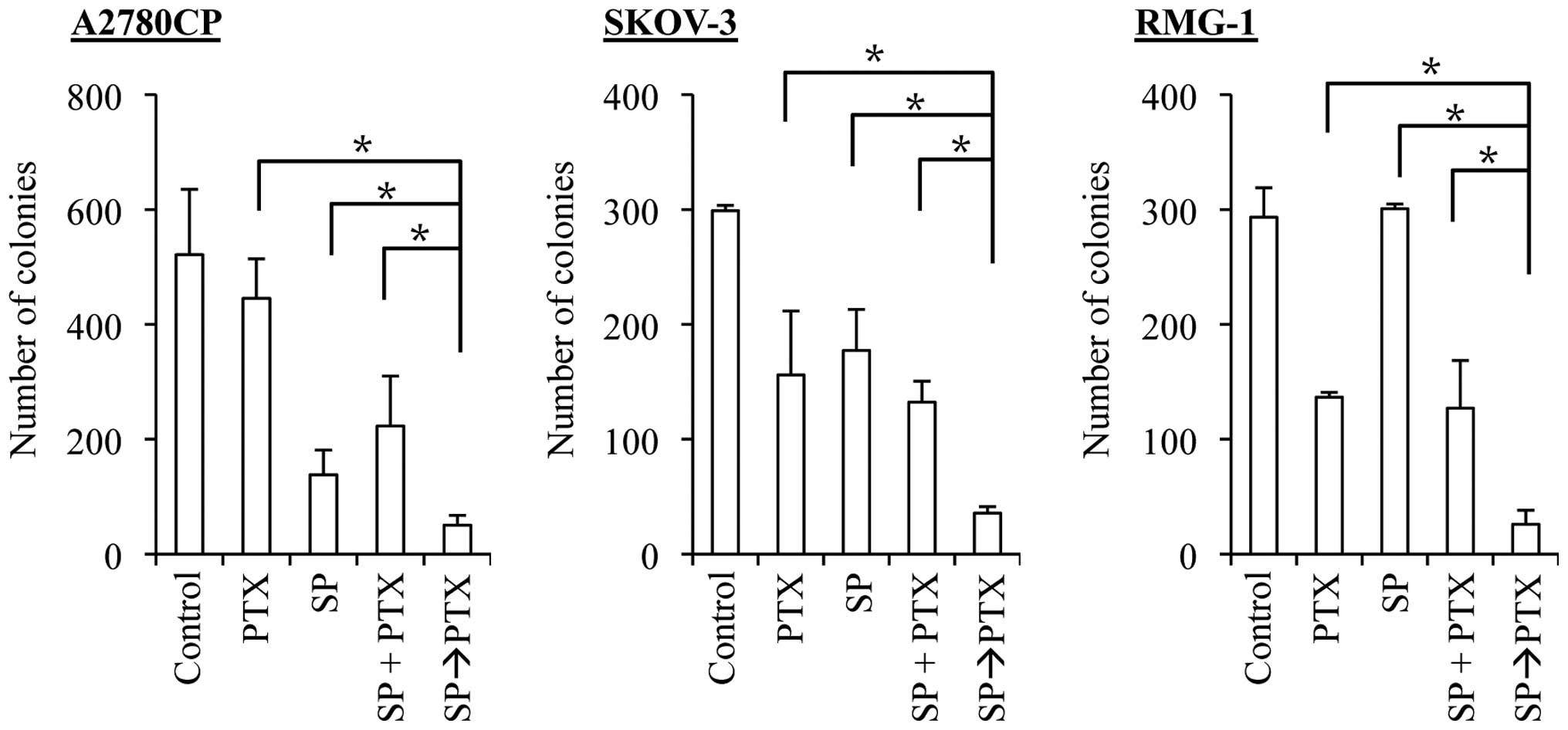
Treatment with SP600125 prior to
paclitaxel enhances the anticancer effects of paclitaxel in ovarian
cancer cells
A number of previous studies showed that paclitaxel
treatment of cancer cells, including ovarian cancer cells, caused
JNK activation and that the JNK activation was required for
paclitaxel-induced cell death (19–23).
Whereas these observations appear to imply that the 'induced' JNK
activation plays an essential role in cell death caused by
paclitaxel treatment, which is quite consistent with the results of
the present study (Fig. 3), the
role of the 'basal' JNK activity in cells treated with paclitaxel
remains unknown. To determine the role of the basal JNK activity in
the sensitivity/resistance of ovarian cancer cells to paclitaxel,
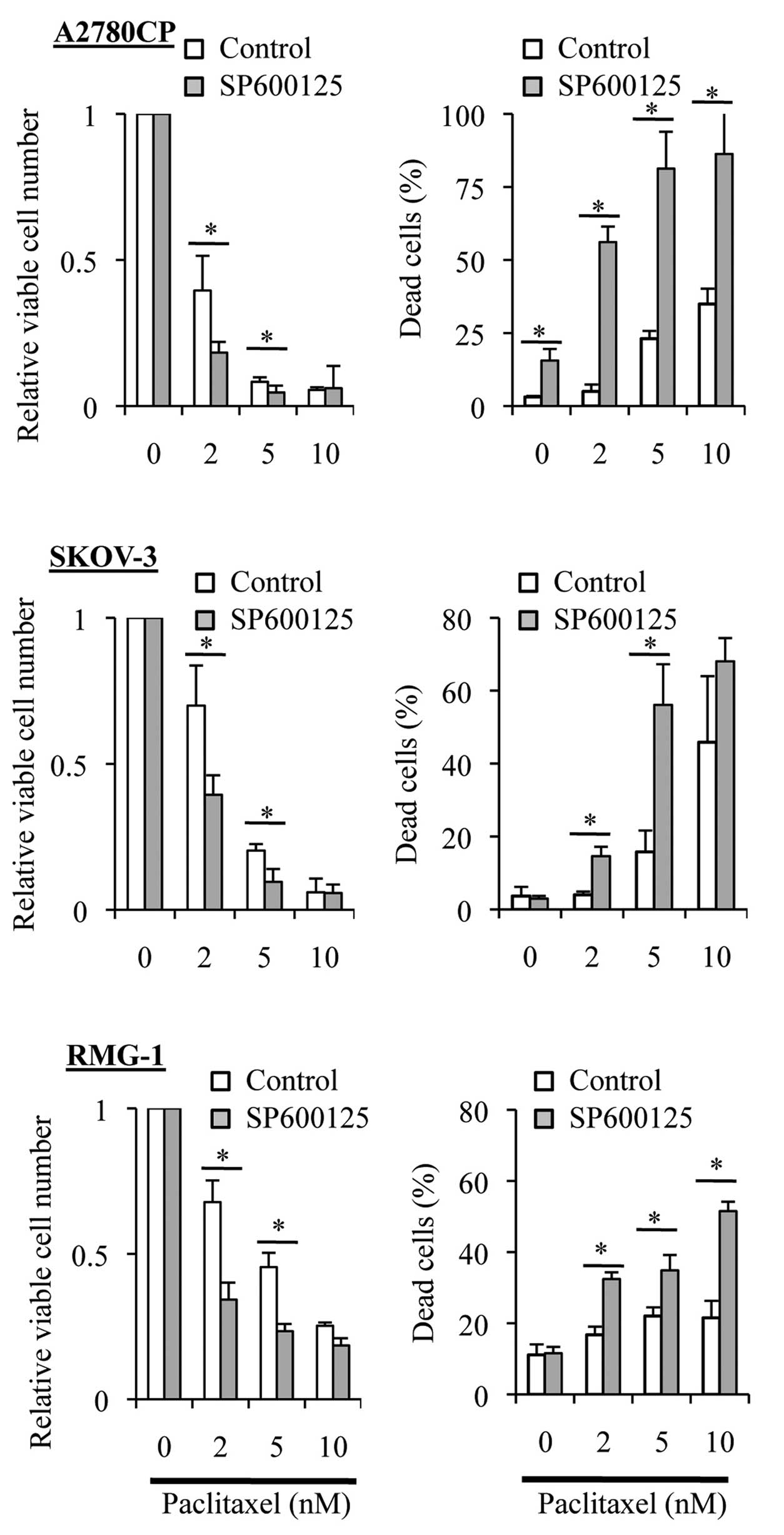
we tested the effect of time-staggered, sequential application of
SP600125 and paclitaxel in the present study. To specifically
inhibit the basal JNK activity without interfering with
paclitaxel-induced activation of JNK, the ovarian cancer cell lines
were first treated for 3 days with SP600125, which is a reversible
inhibitor of JNK, followed by paclitaxel treatment in the entire
absence of SP600125. Strikingly, in sharp contrast to the results
obtained by treating cells simultaneously with SP600125 and
paclitaxel (i.e., SP600125 co-treatment), prior treatment with
SP600125 (i.e., SP600125 pretreatment) significantly enhanced the
growth inhibitory and cell death-inducing effects of paclitaxel
over a range of concentration (Fig.
4). These observations suggested that specific inhibition of
the basal JNK activity may effectively sensitize ovarian cancer
cells to paclitaxel.
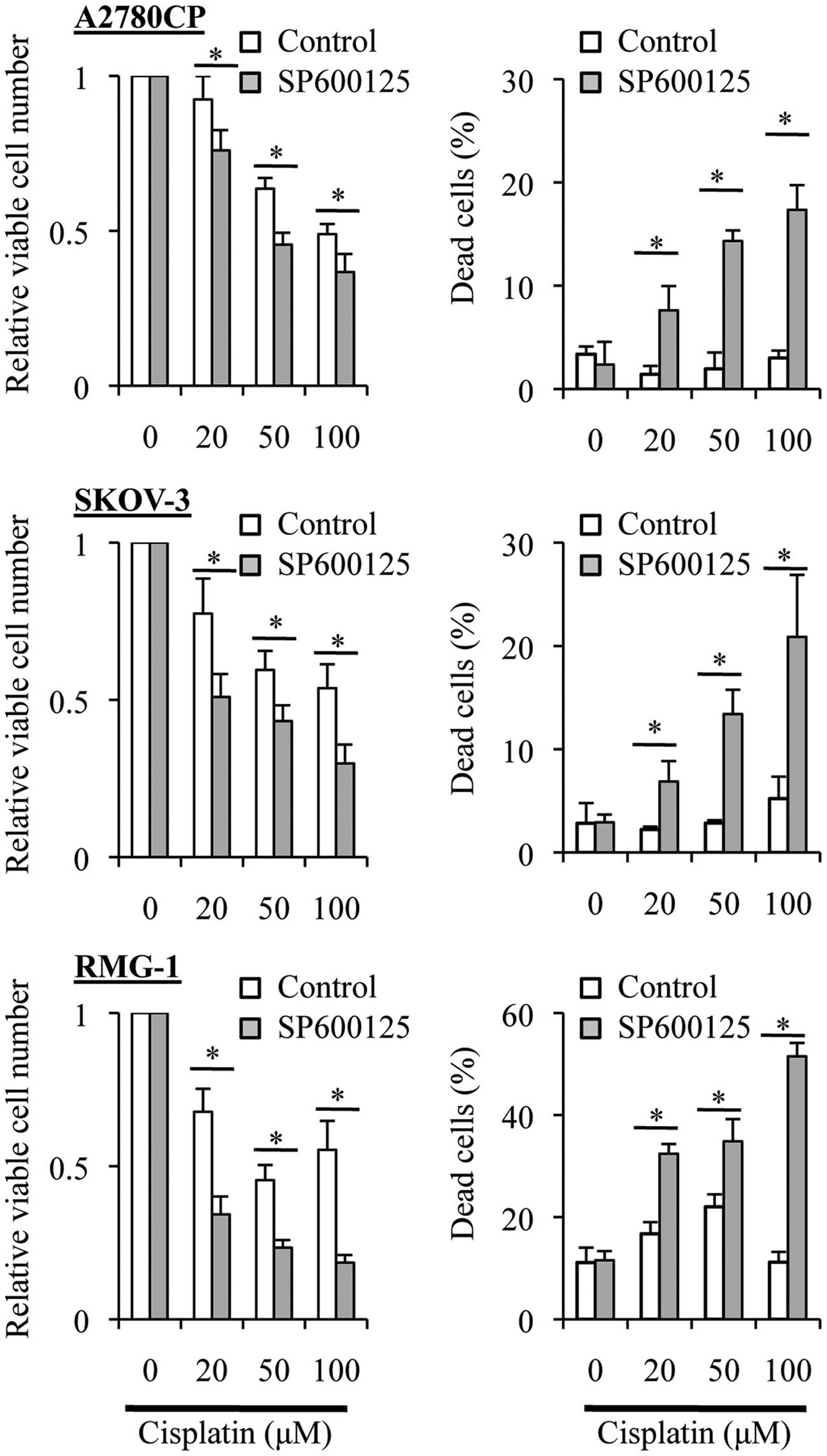
Treatment with SP600125 prior to
cisplatin enhances the anticancer effects of cisplatin in ovarian
cancer cells
Our earlier observations demonstrated that cisplatin
was different from paclitaxel in that SP600125 'co-treatment'
sensitized the ovarian cancer cell lines to cisplatin (Fig. 2) but not to paclitaxel (Fig. 3). We then asked whether or not
SP600125 'pretreatment' sensitizes the ovarian cancer cell lines to
cisplatin as it did to paclitaxel (Fig.
4). When the ovarian cancer cell lines were treated with
varying concentrations of cisplatin in the absence of SP600125
after being exposed or not to SP600125 for 3 days, we found that
the growth inhibitory and cell death-inducing effects of cisplatin
were significantly enhanced by the SP600125 pretreatment (Fig. 5). These results, together with those
obtained from the paclitaxel experiments (Fig. 4), suggested that the basal JNK
activity may be commonly involved in the resistance of the ovarian
cancer cell lines to cisplatin and paclitaxel.
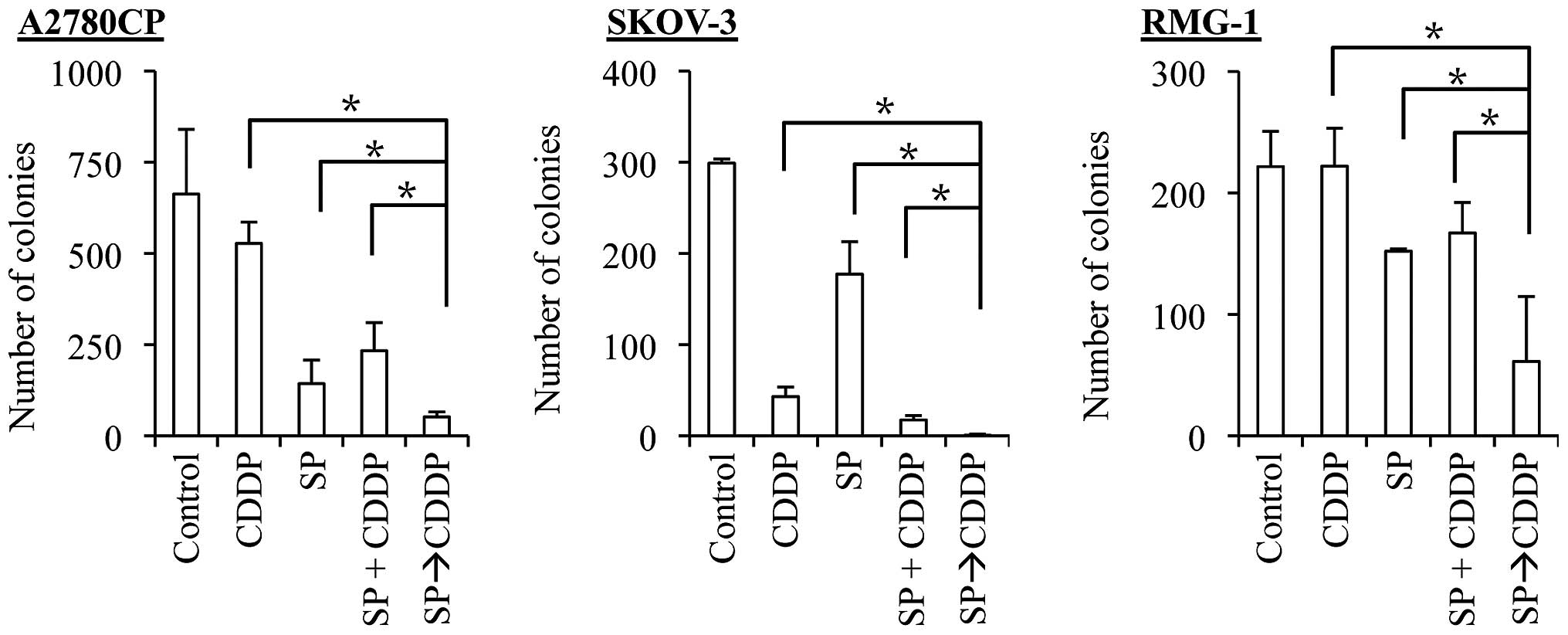
SP600125 pretreatment effectively
synergizes with both cisplatin and paclitaxel to suppress the
clonogenic survival of ovarian cancer cells
The results of the present study suggest that
SP600125 pretreatment effectively enhances the anticancer effects
of cisplatin and paclitaxel. However, we assessed the anticancer
effects of the drugs so far by determining the number and
proportion of viable and dead cells at a relatively early time
point, namely, 3 days after the application of the drugs. It was
therefore possible, for instance, that SP600125 pretreatment was
simply advancing the time-kinetics of cell death that would
eventually occur, making cells destined to die, die earlier. To
exclude such a possibility and determine the therapeutic
significance of SP600125 pretreatment (24), we examined the impact of SP600125
pretreatment on the clonogenic survival of ovarian cancer cells. To
this end, we first exposed the ovarian cancer cell lines to
SP600125 and paclitaxel alone and in combinations and subjected
them to the colony formation assay. For combination treatments,
cells were either exposed to SP600125 and paclitaxel simultaneously
(co-treatment, the SP + PTX protocol), or to SP600125 followed by
paclitaxel (pretreatment, the SP→PTX protocol). The results
indicated that, in all three cell lines examined, whereas the
co-treatment protocol was at best as effective as paclitaxel alone
in inhibiting the clonogenic survival of the cells, the
pretreatment protocol was by far more effective than paclitaxel
alone and the co-treatment protocol (Fig. 6). We then went on to conduct the
same experiment with cisplatin. In line with our earlier results,
the co-treatment protocol tended to be more effective than
cisplatin alone. However, again, the pretreatment protocol was
significantly more effective than the co-treatment protocol, just
as with paclitaxel (Fig. 7). These
findings suggested that pretreatment with SP600125 may be
beneficial in augmenting the therapeutic efficacy of cisplatin and
paclitaxel against ovarian cancer cells.
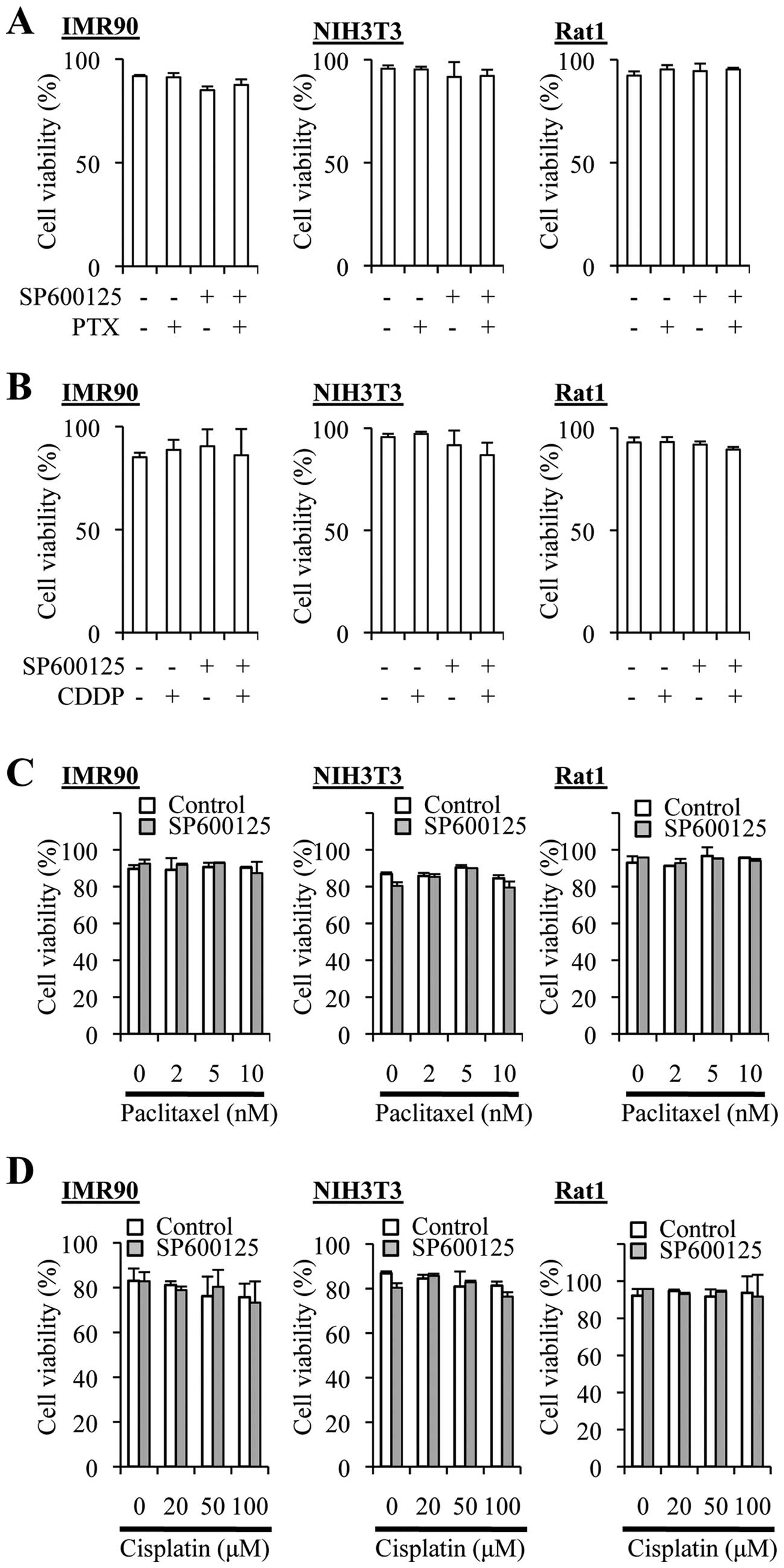
The SP600125 pretreatment protocol does
not augment the cytotoxic effect of cisplatin and paclitaxel on
non-transformed fibroblasts
We next asked whether the combination treatment
protocols might increase the cytotoxic effects of cisplatin and
paclitaxel on normal cells. To this end, we used non-transformed
fibroblasts from human (IMR90), mouse (NIH3T3), and rat (Rat1), and
tested the effects of cisplatin, paclitaxel and SP600125 alone or
in combination. Cisplatin treatment ≤100 µM alone did not
appreciably reduce the viability (=100% − %dead cells) of these
fibroblasts, and neither did paclitaxel alone even at 10 nM
(Fig. 8). SP600125 (20 µM)
alone occasionally reduced the viability of IMR90 cells
significantly, which was nevertheless not reproducible (Fig. 8A and B). Importantly, the
combination protocols (co-treatment and pretreatment protocols for
both cisplatin and paclitaxel) reduced the viability of the cells
significantly in none of the three nontransformed fibroblasts.
Thus, the results suggested that the combination protocols could
enhance the anticancer effects of the drugs without increasing
their toxicity.
Discussion
Chemoresistance eventually develops in the majority
of ovarian cancer cases during the clinical course and is therefore
a major obstacle in realizing the long-term survival of patients
with ovarian cancer. To search for strategies to overcome the
resistance of ovarian cancer to platinum- and taxane-based
chemotherapy, we attempted in this study to identify molecules
and/or pathways that may dictate the sensitivity/resistance of
ovarian cancer cells to cisplatin and paclitaxel. As a result, we
found in the present study that i) there is some parallelism
between the sensitivity/resistance of each human ovarian cancer
cell line to cisplatin and paclitaxel, ii) the JNK pathway activity
(basal activity) is positively correlated with resistance to
cisplatin and paclitaxel, iii) chemotherapeutic agent-induced
activation of JNK is likely involved in the resistance and
sensitivity to cisplatin and paclitaxel, respectively, iv) the
basal JNK activity confers resistance to both cisplatin and
paclitaxel and v) inhibition of the basal JNK activity prior to the
application of cisplatin or paclitaxel effectively augments their
ability to suppress the clonogenic survival of ovarian cancer cells
without increasing their toxicity. These findings not only provide
a useful clue to develop specific measures to sensitize ovarian
cancer to platinum- and taxane-based chemotherapy but also shed
insights into the mechanisms underlying the chemoresistance of
ovarian cancer.
Previous studies have shown that treatment of cancer
cell lines with cisplatin in vitro often leads to the
development of selective resistance to cisplatin without
cross-resistance to paclitaxel and vice versa, most likely because
of their entirely distinct mechanisms of action (25,26).
On the other hand, we observed in the present study a parallelism
between sensitivity/resistance to cisplatin and paclitaxel across
human ovarian cancer cell lines maintained in a drug-naïve culture
condition (A2780, TOV21-G, SKOV-3, SKOV-3ip1, RMG-1) and a subline
established by exposing the parental cell lines to cisplatin but
maintained in a drug-free culture condition (A2780CP).
Collectively, these observations may imply that intrinsic
resistance, which is inherent in cells irrespective of prior drug
treatment, and acquired resistance, which is induced by drug
treatment, involve different underlying mechanisms, the latter
being, as a natural consequence, more specific to the particular
chemotherapeutic agents used to induce resistance. Quite
importantly, we found that the basal JNK pathway activity was
higher in cell lines that were inherently more resistant to both
cisplatin and paclitaxel and that inhibition of the basal JNK
activity prior to the application of these drugs sensitized ovarian
cancer cells to both of them. These findings point to the
intriguing possibility that JNK plays a key role in determining the
intrinsic, basal resistance of ovarian cancer cells to platinum-
and taxane-based chemotherapy, although the mechanism involved in
the intrinsic, basal resistance could nevertheless be induced or
augmented on occasions by treatment with chemotherapeutic agents,
as is evident form the comparison of A2780 and A2780CP in the
present study. Intriguing enough, the JNK pathway has been
demonstrated to be activated in a significant proportion of ovarian
cancers (27,28). More importantly, active JNK
expression level was higher in advanced stage (III and IV) cases
than in early stage (I and II) cases and was inversely associated
with the survival of ovarian cancer patients independently of the
clinical stage (27), suggesting
that the JNK pathway activity may increase with disease progression
and have a negative impact on the clinical outcome of ovarian
cancer patients. Our results, in conjunction with these previous
observations, may give rise to a novel, provocative hypothesis that
ovarian cancers become chemoresistant not simply because they are
treated with chemotherapy but because they progress with time to
more advanced disease with increased JNK activity in their natural
clinical course.
Another important aspect of the present study is
that we delineated and highlighted the distinct roles of JNK in the
intrinsic resistance mechanism of ovarian cancer cells to cisplatin
and paclitaxel in the untreated, steady state condition and in
their acute response to these drugs. The roles of JNK in the acute
response of cancer cells to cisplatin (17,29–33)
and paclitaxel (19–23) have been well documented. In line
with the previous reports demonstrating the critical role of JNK in
the activation of the DNA repair mechanism after cisplatin
treatment (17,29–33),
cisplatin application along with the JNK inhibitor SP600125
resulted in enhanced anticancer effects compared with cisplatin
application alone in the present study. In contrast, paclitaxel
application along with SP600125 resulted in diminished anticancer
effects, again in line with the reported role of JNK in mediating
the death signal elicited by paclitaxel (19–23).
Thus, the results of the present study further strengthen the idea
that JNK has contrasting roles in the acute response of cancer
cells to cisplatin and paclitaxel. On the other hand, the role of
JNK in the mechanism(s) dictating the intrinsic sensitivity and
resistance of cancer cells not yet exposed to these drugs has been
poorly investigated and therefore remains totally obscure. Here in
the present study, we provide evidence supporting the idea that the
basal JNK activity plays a pivotal role in maintaining the
intrinsic resistance of ovarian cancer cells to both cisplatin and
paclitaxel. It still remains to be shown how JNK contributes to the
intrinsic resistance of ovarian cancer cells to both cisplatin and
paclitaxel, however, two major possibilities could be envisaged.
The first possibility is that JNK is involved separately in each of
the distinct mechanisms underlying cisplatin and paclitaxel
resistance. For intrinsic resistance to cisplatin, it may not be
difficult to assume that the basal JNK activity has a role not only
in induced but also in the steady state DNA repair activity,
conferring on cancer cells a higher capacity to repair DNA damages
prior to cisplatin exposure. As for intrinsic paclitaxel
resistance, it has been reported that cisplatin-resistant cancer
cells that are also cross-resistant to palcitaxel often have
upregulation of p-glycoprotein, which is considered to be
responsible for paclitaxel resistance (34–39).
Given the previous reports suggesting a role for JNK in the
regulation of the MDR1 gene that encodes p-glycoprotein (40,41),
it is plausible to assume that p-glycoprotein is involved in the
JNK-dependent paclitaxel resistance observed in this study.
However, the results of our pilot experiment indicated that MDR1
expression was not necessarily decreased in the cell lines
sensitized to paclitaxel by SP600125 treatment (data not shown),
suggesting that at least MDR1 expression alone may not be
accountable for the paclitaxel resistance. The second possibility
is that JNK is involved in a mechanism common to the manifestation
of the anticancer effects of cisplatin and paclitaxel, most likely
in the cell death and cell cycle pathways. In this regard, in so
far as we have examined, we have not yet identified cell death or
cell cycle molecules whose expression reasonably changes upon
SP600125 treatment. Apparently, future studies are warranted to
elucidate the mechanism by which the basal JNK activity contributes
to the intrinsic resistance of ovarian cancer cells to cisplatin
and paclitaxel.
Finally and most importantly, we demonstrated in the
present study that sequential, but not simultaneous, exposure to
SP600125 and cisplatin/paclitaxel in this order effectively
sensitizes ovarian cancer cells to these chemotherapeutic agents,
which has led us to propose that time-staggered inhibition of JNK
in combination with cisplatin and paclitaxel may be beneficial in
the treatment of ovarian cancer. Recently, there has been a growing
awareness that not only the combination and dosage of drugs, but
also the timing, duration, and order of the drugs to be combined
are key to successful combination treatment (42–44).
Our study thus provides a good illustration of the idea,
underscoring the importance of the order, timing, and duration of
drug application in combination treatment. In the present study,
the ovarian cancer cells were treated with the JNK inhibitor for 3
days before exposure to cisplatin or paclitaxel, which we consider
was required and sufficient for rewiring of the JNK signaling
network to mitigate their chemoresistance. It is also important to
emphasize here that the sequential treatment protocols
(SP600125→cisplatin, SP600125→paclitaxel) were no more toxic to
non-transformed cells than treatment with cisplatin or paclitaxel
alone, indicating that SP600125 treatment prior to the application
of the chemotherapeutic agents is quite beneficial in widening
their therapeutic window. All in all, our findings demonstrate for
the first time that, while simultaneous treatment with a JNK
inhibitor could even be hazardous because it may not be without the
risk of reducing the efficacy of taxane-based chemotherapy,
time-staggered inhibition of JNK in combination with platinum- and
taxane-based chemotherapy is highly useful in enhancing the
therapeutic effects of the chemotherapeutic agents. Furthermore, we
recently demonstrated that the JNK activity is required for the
maintenance of ovarian cancer stem cells and, although not in
ovarian but in pancreatic cancer, that time-staggered inhibition of
JNK effectively sensitizes cancer stem cells to chemotherapeutic
agents (11,12). These observations may make
combination therapies involving JNK inhibitors all the more
attractive as an approach to the treatment of ovarian cancer.
Apparently, future preclinical studies are warranted to determine
the therapeutic effects of combination treatment consisting of
time-staggered JNK inhibition and platinum- and taxane-based
chemotherapy, as a pilot to explore the clinical relevance of such
combination treatment.
Acknowledgments
We thank Ms Eriko Watanabe and Ms Asuka Sugai for
their technical and secretarial contributions to this study,
respectively. The present study was supported by Grants-in-Aid for
Scientific Research, for Challenging Exploratory Research, and for
Young Scientists from the Ministry of Education, Culture, Sports,
Science and Technology of Japan.
References
|
1
|
Jayson GC, Kohn EC, Kitchener HC and
Ledermann JA: Ovarian cancer. Lancet. 384:1376–1388. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bookman MA: The addition of new drugs to
standard therapy in the first-line treatment of ovarian cancer. Ann
Oncol. 21(Suppl 7): vii211–vii217. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu X, Gao Y, Lu Y, Zhang J, Li L and Yin
F: Oncogenes associated with drug resistance in ovarian cancer. J
Cancer Res Clin Oncol. 141:381–395. 2015. View Article : Google Scholar
|
|
4
|
Yin F, Liu X, Li D, Wang Q, Zhang W and Li
L: Tumor suppressor genes associated with drug resistance in
ovarian cancer (Review). Oncol Rep. 30:3–10. 2013.PubMed/NCBI
|
|
5
|
Hamilton TC, Winker MA, Louie KG, Batist
G, Behrens BC, Tsuruo T, Grotzinger KR, McKoy WM, Young RC and
Ozols RF: Augmentation of adriamycin, melphalan, and cisplatin
cytotoxicity in drug-resistant and -sensitive human ovarian
carcinoma cell lines by buthionine sulfoximine mediated glutathione
depletion. Biochem Pharmacol. 34:2583–2586. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu D, Wolf JK, Scanlon M, Price JE and
Hung MC: Enhanced c-erbB-2/neu expression in human ovarian cancer
cells correlates with more severe malignancy that can be suppressed
by E1A. Cancer Res. 53:891–898. 1993.PubMed/NCBI
|
|
7
|
Suzuki N, Aoki D, Tamada Y, Susumu N,
Orikawa K, Tsukazaki K, Sakayori M, Suzuki A, Fukuchi T, Mukai M,
et al: HMOCC-1, a human monoclonal antibody that inhibits adhesion
of ovarian cancer cells to human mesothelial cells. Gynecol Oncol.
95:290–298. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu Z, Yamanouchi K, Ohtao T, Matsumura S,
Seino M, Shridhar V, Takahashi T, Takahashi K and Kurachi H: High
levels of Wilms' tumor 1 (WT1) expression were associated with
aggressive clinical features in ovarian cancer. Anticancer Res.
34:2331–2340. 2014.PubMed/NCBI
|
|
9
|
Yamanouchi K, Ohta T, Liu Z, Oji Y,
Sugiyama H, Shridhar V, Matsumura S, Takahashi T, Takahashi K and
Kurachi H: The Wilms' tumor gene WT1 - 17AA/- KTS splice variant
increases tumorigenic activity through up-regulation of vascular
endo-thelial growth factor in an in vivo ovarian cancer model.
Transl Oncol. 7:580–589. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ohta T, Ohmichi M, Shibuya T, Takahashi T,
Tsutsumi S, Takahashi K and Kurachi H: Gefitinib (ZD1839) increases
the efficacy of cisplatin in ovarian cancer cells. Cancer Biol
Ther. 13:408–416. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Seino M, Okada M, Shibuya K, Seino S,
Suzuki S, Ohta T, Kurachi H and Kitanaka C: Requirement of JNK
signaling for self-renewal and tumor-initiating capacity of ovarian
cancer stem cells. Anticancer Res. 34:4723–4731. 2014.PubMed/NCBI
|
|
12
|
Suzuki S, Okada M, Shibuya K, Seino M,
Sato A, Takeda H, Seino S, Yoshioka T and Kitanaka C: JNK
suppression of chemotherapeutic agents-induced ROS confers
chemoresistance on pancreatic cancer stem cells. Oncotarget.
6:458–470. 2015. View Article : Google Scholar :
|
|
13
|
Kiguchi K, Kubota T, Aoki D, Udagawa Y,
Yamanouchi S, Saga M, Amemiya A, Sun FX, Nozawa S, Moossa AR, et
al: A patient-like orthotopic implantation nude mouse model of
highly metastatic human ovarian cancer. Clin Exp Metastasis.
16:751–756. 1998. View Article : Google Scholar
|
|
14
|
Okada M, Shibuya K, Sato A, Seino S,
Suzuki S, Seino M and Kitanaka C: Targeting the K-Ras-JNK axis
eliminates cancer stem-like cells and prevents pancreatic tumor
formation. Oncotarget. 5:5100–5112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Okada M, Sato A, Shibuya K, Watanabe E,
Seino S, Suzuki S, Seino M, Narita Y, Shibui S, Kayama T, et al:
JNK contributes to temozolomide resistance of stem-like
glioblastoma cells via regulation of MGMT expression. Int J Oncol.
44:591–599. 2014.
|
|
16
|
Wang X, Song H, Yu Q, Liu Q, Wang L, Liu Z
and Yu Z: Ad-p53 enhances the sensitivity of triple-negative breast
cancer MDA-MB-468 cells to the EGFR inhibitor gefitinib. Oncol Rep.
33:526–532. 2015.
|
|
17
|
Hayakawa J, Ohmichi M, Kurachi H, Ikegami
H, Kimura A, Matsuoka T, Jikihara H, Mercola D and Murata Y:
Inhibition of extracellular signal-regulated protein kinase or
c-Jun N-terminal protein kinase cascade, differentially activated
by cisplatin, sensitizes human ovarian cancer cell line. J Biol
Chem. 274:31648–31654. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bogoyevitch MA and Arthur PG: Inhibitors
of c-Jun N-terminal kinases: JuNK no more? Biochim Biophys Acta.
1784:76–93. 2008. View Article : Google Scholar
|
|
19
|
Kolomeichuk SN, Terrano DT, Lyle CS,
Sabapathy K and Chambers TC: Distinct signaling pathways of
microtubule inhibitors - vinblastine and Taxol induce JNK-dependent
cell death but through AP-1-dependent and AP-1-independent
mechanisms, respectively. FEBS J. 275:1889–1899. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee LF, Li G, Templeton DJ and Ting JP:
Paclitaxel (Taxol)- induced gene expression and cell death are both
mediated by the activation of c-Jun NH2-terminal kinase (JNK/SAPK).
J Biol Chem. 273:28253–28260. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Selimovic D, Hassan M, Haikel Y and Hengge
UR: Taxol-induced mitochondrial stress in melanoma cells is
mediated by activation of c-Jun N-terminal kinase (JNK) and p38
pathways via uncoupling protein 2. Cell Signal. 20:311–322. 2008.
View Article : Google Scholar
|
|
22
|
Sunters A, Madureira PA, Pomeranz KM,
Aubert M, Brosens JJ, Cook SJ, Burgering BM, Coombes RC and Lam EW:
Paclitaxel-induced nuclear translocation of FOXO3a in breast cancer
cells is mediated by c-Jun NH2-terminal kinase and Akt. Cancer Res.
66:212–220. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang TH, Popp DM, Wang HS, Saitoh M, Mural
JG, Henley DC, Ichijo H and Wimalasena J: Microtubule dysfunction
induced by paclitaxel initiates apoptosis through both c-Jun
N-terminal kinase (JNK)-dependent and -independent pathways in
ovarian cancer cells. J Biol Chem. 274:8208–8216. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Abend M: Reasons to reconsider the
significance of apoptosis for cancer therapy. Int J Radiat Biol.
79:927–941. 2003. View Article : Google Scholar
|
|
25
|
Stordal B and Davey R: A systematic review
of genes involved in the inverse resistance relationship between
cisplatin and paclitaxel chemotherapy: Role of BRCA1. Curr Cancer
Drug Targets. 9:354–365. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stordal B, Pavlakis N and Davey R: A
systematic review of platinum and taxane resistance from bench to
clinic: An inverse relationship. Cancer Treat Rev. 33:688–703.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Eckhoff K, Flurschütz R, Trillsch F,
Mahner S, Jänicke F and Milde-Langosch K: The prognostic
significance of Jun transcription factors in ovarian cancer. J
Cancer Res Clin Oncol. 139:1673–1680. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vivas-Mejia P, Benito JM, Fernandez A, Han
HD, Mangala L, Rodriguez-Aguayo C, Chavez-Reyes A, Lin YG, Carey
MS, Nick AM, et al: c-Jun-NH2-kinase-1 inhibition leads to
antitumor activity in ovarian cancer. Clin Cancer Res. 16:184–194.
2010. View Article : Google Scholar
|
|
29
|
Hayakawa J, Depatie C, Ohmichi M and
Mercola D: The activation of c-Jun NH2-terminal kinase (JNK) by
DNA-damaging agents serves to promote drug resistance via
activating transcription factor 2 (ATF2)-dependent enhanced DNA
repair. J Biol Chem. 278:20582–20592. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hayakawa J, Mittal S, Wang Y, Korkmaz KS,
Adamson E, English C, Ohmichi M, McClelland M and Mercola D:
Identification of promoters bound by c-Jun/ATF2 during rapid
large-scale gene activation following genotoxic stress. Mol Cell.
16:521–535. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li QQ, Lee RX, Liang H, Wang G, Li JM,
Zhong Y and Reed E: β-Elemene enhances susceptibility to cisplatin
in resistant ovarian carcinoma cells via downregulation of ERCC-1
and XIAP and inactivation of JNK. Int J Oncol. 43:721–728.
2013.PubMed/NCBI
|
|
32
|
Ohmichi M, Hayakawa J, Tasaka K, Kurachi H
and Murata Y: Mechanisms of platinum drug resistance. Trends
Pharmacol Sci. 26:113–116. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Potapova O, Haghighi A, Bost F, Liu C,
Birrer MJ, Gjerset R and Mercola D: The Jun kinase/stress-activated
protein kinase pathway functions to regulate DNA repair and
inhibition of the pathway sensitizes tumor cells to cisplatin. J
Biol Chem. 272:14041–14044. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Parekh H and Simpkins H: Cross-resistance
and collateral sensitivity to natural product drugs in
cisplatin-sensitive and -resistant rat lymphoma and human ovarian
carcinoma cells. Cancer Chemother Pharmacol. 37:457–462. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stordal B, Hamon M, McEneaney V, Roche S,
Gillet JP, O'Leary JJ, Gottesman M and Clynes M: Resistance to
paclitaxel in a cisplatin-resistant ovarian cancer cell line is
mediated by P-glycoprotein. PLoS One. 7:e407172012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xu H, Choi SM, An CS, Min YD, Kim KC, Kim
KJ and Choi CH: Concentration-dependent collateral sensitivity of
cisplatin-resistant gastric cancer cell sublines. Biochem Biophys
Res Commun. 328:618–622. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang H, Zou W, Li Y, Chen B and Xin X:
Bridge linkage role played by CD98hc of anti-tumor drug resistance
and cancer metastasis on cisplatin-resistant ovarian cancer cells.
Cancer Biol Ther. 6:942–947. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang LY, Trujillo JM, Siciliano MJ, Kido
Y, Siddik ZH and Su YZ: Distinct P-glycoprotein expression in two
subclones simultaneously selected from a human colon carcinoma cell
line by cis-diamminedichloroplatinum (II). Int J Cancer.
53:478–485. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang X and Pagé M: P-glycoprotein
expression in ovarian cancer cell line following treatment with
cisplatin. Oncol Res. 7:619–624. 1995.PubMed/NCBI
|
|
40
|
Comerford KM, Cummins EP and Taylor CT:
c-Jun NH2-terminal kinase activation contributes to
hypoxia-inducible factor 1alpha-dependent P-glycoprotein expression
in hypoxia. Cancer Res. 64:9057–9061. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu M, Li D, Aneja R, Joshi HC, Xie S,
Zhang C and Zhou J: PO(2)-dependent differential regulation of
multidrug resistance 1 gene expression by the c-Jun NH2-terminal
kinase pathway. J Biol Chem. 282:17581–17586. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fitzgerald JB, Schoeberl B, Nielsen UB and
Sorger PK: Systems biology and combination therapy in the quest for
clinical efficacy. Nat Chem Biol. 2:458–466. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lee MJ, Ye AS, Gardino AK, Heijink AM,
Sorger PK, MacBeath G and Yaffe MB: Sequential application of
anticancer drugs enhances cell death by rewiring apoptotic
signaling networks. Cell. 149:780–794. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Morton SW, Lee MJ, Deng ZJ, Dreaden EC,
Siouve E, Shopsowitz KE, Shah NJ, Yaffe MB and Hammond PT: A
nanoparticle-based combination chemotherapy delivery system for
enhanced tumor killing by dynamic rewiring of signaling pathways.
Sci Signal. 7:ra442014. View Article : Google Scholar : PubMed/NCBI
|