Introduction
Lung cancer is the most common malignancy and is
still increasing both in incidence and mortality worldwide
(1,2). Non-small cell lung cancer (NSCLC)
comprises 85% of all lung cancer cases (3). Therefore, development of early
diagnosis and effective therapies for NSCLC are urgently
needed.
Polo-like kinase 1 (PLK1), belongs to the polo-like
kinase protein family, is a serine/threonine kinase that regulates
a multitude of mitotic processes (4,5).
Importantly, PLK1 is highly expressed in a wide range of human
malignancies, such as lung, prostate, esophageal and colon cancer
(6–9). In addition, it is often correlated
with poor prognosis in patients suffering from NSCLC (5,10,11).
Previous studies in vitro have shown that downregulation of
PLK1 could inhibit the growth of lung cancer cells (11). Given the increasing appreciation of
PLK1 as an important oncogene, the mechanisms underlying the
upregulation of PLK1 expression in cancer cells have been
investigated.
MicroRNAs (miRNAs) are a conserved class of
non-coding RNAs that regulate protein expression by binding to the
mRNA 3′-untranslated region (3′-UTR) (12,13).
miRNAs likely have been implicated in diverse biological processes,
and have contributing roles in cancer initiation and progression
(14,15). Changes in miRNA expression have
profound effects on human carcinogenesis and cancer progression
(16). In this regard, the role of
miRNAs in regulating PLK1 expression in cancer cells has also been
investigated. Ito et al (17) demonstrated that miR-593* has an
important role in downregulating PLK1 expression, and is
responsible for the increased expression of PLK1 in human
esophageal cancer. Another miRNA, miR-100 acts as a tumor
suppressor by targeting PLK1 and downregulates its expression in
NSCLC (18). However, whether there
are additional miRNAs which are responsible for the increase of
PLK1 expression in NSCLC have not been reported.
To address this problem, we identified miR-296-5p as
a potential miRNA which could target PLK1 mRNA. Although the tumor
suppressor function of miR-296-5p has been proved in previous
studies (19,20), the oncogenic function of miR-296-5p
has not been fully investigated. miR-296-5p expression is
downregulated in a diverse array of tumors including lung cancer
(21–23).
In the present study, the miR-296-5p/PLK1 pathway
was investigated, in addition to the mechanistic roles of
miR-296-5p and PLK1 in NSCLC cells.
Materials and methods
Human samples
Twenty-four paires of NSCLC and corresponding normal
lung tissue specimens were obtained from the First Affiliated
Hospital of Soochow University. The adjacent macroscopically
non-tumor tissues were taken at least 6 cm distant from the tumor.
The patients with NSCLC had received neither radiotherapy nor
chemotherapy prior to tissue sampling. All tissues were snap-frozen
in liquid nitrogen and stored at −80°C until used. The study
protocol was approved by the ethics committee of Soochow
University.
Cell culture
Human non-small cell lung cancer cell lines A549,
H1299, LTEP-α-2, 95C and 95D were obtained from the Chinese Academy
of Sciences (Shanghai, China). Human bronchial epithelial (HBE)
cells were obtained from ScienCell Research Laboratories (Carlsbad,
CA, USA). All cells were cultured in RPMI-1640 medium (Gibco, Grand
Island, NY, USA) containing 10% fetal bovine serum (FBS; Gibco) and
maintained in a 5% CO2 humidified sterile atmosphere at
37°C.
RNA extraction, cDNA synthesis and
real-time quantitative reverse transcriptase-polymerase chain
reaction (qRT-PCR)
Total RNA from NSCLC tissues and cell lines were
extracted using TRIzol reagent (Invitrogen; Life Technologies) and
RNA concentration was measured on a NanoDrop (Thermo Fisher
Scientific, Waltham, MA, USA) according to the manufacturer's
instructions. Synthesis of cDNA with reverse transcriptase (RT) was
performed with an M-MLV first strand kit (Invitrogen; Life
Technologies). For miR-296-5p quantification, RNA was
reverse-transcribed with stem-loop RT primer:
GTCGTATCCAGTGCAGGGTCCGAGGTATTCG CACTGGATACGACACAGGATTG, and the
amplification primers were as follows: forward,
5′-GTATCCAGTGCAGGGTCCGA-3′ and reverse, 5′-CGACGAGGGCCCCCCCT-3′.
For PLK1 mRNA quantification, RNA was reverse-transcribed with a
random primer. The amplification primers were as follows: forward,
5′-GCCTAAGTCTCTGCTGCTCAA-3′ and reverse,
5′-CAACACCACGAACACGAAGT-3′.
Quantitative real-time PCR (qRT-PCR) of miR-296-5p
and PLK1 mRNA were performed using Takara SYBR-Green PCR kit
(Takara, Dalian, China) and an ABI 7500 Real-Time system (Applied
Biosystems, Carlsbad, CA, USA) was used to analyze the expression
of RNA.
We used U6 small nuclear RNA (U6 snRNA) and GAPDH
mRNA as an endogenous control to normalize miR-296-5p and PLK1 mRNA
expression level separately. Primer sequences for U6 detection
were: forward, 5′-CGAGCA CAGAATCGCTTCA-3′ and reverse,
5′-CTCGCTTCGGC AGCACATAT-3′. Primer sequences for GAPDH detection
were: forward, 5′-GAAGGTGAAGGTCGGAGTC-3′ and reverse,
5′-GAAGATGGTGATGGGATTTC-3′. Relative expression was calculated
using the comparative cycle threshold (Ct) method.
Plasmid construction and transient
transfection of A549 cells
The genome cDNA was generated by reverse
transcription-polymerase chain reaction (RT-PCR). The cDNA encoding
PLK1 (without 3′-UTR) was further amplified by PCR with sense
primer (5′-CCGGAATTCGCAGCTTCGGGAGCATGAGTGC-3′) containing a
BamHI restriction site and anti-sense primer
(5′-CCGCTCGAGCGGGAGCCAACCAGTATGGG-3′) containing an XhoI
restriction site. The PCR products were purified and then cloned
into the BamHI/XhoI sites of pcDNA3.1(+) (Invitrogen)
to create pcDNA3.1(+)-PLK1. The identification of plasmid was
verified by DNA sequencing. A549 cells were seeded at
2×105/well in a 6-well plate 24 h prior to transfection.
The cells were transfected using Lipofectamine 2000 reagent
(Invitrogen) according to the manufacturer's protocols. Transfected
cells were selected in the presence of G418 (600 µg/ml).
miRNA and siRNA transfection
A549 cells were transfected with miRNA or siRNA 24 h
after being seeded in 6-well plates. miR-296-5p mimics or PLK1
siRNA (100 pmol) and Lipofectamine 2000 (5 µl) were added
into 250 µl of serum-free medium separately and then mixed
at room temperature for 20 min. The complexes were then added
dropwise to the cells and mixed gently. The cells were incubated
for 48 h for further study.
Western blot assay
Cells were lysed in a RIPA buffer (Cell Signaling
Technology, Boston, MA, USA) with protease inhibitor
(Sigma-Aldrich, Dorset, UK) at 48 h post-transfection. The total
proteins were then separated on 12% SDS-PAGE and blotted onto PVDF
membranes. The membranes were blocked by 1% BSA for 30 min and
incubated with the primary antibody (anti-PLK1 or anti-GAPDH; Santa
Cruz Biotechnology, Santa Cruz, CA and Bioworld Technology, Inc.,
St. Louis Park, MN, USA, respectively) overnight at 4°C. After 3
washes with TBST, the membranes were incubated with anti-mouse IgG
(Santa Cruz Biotechnology, Dallas, TX, USA) or anti-rabbit IgG
(Santa Cruz Biotechnology) at room temperature for 2 h. Proteins
were visualized using ECL detection system (Pierce, Rockford, IL,
USA). PLK1 protein level was normalized to GAPDH protein and the
density was quantified using Quantity One 4.6 software (Bio-Rad
Laboratories, Hercules, CA, USA).
Luciferase activity assay
The potential binding site of miR-296-5p in the
PLK1-3′UTR was predicted at the position 51–58 of the 3′-UTR of
PLK1 (NM_005030) by TargetScan software (www.targetscan.org). PsiCHECK-2 vector (Promega,
Madison, WI, USA) was used to construct the plasmid containing
3′-untranslated region (3′-UTR) of PLK1. The fragments containing
the predicted wild and mutant sites were directly synthesized by
Genewiz (Suzhou, China) and then subcloned into psiCHECK-2 vector.
After 16 h of culture in a 24-well plate, A549 cells were
co-transfected with psiCHECK-2-PLK1-3′-UTR-wild/mutant vector (50
ng) and miR-296-5p mimics or scrambled microRNA negative control
(miR-NC). After 48 h of culture, the luciferase activity was
measured using a Dual-luciferase assay kit (Promega) on a TD20/20
Luminometer (Turner Designs, Westport, MA, USA). Expression values
were normalized to Renilla luciferase. All assays were
performed in triplicate and repeated at least three times.
Cell proliferation assay
A549 cells were seeded into 96-well plates with the
density of 5×103 cells/well after transfected with
miR-296-5p or PLK1-siRNA for 24 h. Cell proliferation was detected
by CCK-8 and EdU assay.
For the CCK-8 assay, cells were harvested over four
consecutive days and incubated with 20 µl of the Cell
Counting kit-8 (CCK-8; Beyotime Institute of Biotechnology,
Shanghai, China) reagent at 37°C for 2 h. The OD value was measured
at 450 nm.
5-Ethynyl-2′-deoxyuridine (EdU) assay (Guangzhou
RiboBio, Co., Ltd., Guangzhou, China) was carried out to label
cells undergoing DNA replication. Cells were planted in 96-well
plates and treated, and then were incubated with EdU for 2 h before
fixation. The cells were fixed in 4% paraformaldehyde for 30 min
and permeabilized in 0.5% Triton X-100 for 25 min at room
temperature. Apollo dyeing reaction buffer was added and shaken in
the dark for 30 min. Then, the DNA was stained with Hoechst 33342
for 30 min. The proportion of nucleated cells incorporating EdU was
observed by fluorescence microscopy. Cell proliferation rate was
calculated as the percentage of EdU-positive nuclei to total nuclei
in five high-power fields/well. The assay was performed in
triplicate and repeated three times.
Statistical analysis
All data are presented as means ± SEM from at least
three separate experiments. Statistical analysis was performed by
non-parametric tests (Mann-Whitney U test for 2 groups). For cell
lines, unpaired t-test (2-tailed) was used to assess the
differences between groups. Data analysis was performed using the
SPSS 17.0 software package. Differences were considered
statistically significant at P<0.05.
Results
PLK1 expression is increased in NSCLC
samples and cell lines
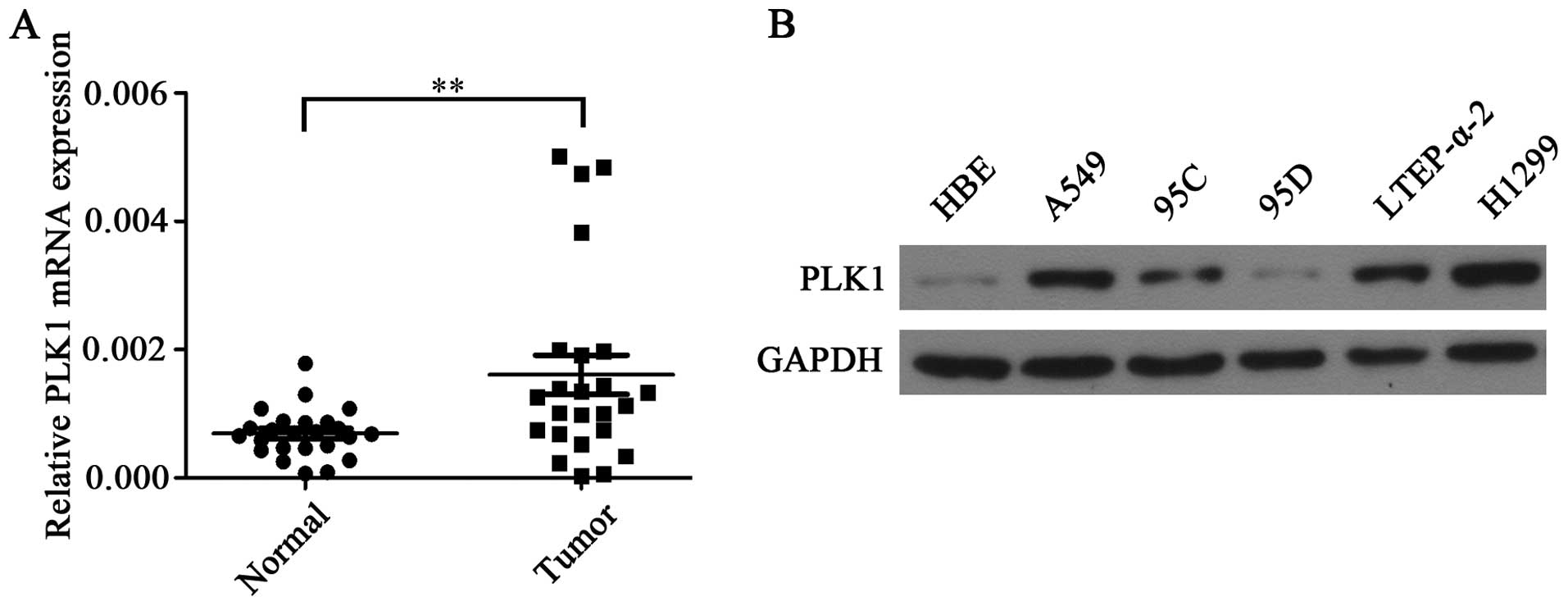
To determine the role of PLK1 in NSCLC, PLK1 mRNA
expression levels were determined in 24 paired NSCLC tissues and
adjacent non-tumor tissues. It is shown that the average expression
of PLK1 mRNA was significantly higher in NSCLC tissues when
compared with adjacent non-tumor tissues (Fig. 1A). In addition, the higher levels of
PLK1 protein were also detected in NSCLC cells lines including
A549, 95C, 95D, LTEP-α-2 and H1299 when compared with HBE cells
(Fig. 1B).
Functional role of PLK1 in NSCLC
cells
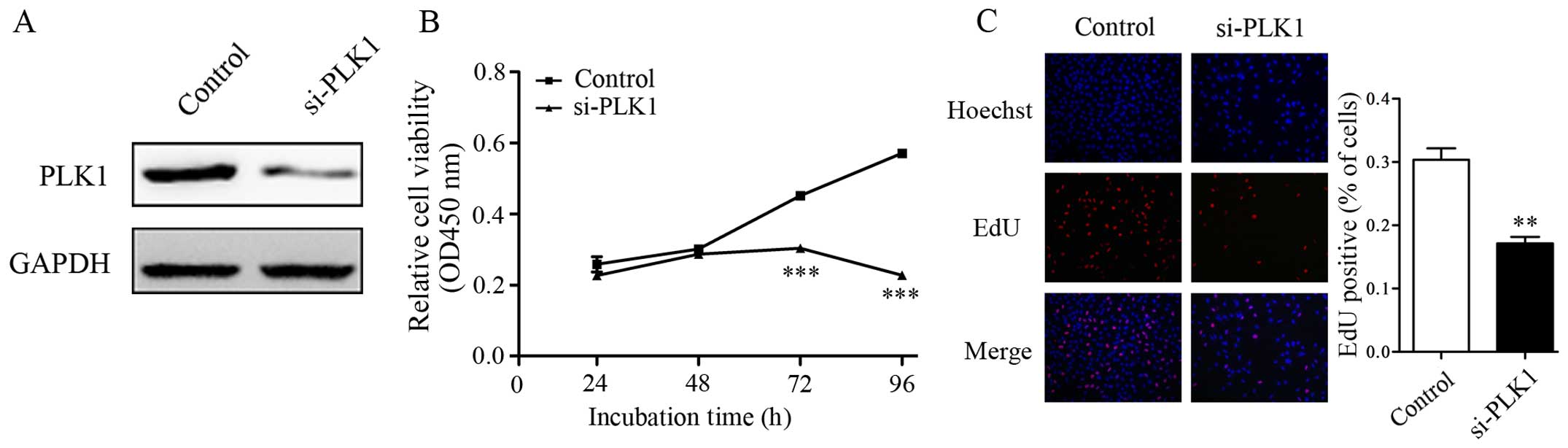
It is shown that PLK1 acted as an oncogene, since
the increased expression of PLK1 was examined in NSCLC tissues and
cell lines. We then investigated the promotion effect of PLK1 on
cell proliferation in NSCLC. siRNA which could downregulate PLK1
expression was introduced into A549 cells (Fig. 2A). CCK-8 results showed that the
proliferation rate of A549 cells was significantly lower in the
PLK1 siRNA treated group than control (Fig. 2B). EdU assay also showed that A549
cells treated with miR-296-5p mimics had significantly lower rates
of proliferation (Fig. 2C). These
results suggested that downregulation of PLK1 significantly
inhibited the proliferation of A549 cells.
PLK1 protein expression is downregulated
by miR-296-5p in NSCLC cells
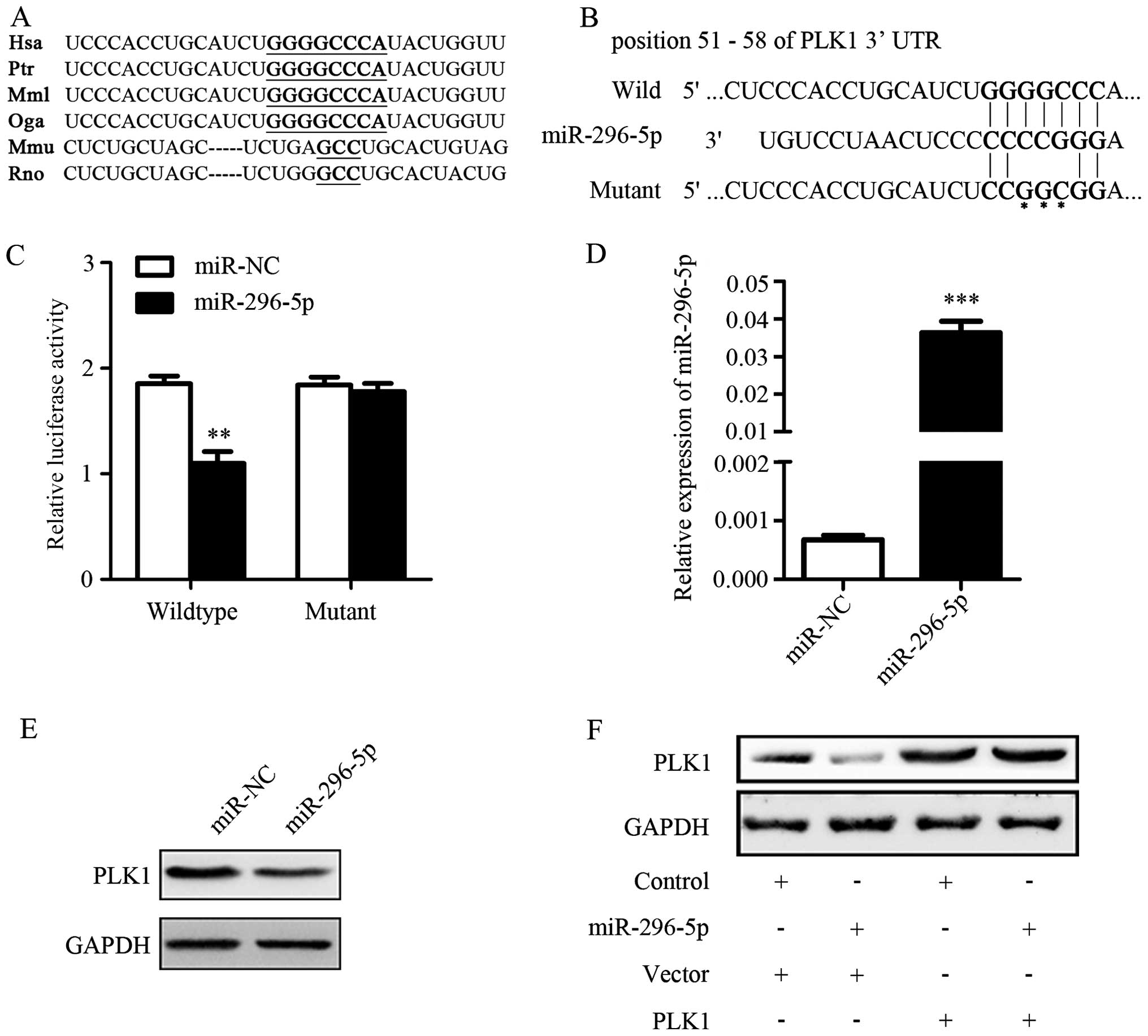
To explore the underlying mechanism of PLK1
regulation, we used TargetScan software to predict the possible
microRNAs that targeted PLK1 mRNA. The conserved predicted target
site among species on the 3′-UTR of PLK1 mRNA was identified
(Fig. 3A). It showed that
miR-296-5p was one of the predicted microRNAs which targeted PLK1
mRNA 3′-UTR (Fig. 3B). The results
of luciferase reporter assays showed that the luciferase activity
decreased significantly in cells co-transfected with
psiCHECK-2-PLK1-3′-UTR-wild vector and miR-296-5p mimics when
compared with control. However, the luciferase activities did not
change much in A549 cells with the mutant construct (Fig. 3C). The above results suggested that
miR-296-5p could directly target 3′-UTR of PLK1.
Next, we detected whether PLK1 protein expression
could be suppressed by miR-296-5p. The transfection efficiency of
miR-296-5p was determined by qRT-PCR (Fig. 3D). The effect of miR-296-5p on
levels of endogenous PLK1 protein was determined by western blot
assay in A549 cells. As expected, PLK1 protein expression was
significantly down-regulated in miR-296-5p transfected group when
compared with miR-NC transfected group (Fig. 3E). Furthermore, A549 cells were
transfected with pcDNA3.1(+)-PLK1 in combination with miR-296-5p
transduction. The results of western blotting assay revealed that
miR-296-5p downregulated PLK1 expression, but the effect could be
rescued by transfection with PLK1 plasmid (Fig. 3F). These results further proved that
PLK1 expression could be mediated by miR-296-5p through targeting
its mRNA 3′-UTR.
miR-296-5p expression is decreased in
NSCLC samples and cell lines
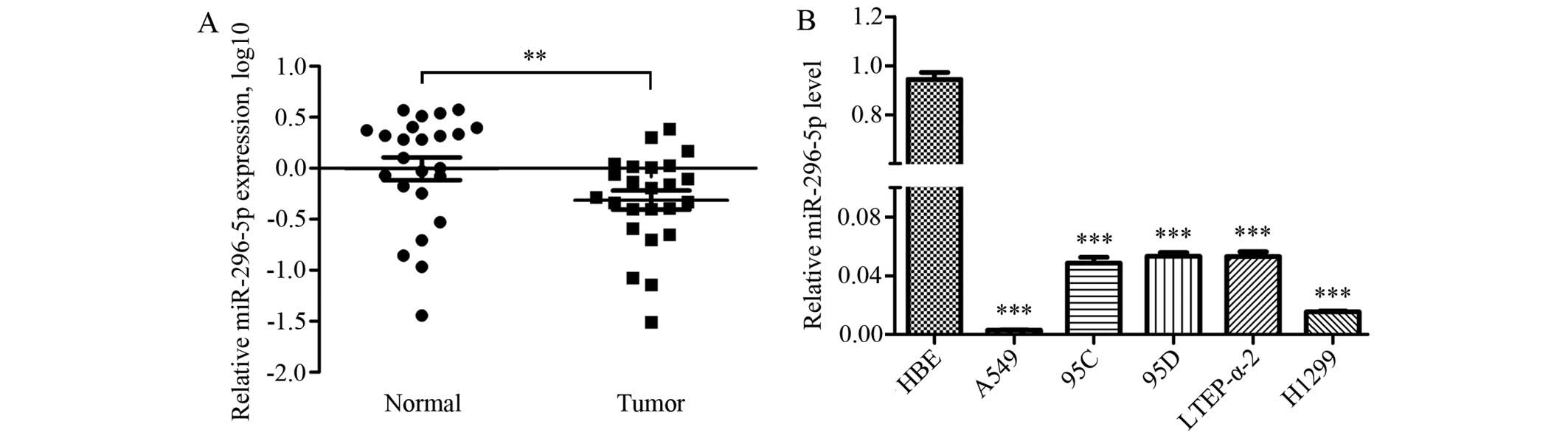
The expression levels of miR-296-5p were further
investigated in 24 pairs of NSCLC samples by qRT-PCR. The results
showed that the expression levels of miR-296-5p were significantly
reduced in NSCLC specimens when compared with paired normal tissues
(Fig. 4A). Furthermore, the
decreased expression levels of miR-296-5p were also detected in
NSCLC cell lines including A549, 95C, 95D, LTEP-α-2 and H1299 when
compared with HBE cells (Fig.
4B).
miR-296-5p inhibits NSCLC cell growth in
vitro
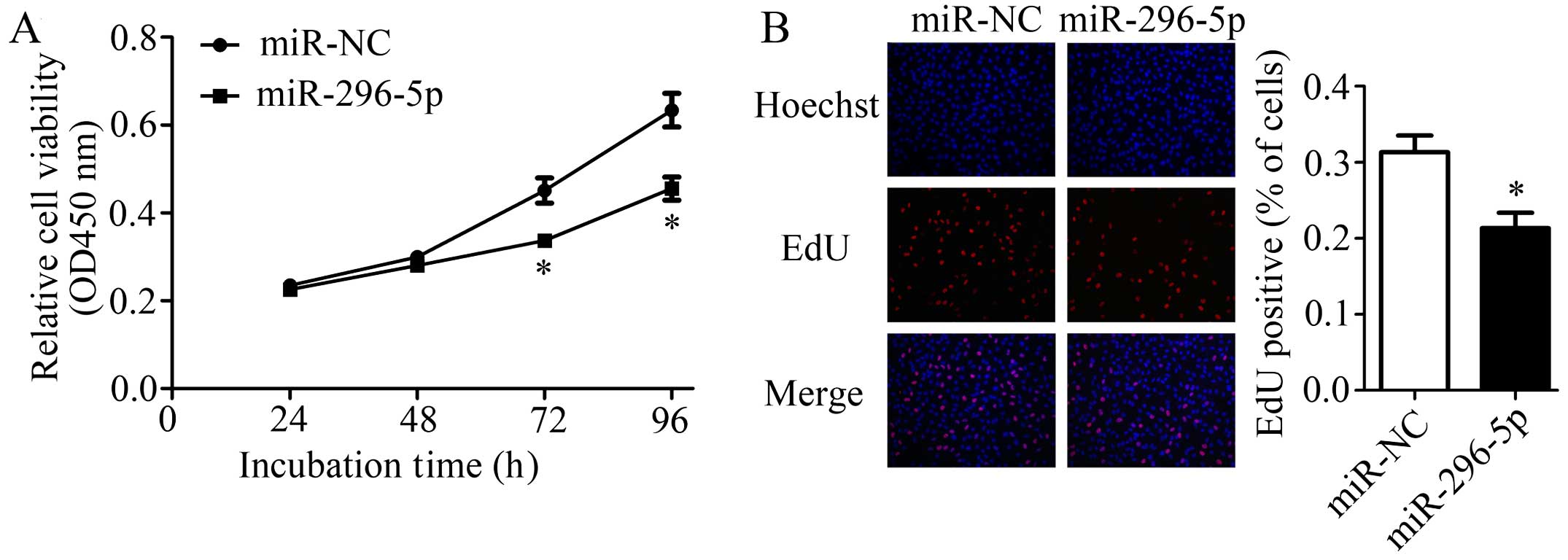
Since reduced expression of miR-296-5p was observed
in NSCLC tissues and cells, we explored whether restoration of
miR-296-5p had similar effect as PLK1 knockdown in A549 cells.
After transfected with miR-296-5p mimics or miR-NC, cell viability
was measured by CCK-8 and EdU assays. The results of CCK-8 assay
indicated that cells treated with miR-296-5p mimics showed a
significantly decreased proliferation rate when compared with
control (Fig. 5A). Similar results
were also observed in EdU assay (Fig.
5B). These results implied that miR-296-5p has a strong ability
to suppress NSCLC cell proliferation.
Discussion
PLK1 is an important member of the Polo-like kinases
(PLKs), which are a family of highly conserved serine/threonine
kinases (24). It is involved in
several crucial physiological process including cell cycle
progression, spindle formation and chromosome segregation during
mitosis (25,26). Additionally, PLK1 is upregulated in
various human malignancies, such as non-small cell lung cancer,
breast cancer, colorectal cancer, and melanomas (27–30).
In view of the important role of PLK1 in tumors, it is necessary to
study the mechanism of aberrantly expressed PLK1 in NSCLC.
In general, miRNAs have been suggested to be
classified into two main classes: oncosuppressor genes and
oncogenes. Some low abundance miRNAs act as oncosuppressor genes by
negatively regulating oncogenes, whereas some highly expressed
miRNAs act as oncogenes by repressing tumor suppressor genes
(31,32). Sometimes the same microRNA performs
distinct functions in different tumor circumstances. For example,
miR-296-5p is upregulated in esophageal squamous cell cancer
tissues and downregulation of miR-296-5p is shown to suppress
esophageal cancer cell progression (33). However, more studies have found that
miR-296-5p acted as a tumor suppressor and was downregulated in
breast, prostate and lung cancer (19–21).
These controversial results indicate that the roles of miR-296-5p
were highly dependent on its targets in different cancer cells.
PLK1 has been shown to be a target of several miRNAs
including miR-100 and miR-593*, and these miRNAs are known to be
responsible for the regulation of cancer cell proliferation,
differentiation and invasion (17,18).
In the present study, we investigated whether miR-296-5p suppresses
NSCLC cell viability by targeting PLK1. Using a luciferase reporter
assay, we demonstrated conclusively that miR-296-5p directly
targeted PLK1 mRNA by binding to the potential 3′-UTR binding
site.
It is known that one transcript can be regulated by
one or more miRNAs while each miRNA also can have multiple target
transcripts (34). For instance,
miR-128 acts as a tumor suppressor by directly targeting Bmi1 and
CYP2C9 in prostate cancer and hepatocellular carcinoma,
respectively (35,36). Bmi1 can be targeted by miR-203,
miR-218 and miR-135a in different malignancies (37–39).
Although abundant transcriptional targets are predicted, most of
these have not been fully elucidated. In the present study, we
found that PLK1 mRNA could be targeted by miR-296-5p, and PLK1
protein could be downregulated by miR-296-5p transfection.
Our analysis confirmed the downregulation of
miR-296-5p expression in all the NSCLC cell lines tested compared
with HBE. To confirm this result, we examined primary tissues from
24 cases of NSCLC and compared them with paired non-tumor tissues,
and found that miR-296-5p expression in NSCLC was significantly
decreased.
Next, we investigated whether the downregulation of
miR-296-5p was responsible for the uncontrolled growth of tumor
cells. Our results showed that transfection of miR-296-5p mimics in
A549 cells resulted in a significant suppression in tumor growth,
and this result is in agreement with a previous study in breast
cancer (40). Vaira et al
(21) also revealed that miR-296-5p
could inhibit lung cancer cell migration and invasion.
Our research indicates that miR-296-5p is involved
in the PLK1 oncogene network, and the introduction of miR-296-5p is
able to rebuild the tumor-suppressing signaling pathway in A549
cells. Importantly, miR-296-5p potently inhibites cell viability in
PLK1-overexpressing NSCLC cells, providing the first evidence that
there is a potential link between the tumor suppressor miR-296-5p
and NSCLC cell self-renewal. The present study also suggests that
miR-296-5p could contribute to human NSCLC therapy.
Acknowledgments
We wish to thank all the NSCLC patients for their
participation and cooperation. The present study was supported in
part by the grants from the Jiangsu Province's Key Provincial
Talents Program (RC2011106 to J.Z.), the Natural Science Fundation
of Jiangsu Province (BK20131159 to J.Z.), the Natural Science
Research Foundation of the Jiangsu Higher Education Institutions of
China (14KJB320012 to C.L.) the Suzhou City's Municipal Youth Fund
of Science and Education (KJXW2013007 to C.X.) and the Suzhou Key
Laboratory for Molecular Cancer Genetics (SZS201209 to J.Z. and
H.-T.Z.).
References
|
1
|
Reck M, Popat S, Reinmuth N, De Ruysscher
D, Kerr KM and Peters S; ESMO Guidelines Working Group: Metastatic
non-small cell lung cancer (NSCLC): ESMO Clinical Practice
Guidelines for diagnosis, treatment and follow-up. Ann Oncol.
25(Suppl 3): iii27–39. 2014. View Article : Google Scholar
|
|
2
|
Field JK, Oudkerk M, Pedersen JH and Duffy
SW: Prospects for population screening and diagnosis of lung
cancer. Lancet. 382:732–741. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Curioni-Fontecedro A, Husmann L, Soldini D
and Stahel RA: Primary non-small cell lung cancer response upon
treatment with denosumab. Lung Cancer. 82:506–508. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barr FA, Silljé HH and Nigg EA: Polo-like
kinases and the orchestration of cell division. Nat Rev Mol Cell
Biol. 5:429–440. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cholewa BD, Liu X and Ahmad N: The role of
polo-like kinase 1 in carcinogenesis: Cause or consequence? Cancer
Res. 73:6848–6855. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Takai N, Hamanaka R, Yoshimatsu J and
Miyakawa I: Polo-like kinases (Plks) and cancer. Oncogene.
24:287–291. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Weichert W, Schmidt M, Gekeler V, Denkert
C, Stephan C, Jung K, Loening S, Dietel M and Kristiansen G:
Polo-like kinase 1 is overexpressed in prostate cancer and linked
to higher tumor grades. Prostate. 60:240–245. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Feng YB, Lin DC, Shi ZZ, Wang XC, Shen XM,
Zhang Y, Du XL, Luo ML, Xu X, Han YL, et al: Overexpression of PLK1
is associated with poor survival by inhibiting apoptosis via
enhancement of survivin level in esophageal squamous cell
carcinoma. Int J Cancer. 124:578–588. 2009. View Article : Google Scholar
|
|
9
|
Weichert W, Kristiansen G, Schmidt M,
Gekeler V, Noske A, Niesporek S, Dietel M and Denkert C: Polo-like
kinase 1 expression is a prognostic factor in human colon cancer.
World J Gastroenterol. 11:5644–5650. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang ZX, Xue D, Liu ZL, Lu BB, Bian HB,
Pan X and Yin YM: Overexpression of polo-like kinase 1 and its
clinical significance in human non-small cell lung cancer. Int J
Biochem Cell Biol. 44:200–210. 2012. View Article : Google Scholar
|
|
11
|
Spänkuch-Schmitt B, Wolf G, Solbach C,
Loibl S, Knecht R, Stegmüller M, von Minckwitz G, Kaufmann M and
Strebhardt K: Downregulation of human polo-like kinase activity by
antisense oligonucleotides induces growth inhibition in cancer
cells. Oncogene. 21:3162–3171. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yates LA, Norbury CJ and Gilbert RJ: The
long and short of microRNA. Cell. 153:516–519. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ha M and Kim VN: Regulation of microRNA
biogenesis. Nat Rev Mol Cell Biol. 15:509–524. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang W, Dahlberg JE and Tam W: MicroRNAs
in tumorigenesis: A primer. Am J Pathol. 171:728–738. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Papagiannakopoulos T and Kosik KS:
MicroRNAs: Regulators of oncogenesis and stemness. BMC Med.
6:152008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bowen T, Jenkins RH and Fraser DJ:
MicroRNAs, transforming growth factor beta-1, and tissue fibrosis.
J Pathol. 229:274–285. 2013. View Article : Google Scholar
|
|
17
|
Ito T, Sato F, Kan T, Cheng Y, David S,
Agarwal R, Paun BC, Jin Z, Olaru AV, Hamilton JP, et al: Polo-like
kinase 1 regulates cell proliferation and is targeted by miR-593*
in esophageal cancer. International journal of cancer. Int J
Cancer. 129:2134–2146. 2011. View Article : Google Scholar :
|
|
18
|
Liu J, Lu KH, Liu ZL, Sun M, De W and Wang
ZX: MicroRNA-100 is a potential molecular marker of non-small cell
lung cancer and functions as a tumor suppressor by targeting
polo-like kinase 1. BMC Cancer. 12:5192012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Savi F, Forno I, Faversani A, Luciani A,
Caldiera S, Gatti S, Foa P, Ricca D, Bulfamante G, Vaira V, et al:
miR-296/Scribble axis is deregulated in human breast cancer and
miR-296 restoration reduces tumour growth in vivo. Clin Sci (Lond).
127:233–242. 2014. View Article : Google Scholar
|
|
20
|
Lee KH, Lin FC, Hsu TI, Lin JT, Guo JH,
Tsai CH, Lee YC, Lee YC, Chen CL, Hsiao M, et al: MicroRNA-296-5p
(miR-296-5p) functions as a tumor suppressor in prostate cancer by
directly targeting Pin1. Biochim Biophys Acta. 1843:2055–2066.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vaira V, Faversani A, Martin NM, Garlick
DS, Ferrero S, Nosotti M, Kissil JL, Bosari S and Altieri DC:
Regulation of lung cancer metastasis by Klf4-Numb-like signaling.
Cancer Res. 73:2695–2705. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu J, Li A, Hong SM, Hruban RH and Goggins
M: MicroRNA alterations of pancreatic intraepithelial neoplasias.
Clin Cancer Res. 18:981–992. 2012. View Article : Google Scholar :
|
|
23
|
Corbetta S, Vaira V, Guarnieri V,
Scillitani A, Eller-Vainicher C, Ferrero S, Vicentini L, Chiodini
I, Bisceglia M, Beck-Peccoz P, et al: Differential expression of
microRNAs in human parathyroid carcinomas compared with normal
parathyroid tissue. Endocr Relat Cancer. 17:135–146. 2010.
View Article : Google Scholar
|
|
24
|
Weitzer S and Uhlmann F: Chromosome
segregation: Playing polo in prophase. Dev Cell. 2:381–382. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xie S, Xie B, Lee MY and Dai W: Regulation
of cell cycle checkpoints by polo-like kinases. Oncogene.
24:277–286. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
McInnes C and Wyatt MD: PLK1 as an
oncology target: Current status and future potential. Drug Discov
Today. 16:619–625. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
McCarroll JA, Dwarte T, Baigude H, Dang J,
Yang L, Erlich RB, Kimpton K, Teo J, Sagnella SM, Akerfeldt MC, et
al: Therapeutic targeting of polo-like kinase 1 using
RNA-interfering nanoparticles (iNOPs) for the treatment of
non-small cell lung cancer. Oncotarget. 6:12020–12034. 2014.
View Article : Google Scholar
|
|
28
|
Bhola NE, Jansen VM, Bafna S, Giltnane JM,
Balko JM, Estrada MV, Meszoely I, Mayer I, Abramson V, Ye F, et al:
Kinome-wide functional screen identifies role of PLK1 in
hormone-independent, ER-positive breast cancer. Cancer Res.
75:405–414. 2015. View Article : Google Scholar
|
|
29
|
Degenhardt Y and Lampkin T: Targeting
Polo-like kinase in cancer therapy. Clin Cancer Res. 16:384–389.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cholewa BD, Pellitteri-Hahn MC, Scarlett
CO and Ahmad N: Large-scale label-free comparative proteomics
analysis of polo-like kinase 1 inhibition via the small-molecule
inhibitor BI 6727 (Volasertib) in BRAF(V600E) mutant melanoma
cells. J Proteome Res. 13:5041–5050. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Davis-Dusenbery BN and Hata A: MicroRNA in
cancer: The involvement of aberrant microRNA biogenesis regulatory
pathways. Genes Cancer. 1:1100–1114. 2010. View Article : Google Scholar
|
|
32
|
Mavrakis KJ, Leslie CS and Wendel HG:
Cooperative control of tumor suppressor genes by a network of
oncogenic microRNAs. Cell Cycle. 10:2845–2849. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yoon AR, Gao R, Kaul Z, Choi IK, Ryu J,
Noble JR, Kato Y, Saito S, Hirano T, Ishii T, et al: MicroRNA-296
is enriched in cancer cells and downregulates p21WAF1 mRNA
expression via interaction with its 3′ untranslated region. Nucleic
Acids Res. 39:8078–8091. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vasilatou D, Papageorgiou S, Pappa V,
Papageorgiou E and Dervenoulas J: The role of microRNAs in normal
and malignant hematopoiesis. Eur J Haematol. 84:1–16. 2010.
View Article : Google Scholar
|
|
35
|
Godlewski J, Nowicki MO, Bronisz A,
Williams S, Otsuki A, Nuovo G, Raychaudhury A, Newton HB, Chiocca
EA and Lawler S: Targeting of the Bmi-1 oncogene/stem cell renewal
factor by microRNA-128 inhibits glioma proliferation and
self-renewal. Cancer Res. 68:9125–9130. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yu D, Green B, Marrone A, Guo Y, Kadlubar
S, Lin D, Fuscoe J, Pogribny I and Ning B: Suppression of CYP2C9 by
microRNA hsa-miR-128-3p in human liver cells and association with
hepatocellular carcinoma. Sci Rep. 5:85342015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Okumura T, Shimada Y, Moriyama M, Takei Y,
Omura T, Sekine S, Nagata T, Shimizu K and Tsukada K: MicroRNA-203
inhibits the progression of esophageal squamous cell carcinoma with
restored epithelial tissue architecture in vivo. Int J Oncol.
44:1923–1932. 2014.PubMed/NCBI
|
|
38
|
Wei Y, Du Y, Chen X, Li P, Wang Y, Zang W,
Zhao L, Li Z and Zhao G: Expression patterns of microRNA-218 and
its potential functions by targeting CIP2A and BMI1 genes in
melanoma. Tumour Biol. 35:8007–8015. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dang Z, Xu WH, Lu P, Wu N, Liu J, Ruan B,
Zhou L, Song WJ and Dou KF: MicroRNA-135a inhibits cell
proliferation by targeting Bmi1 in pancreatic ductal
adenocarcinoma. Int J Biol Sci. 10:733–745. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vaira V, Faversani A, Dohi T, Montorsi M,
Augello C, Gatti S, Coggi G, Altieri DC and Bosari S: miR-296
regulation of a cell polarity-cell plasticity module controls tumor
progression. Oncogene. 31:27–38. 2012. View Article : Google Scholar :
|