Introduction
Hypoxia is an event that allows neoplastic cells
from the primary tumor to become metastatic cells (1,2). It
also influences the formation of new vessels by upregulating the
expression of VEGF (3). It has been
established that cellular response to hypoxia is mediated by
hypoxia-inducible factors (HIFs), which are transcriptional factors
that promote the expression of genes involved in cell survival
under hypoxic conditions. They also participate directly in other
processes of tumoral development such as neoplastic glucose/energy
metabolism, cellular growth and apoptosis (4,5). HIFs
are heterodimers consisting of either HIF-1α or HIF-2α bound to the
HIF-1β subunit. In normoxic conditions, the α subunit is
constitutively expressed but rapidly degraded. In a low-oxygen
environment, the α subunit is stabilized and translocated to the
nucleus (6,7). Therefore, both HIF-α subunits are
regulated by O2 availability, while HIF-1β is
constitutively expressed.
2-Methoxyestradiol (2-ME) is an anti-angiogenic,
anti-proliferative and pro-apoptotic agent that suppresses HIF-1α
protein levels and its transcriptional activity (8,9). Its
effect correlates with a decrease in tubulin polymerization
(10) and it also disrupts normal
microtubule function and stability (11). 2-ME binds directly to the colchicine
binding site and does not interact with estrogen receptors,
lowering therefore its probable side-effects (12). 2-ME may have potential clinical
benefit in the treatment of cancer since it inhibits the
proliferation of many human cancer cell lines in vitro
(13,14). There is evidence that HIF-1α
mediates tumoral cell survival and apoptosis resistance under
hypoxic and normoxic conditions (15,16–18).
Furthermore, the pharmacological inhibition of HIF-1α, and
particularly HIF-regulated genes that are important for cancer cell
survival, may be more advantageous than HIF-gene inactivation
therapeutic approaches (19).
Hypoxic tumor cells are known to be more resistant
to current treatment modalities and to radiation than normoxic
cells (20). Hypoxia can also
confer resistance against chemotherapy-induced apoptosis in
numerous solid tumors such as breast and non-small cell lung cancer
and pancreatic ductal adenocarcinoma (21–23).
Therefore, and considering hypoxia as an important factor leading
cancer cells to enhanced resistance to cytotoxic drugs, we studied
the effects of 2-ME on cell growth, apoptosis, and HIF-1α and
HIF-2α gene and protein expression in human lung adenocarcinoma
A549 cells grown under normoxic and hypoxic conditions.
Materials and methods
The protocol was approved by the local Ethics and
Research Committees.
Cell culture
The A549 human lung adenocarcinoma cell line was
obtained from the American Type Culture Collection (ATTC;
Rockville, MD, USA). Cells were cultured in Dulbecco's modified
Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) and
supplemented with non-essential amino acids, 50 U/ml penicillin and
50 µg/ml streptomycin at 37°C in a humidified atmosphere
with 5% CO2/95% air.
Cell growth assay
After cells reached 80% confluence, 2×104
cells/well were cultured into 48-well plates for 24 h. A stock
solution of 33 mM 2-ME (Sigma-Aldrich, St. Louis, MO, USA) was
prepared in dimethyl sulfoxide (DMSO). Final 2-ME concentrations
were prepared by diluting the stock solution with DMEM. Medium was
replaced with fresh medium with 10% FBS and with the corresponding
2-ME concentrations (0.001, 0.1, 0.1, 1 and 10 µM final
concentrations). A549 cells with 10% FBS medium (non-stimulated
cells) were used as growth control. Cells were also cultured with
10% FBS plus 0.03% DMSO (vehicle-control group for all
experiments). Cells were cultured for 12, 24, 48, 72 and 96 h in
normoxia (5% CO2 and 95% air) and hypoxia (1% oxygen and
5% CO2) conditions, at 37°C in a humidified incubator.
The culture medium, with and without 2-ME, was not changed during
the assay.
Cells were placed into a chamber MIC-101
(Billups-Rothenberg, Del Mar, CA, USA) to expose them to hypoxic
conditions. Briefly, a mixture of 95% nitrogen and 5%
CO2 gas that displaces the oxygen into the chamber was
injected. The oxygen concentration was measured by an oxygen sensor
(Vascular Technology, Nahua, NH, USA) and maintained at 1%. At the
end of the incubation period, the media were discarded and the
cells were washed with phosphate-buffered solution. Five hundred
microliters of 1% glutaraldehyde solution were added to each well
and incubated for 20 min at room temperature; subsequently the
glutaraldehyde solution was discarded and 0.1% crystal violet
(N-hexamethylpararosaniline) (Sigma-Aldrich) solution was added to
each well and incubated under constant stirring for 15 min at room
temperature. Crystal violet was washed exhaustively, allowed to dry
and afterwards 400 µl of 10% acetic acid solution were added
to each well. The sample absorbance was measured at 590 nm in
96-well microtiter plates with an ELISA reader (Molecular Devices,
Sunnyvale, CA, USA). All experiments were carried out in
triplicate. Cell growth was expressed as the relative increase in
light absorbance at 590 nm with respect to the value at 0 h.
Apoptosis assay
A549 cells were cultured in 6-well plates
(3×105 cells/well) under normoxic or hypoxic conditions
for 72 h. Three different media were used: DMEM (control), DMEM
with DMSO (without 2-ME) and DMEM with 10 µM 2-ME. Apoptosis
was examined by flow cytometry using the Annexin V
(Becton-Dickinson Biosciences, Franklin Lakes, NJ, USA) staining
kit. Briefly, 1×106 cells in 100 µl Annexin
buffer were stained with propidium iodide and FITC-Annexin V
staining solutions. Cells were incubated at room temperature in the
dark for 15 min, and the data were subsequently acquired through a
FACSAria flow cytometer (Becton-Dickinson Biosciences). Data were
analyzed using the FlowJo X.0.7 software (Stanford University,
Stanford, CA, USA). Results are expressed as percentage; 100% =
5,000 cells.
Western blotting
Cells were plated into 6-well cell culture plates
and were grown until they reached 60% confluence. Cells were then
exposed to hypoxic conditions and lysed with 0.1% Triton
(Sigma-Aldrich) in PBS without calcium to obtain total cell
extracts. Protein quantification was performed by the bicinchoninic
acid protein assay (BCA protein assay kit; Pierce Biotechnology,
Rockford, IL, USA). Western blotting was carried out using 30
µg of cell extract proteins in 8% SDS-polyacrylamide gels
(PAGE) under reducing conditions with 5% 2-mercaptoethanol boiled
for 10 min. After electrophoresis, the proteins were transferred to
PVDF membranes and blocked with 2.5% non-fat dry milk in 100 mM
Tris-HCl buffer, pH 7.5 with 150 mM NaCl and 0.1% Tween-20 (TTBS
buffer); they were then incubated for 90 min at room temperature
with the corresponding antibody: 1:500 anti-HIF-1α (mouse
monoclonal antibody, NB100-479) or 1:500 anti-HIF-2α (rabbit
polyclonal antibody, NB100-122) (both from Novus Biologicals,
Littleton, CO, USA). β-tubulin antibody (1:500) was used as a
loading control (sc-53140; Santa Cruz Biotechnology, Santa Cruz,
CA, USA). Unbound antibodies were washed with TTBS buffer and bands
were detected using the Vectastain® ABC kit (Vector
Laboratories, Burlingame, CA, USA). Western blotting bands were
analyzed by densitometry scanning using the Kodak Digital Science
ID Image analysis software (Eastman Kodak, Rochester, NY, USA). The
results are expressed as densitometry units (DU).
Immunocytochemistry
Five hundred thousand cells were cultured in a Nunc
Lab-Tek chamber slide system (Thermo Fisher Scientific, Carlsbad,
CA, USA) and allowed to grow in normoxia or hypoxia with or without
10 µM 2-ME treatment for 72 h. The cells were fixed with 1%
glutaraldehyde (Sigma-Aldrich) and washed thrice with distilled
water for 5 min. Antigen retrieval was performed with 1:10 citrate
buffer (Sigma-Aldrich). The slides were heated during 5 min in a
microwave and they were then allowed to cool for 20 min. Endogenous
peroxidase activity was quenched with 2% H2O2
solution (Sigma-Aldrich). Antibody blockade and incubation were
performed in a humid chamber with 100 µl of blocking
solution with serum, followed by the blockade of non-specific sites
with 4% (wt/vol) non-fat dry milk in PBS at 4°C overnight. Cells
were incubated with 100 µl of the corresponding antibody for
90 min at room temperature: 1:100 anti-HIF-1α (mouse monoclonal
antibody, 1NB100-479) or 1:100 anti-HIF-2α (rabbit polyclonal
antibody, NB100-122; Novus Biologicals). In another set of
experiments, cells were processed with a non-immune IgG instead of
the primary antibody as a negative control. Visualization of
antibody localization was achieved with a Vectastain®
ABC detection kit. Between each step, the slides were thoroughly
washed with distilled water. Finally, the slides were manually
counterstained with Harris hematoxylin and mounted with non-aqueous
medium. Images were captured with an Evos-FL Auto microscope
(Thermo Fisher Scientific).
RT-PCR and quantitative real-time PCR of
HIF-1α and HIF-2α
Cells were incubated under normoxic or hypoxic
conditions with or without 10 µM 2-ME for 72 h. After
incubation, total RNA and protein were extracted. RNA was extracted
using TRIzol reagent (Invitrogen Life Technologies, Thermo Fisher
Scientific) and reverse transcribed into cDNA (Advantage RT-for-PCR
kit; Clontech, Palo Alto, CA, USA). One microgram of total RNA was
reverse transcribed using 2 µg of random primers and Moloney
murine leukemia virus reverse transcriptase according to the
manufacturer's protocol (Advantage RT-for-PCR kit). Real-time PCR
was carried out on a One-Step system (Applied Biosystems, Thermo
Fisher Scientific). The expression assay was carried out using
pre-designed TaqMan gene expression assay Hs00153153 for HIF-1α
labeled with FAM, Hs01026149_m1 for HIF-2α labeled with FAM and
normalized with Hs99999901_s1 for 18S ribosomal RNA labeled with
VIC (Applied Biosystems). The PCR duplex reactions were performed
in a 20-µl reaction volume containing 10 µl of TaqMan
Universal PCR Master Mix 2X, 0.5 µl of TaqMan gene
expression assay Hs99999901_s1 20X used as an endogenous control
(18S rRNA), 1 µl TaqMan gene expression assay of the target
gene (Hs00153153 or Hs01026149_m1), 50 ng of cDNA (4 µl) and
4 µl of RNAse-free water.
Relative quantitation method was used to analyze the
results of two independent experiments made in triplicate. For each
experimental sample, a gene was considered as not expressed if
amplification was not detected by threshold cycle Ct = 40. The
results are expressed in arbitrary units of ΔCt, where ΔCt =
Cttarget - Ct18S. ΔCt values represent mRNA
transcripts.
Statistical analysis
Cell growth was expressed as a percentage of their
relative controls. The mean and standard deviation (SD) were
obtained in triplicate. Differences between experimental assays in
hypoxia, normoxia (0, 12, 24, 48, 72 and 96 h) and apoptosis assays
were analyzed using the Student's t-test. Statistical analysis was
conducted using the statistical software SPSS version 20.0 (IBM
SPSS). p≤0.05 was considered to indicate a statistically
significant result.
Results
2-ME inhibits cell growth and induces
apoptosis in A549 cells under normoxic but not hypoxic
conditions
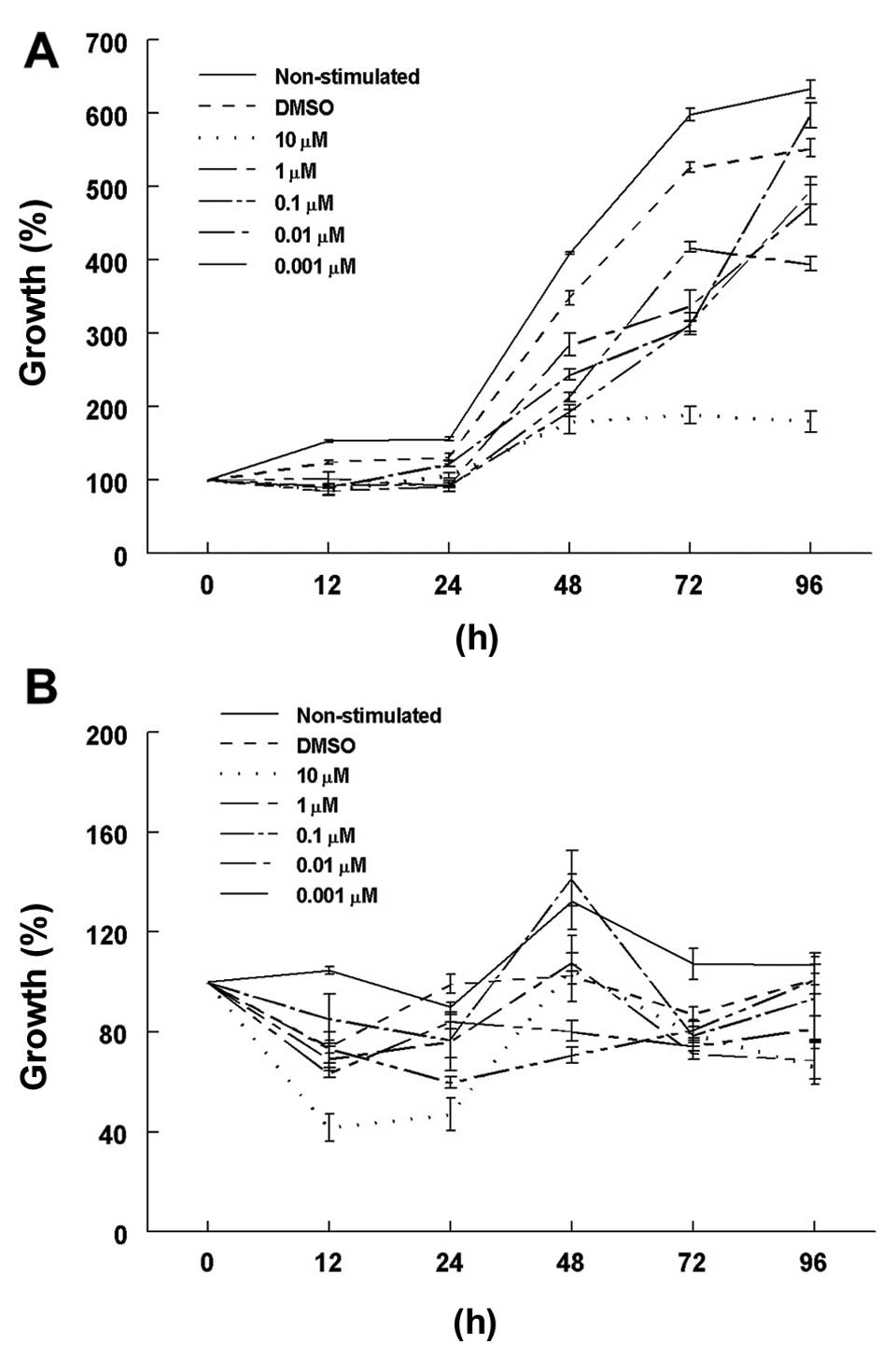
Cell growth rates in the 2-ME-stimulated cells were
decreased in comparison to the rates in the DMSO-incubated control
cells, mainly at 48 and 72 h under normoxic conditions (Fig. 1A). The lowest cell growth rate was
observed using a 10 µM concentration of 2-ME in comparison
with the DMSO control cells: 179.4±16.6 and 347.7±9.6%,
respectively (p<0.0001) at 48 h; 189.0±11.6 and 526.3±7.2%,
respectively (p<0.0001) at 72 h, and 179.7±14.2 and 552.9±12.1%,
respectively (p<0.0001) at 96 h. In contrast, there were no
significant differences among the growth rates between the control
cells and those treated with 10 µM 2-ME at 72 and 48 h under
hypoxic conditions (Fig. 1B).
However, a significant decrease in the growth rate was found in the
10 µM 2-ME-treated cells in comparison with the DMSO-treated
cells (66.2±7.2 and 101.2±2.3%, respectively; p=0.04) at 96 h. An
exponential cell growth was observed in the DMSO medium without
stimulation, as expected.
2-ME at a concentration of 10 µM was used for
the apoptosis and HIF-1α and HIF-2α expression assays, due to the
significance found for this concentration when cells were incubated
under normoxic conditions at 72 h. Longer incubation periods were
not used since absence of nutrients could have biased the results.
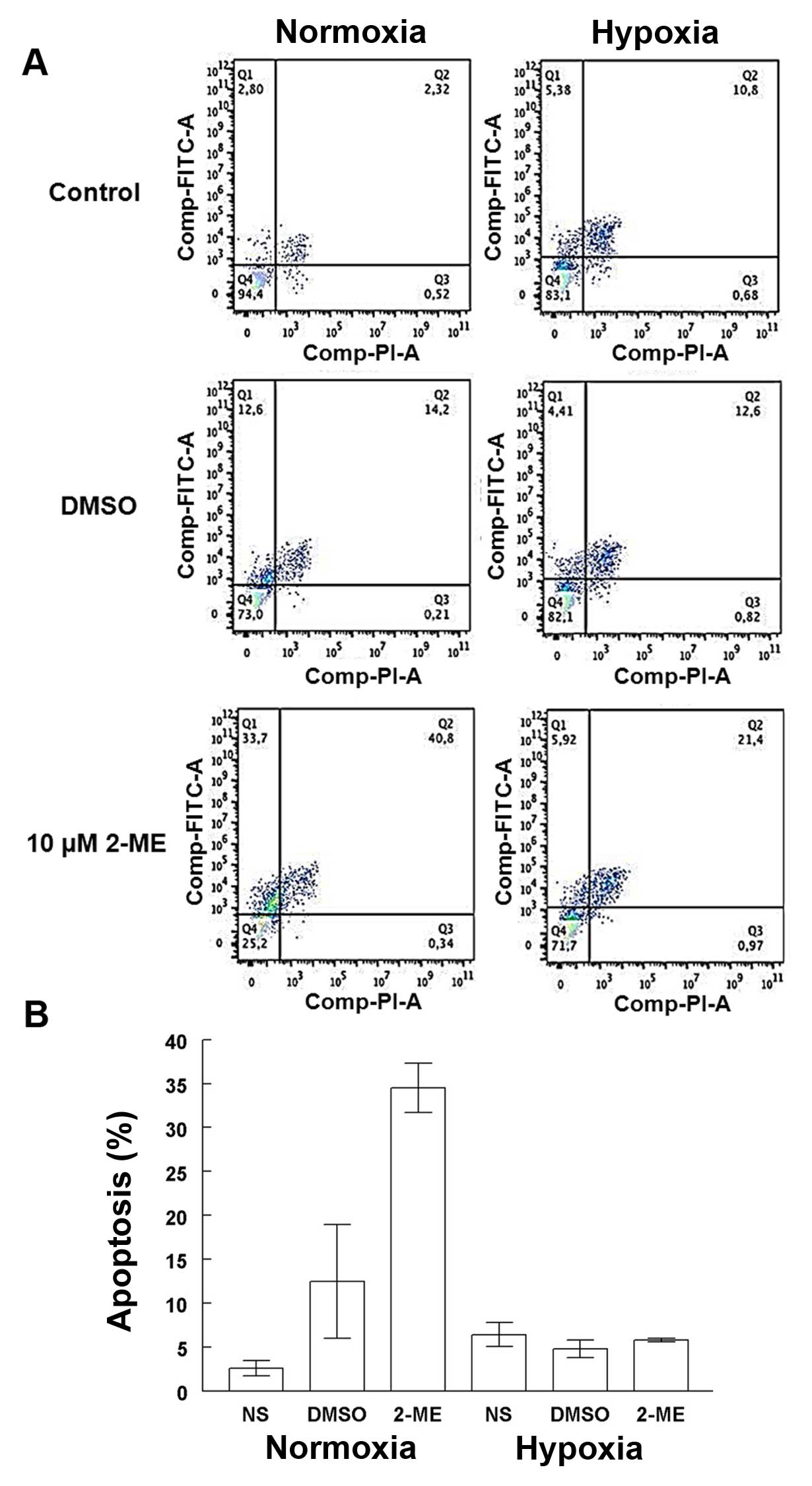
Apoptosis induced by 2-ME in the A549 cells was differentially
affected by the oxygenation conditions (Fig. 2A). The presence of 10 µM 2-ME
significantly increased the percentage of apoptosis (34.5±2.8%) in
comparison with the DMSO control cells (12.5±6.5%) (p=0.006) and
non-stimulated cells (NS) (p=4.8×10−5) in a normoxic
condition. There were no significant differences among 10 µM
2-ME-treated and control cells grown under hypoxic conditions. A
significant increase in apoptosis was observed in cells treated
with 10 µM 2-ME in a normoxic condition in comparison with
cells under lower O2 concentration (5.8±0.2%; p=0.003)
(Fig. 2B).
Western blotting for HIF-1α and
HIF-2α
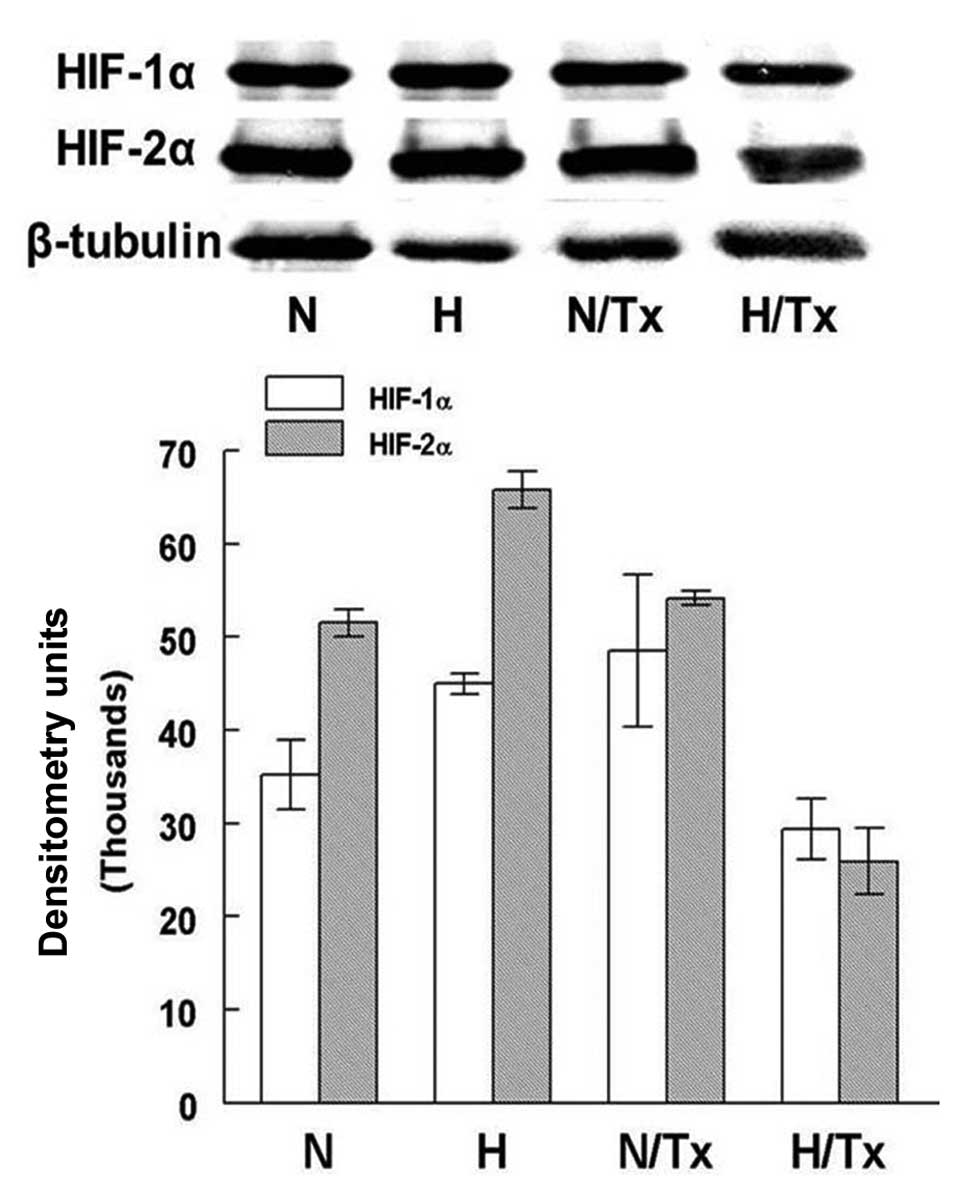
Western blot densitometry analysis showed
differences in the protein expression of HIF-1α and HIF-2α under
hypoxic conditions (Fig. 3).
HIF-1α was significantly increased in hypoxic cells
(44,998.1±1,079.3 DU) in comparison with cells cultured in normoxic
conditions (35,200.8±3,726.9 DU; p=0.01). HIF-1α protein expression
was not modified when the cells were treated with 2-ME in a
normoxic condition. In contrast, there was a decrease in HIF-1α
when cells were treated with 2-ME under hypoxic conditions
(29,390.1±3,542.9 DU; p=0.001).
HIF-2α protein levels were significantly increased
in the cells cultured under hypoxia (65,834.3±1,957.7 DU) in
comparison with cells incubated under a normoxic condition
(51,537±1,451.3 DU, p=0.001). The synthesis of this protein was not
significantly modified by the exposure to 2-ME under a normoxic
condition. In contrast, the HIF-2α level was significantly
decreased in cells treated with 10 µM 2-ME under hypoxic
conditions (25,921±3,5442.9 DU; p=6.8×10−5).
Significant differences were also found when HIF-1α
and HIF-2α levels of cells grown under normoxic (p=0.02) and
hypoxic conditions (p=8.6×10−3) were compared, but not
among the same cell groups treated with 2-ME.
HIF-1α and HIF-2α immunocytochemistry in
2-ME-treated cells
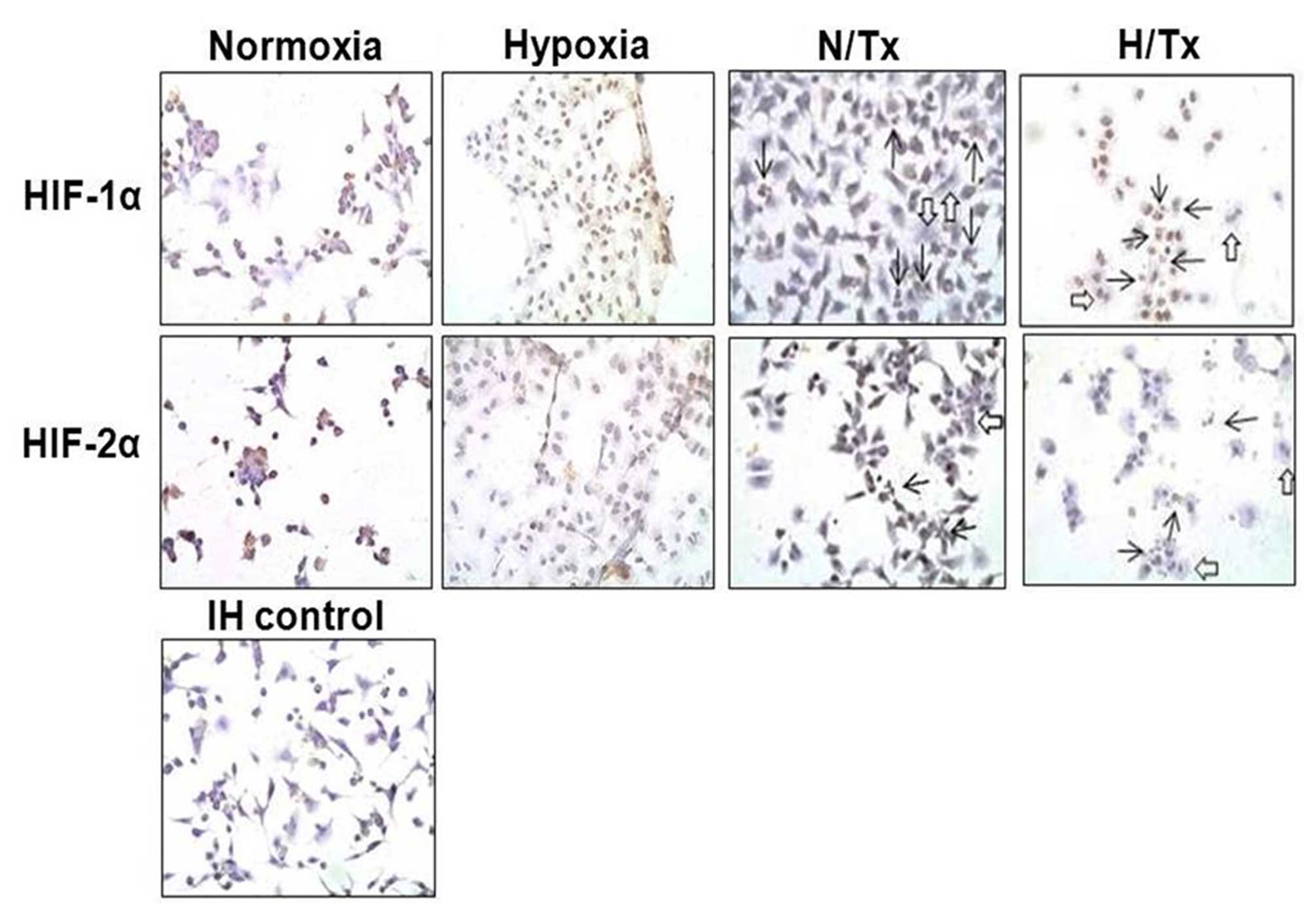
HIF-1α and HIF-2α were detected in the
normoxic-grown cell cytoplasm, particularly HIF-2α, using
immunoperoxidase staining (Fig. 4).
When cells were cultured under hypoxic conditions, HIF-1α was
observed with higher intensity in the cell nucleus; however,
staining for HIF-2α was low. Neither HIF-1α nor HIF-2α were noted
in the cells treated with 2-ME under a normoxic condition. The
incubation with 2-ME decreased the nuclear staining for HIF-1α in
cells under hypoxia, while no staining for HIF-2α was observed
under the same experimental conditions.
Apart from the effects of 2-ME on HIF-1α and HIF-2α
protein expression in both experimental conditions, some
morphological and physiological effects were also evident. Nuclear
lobation (open arrows) and nuclear fragmentation (apoptosis, solid
arrows) were present in cells treated with 2-ME under a normoxic
condition; these phenomena were more frequent in cells exposed
concomitantly to hypoxia and 2-ME.
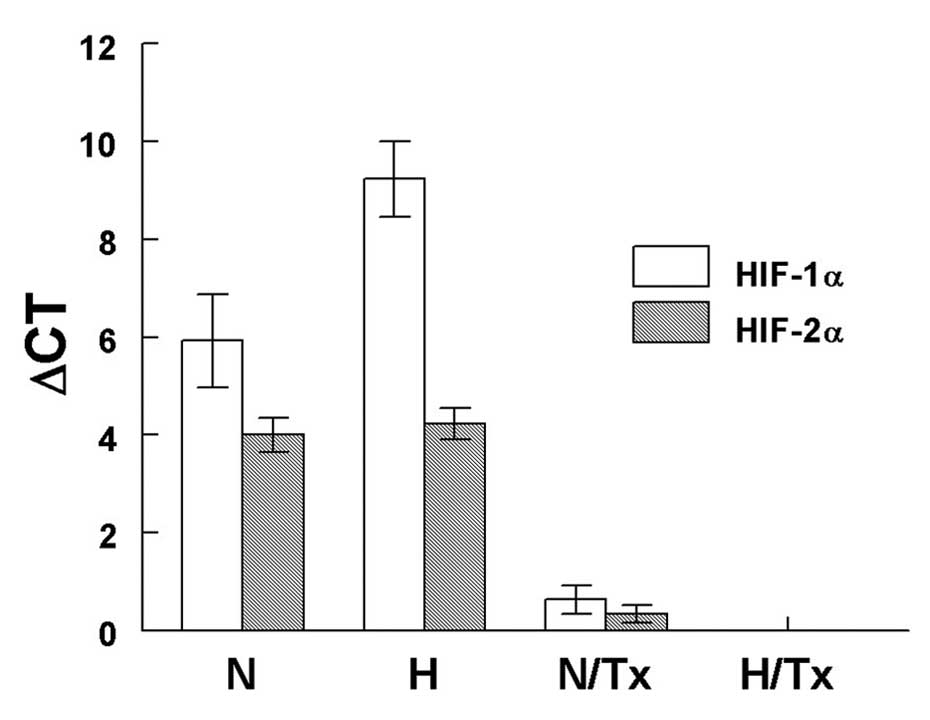
HIF-1α and HIF-2α gene expression
The gene expression assay revealed that there were
significant differences in HIF-1α mRNA expression among normoxic
(5.9±0.9 ΔCt values) and hypoxic (9.2±0.8 ΔCt values; p=0.0004)
cells (Fig. 5). A significant
decrease in HIF-1α mRNA expression was observed when cells were
cultured with 2-ME under a normoxic (0.63±0.3 ΔCt values; p=0.03)
condition. HIF-1α mRNA was untraceable in cells exposed to 2-ME
under hypoxia.
There were no differences in HIF-2α mRNA expression
among cells cultured under normoxic and hypoxic conditions. When
normoxic-grown cells were exposed to 2-ME, HIF-2α gene expression
was significantly decreased (0.3±0.1 ΔCt values) in comparison with
the untreated cells (3.9±0.4 ΔCt values; p=0.02). Neither HIF-1α
nor HIF-2α mRNA was detected in hypoxic cells exposed to 2-ME.
The mRNA expression of HIF-1α was significantly
higher than HIF-2α gene expression under hypoxia (p=0.04); but no
differences were noted when mRNA expression for HIF-1α and HIF-2α
grown in normoxic conditions was compared.
Discussion
2-Methoxyestradiol (2-ME) is a natural metabolite of
17β-estradiol that exerts antiproliferative action in carcinoma
cell lines and may play a possible antitumor and anti-angiogenesis
role in vivo (8,24–26).
Furthermore, it has also been used in a number of preclinical and
clinical studies for the treatment of solid tumors (27–29).
Likewise, it is widely known that hypoxic microenvironments inside
solid tumors are one of the major causes of drug resistance
(15,30,31),
and that the extent of tumor hypoxia is an important prognostic
factor for assessing tumor progression as well as resistance to
therapy and overall patient survival (32–34).
Under hypoxic conditions, some genes, such as the hypoxia-inducible
factors (HIFs), are activated and their products favor tumor
progression. In the present study, we analyzed whether 2-ME could
inhibit the expression of HIF genes in lung carcinoma cells
simultaneously exposed to this drug and hypoxia. The effect of
different 2-ME concentrations on cell growth rates of human
adenocarcinoma A549 cells grown under normoxia or hypoxia was
analyzed first. Our results showed a dose-dependent inhibition of
cell growth for 2-ME-treated normoxic cells. In contrast, a strong
cell growth inhibition, probably due to the hypoxia rather than to
2-ME treatment, was observed. The effects of this compound on
apoptosis were also examined, since there is evidence that HIF-1α
mediates tumoral cell survival and apoptotic resistance under
hypoxic and normoxic conditions (15-18).
We observed that, under a normoxic condition, 2-ME stimulated
apoptosis, an effect probably due to Bcl-2 and Bcl-xL
phosphorylation and the subsequent inhibition of the anti-apoptotic
effects (35). Contrastingly, 2-ME
had no effect on cells grown under hypoxia. Notably, this 2-ME lack
of effect was concentration independent. This finding correlates
with the observation that hypoxia by itself prevents several agents
such as the ones used in chemotherapy from inducing apoptosis
(17,36,37).
Likewise, the possibility that HIF-1α could display either a
pro-apoptotic or an anti-apoptotic role has been raised and it has
been proposed that HIF-1α acting one way or the other is probably
related to the severity of the hypoxic conditions (36).
It has been determined that 2-ME inhibits the
expression of HIF-1α and HIF-2α proteins and their nuclear
translocation in hepatocellular carcinoma cells (31). Accordingly, we also found a
significant decrease in HIF-1α and HIF-2α mRNA and protein
expression in lung adenocarcinoma cells treated with 2-ME. Even
though HIF-1α protein expression decreased noticeably in these
cells after treatment, it was still possible to observe it in the
nuclei. These findings suggest that although 2-ME stimulates
apoptosis by Bcl molecule phosphorylation and HIF expression
inhibition, the decline in apoptosis could be due to the HIF
molecules present in the nuclei in hypoxia; it is possible that
nuclear HIFs may contribute to apoptosis inhibition through the
activation of cell-stress response genes (38).
According to our data, the 2-ME therapy may not be
effective during the early stages of cancer in which neoplastic
cells grown in a hypoxic environment. However, due to its effect on
HIF expression, 2-ME may still be effective as an antimetastatic
agent and it could be used in combination with other therapeutic
agents. In this regard, some 2-ME analogues have been synthesized
and tested in an attempt to develop drugs with improved oral
bioavailability and efficacy, for example:
2-methoxyestradiol-bis-sulphamate (2-ME-BM),
2-methoxyoestradiol-3,17-O,O-bis-sulphamate
(2-MeOE2bisMATE), NanoCrystal Dispersion formulation of
2-ME2 (2-ME2 NCD) and sulphamoylated 2-ME analogues (29,39–44).
These 2-ME analogues were probed in combination with other drugs,
for example: docetaxel in breast cancer (28) or paclitaxel in head and neck
squamous cell carcinoma with good results (45). However, these analogues need to be
tested under hypoxia, as the oxygenation levels could determine
treatment response.
Finally, although HIF-1α is the best-known and
widely described isoform, many data suggest that HIF-2α is as
important as HIF-1α. In the present study, we found an increase in
HIF-2α protein expression in comparison with HIF-1α when cells were
cultured for 72 h under hypoxia. Unfortunately, it was not possible
to confirm this observation by immunoperoxidase staining since this
technique is not as sensitive as the western blot assay. Regarding
our results, some authors report a decrease in HIF-1α protein
whereas HIF-2α levels remained stable when A549 cells were
incubated for >6 h under hypoxic conditions (46,47).
Moreover, a high HIF-2α expression was observed in patients with
advanced stage cancer, therefore this molecule was considered as a
negative prognostic factor associated with a mutant form of Kras in
non-small cell lung cancer (48,49).
In summary, treatment of A549 cells with 2-ME may be
ineffective to increase apoptosis under hypoxic conditions,
although it could be useful to treat advanced stage cancer due to
its effects on HIF expression. Understandably, further drug tests
should carefully consider the conditions of normoxia and hypoxia to
accurately assess whether the drug will be beneficial for the
patient with a hypoxic tumor.
Acknowledgments
The present study was partially supported by grants
from the Consejo Nacional de Ciencia y Tecnología (CONACYT,
SALUD-2010-1-141991), and the Instituto de Ciencia y Tecnología del
Distrito Federal (ICyT, PIFUTP09-281) of Mexico. The authors are
grateful to Hugo Olivera, Cristina Tejas and Cuauhtémoc Sandoval
for their technical support.
References
|
1
|
Lu X and Kang Y: Hypoxia and
hypoxia-inducible factors: Master regulators of metastasis. Clin
Cancer Res. 16:5928–5935. 2010.
|
|
2
|
Ye J, Wu D, Wu P, Chen Z and Huang J: The
cancer stem cell niche: Cross talk between cancer stem cells and
their microenvironment. Tumour Biol. 35:3945–3951. 2014.
|
|
3
|
Carmeliet P: VEGF as a key mediator of
angiogenesis in cancer. Oncology. 69(Suppl 3): S4–S10. 2005.
|
|
4
|
Semenza GL: Hypoxia-inducible factor 1 and
the molecular physiology of oxygen homeostasis. J Lab Clin Med.
131:207–214. 1998.
|
|
5
|
Semenza GL: HIF-1 and human disease: One
highly involved factor. Genes Dev. 14:1983–1991. 2000.
|
|
6
|
Bertout JA, Patel SA and Simon MC: The
impact of O2 availability on human cancer. Nat Rev
Cancer. 8:967–975. 2008.
|
|
7
|
Clerici C and Planès C: Gene regulation in
the adaptive process to hypoxia in lung epithelial cells. Am J
Physiol Lung Cell Mol Physiol. 296:L267–L274. 2009.
|
|
8
|
Fotsis T, Zhang Y, Pepper MS, Adlercreutz
H, Montesano R, Nawroth PP and Schweigerer L: The endogenous
oestrogen metabolite 2-methoxyoestradiol inhibits angiogenesis and
suppresses tumour growth. Nature. 368:237–239. 1994.
|
|
9
|
Becker CM, Rohwer N, Funakoshi T, Cramer
T, Bernhardt W, Birsner A, Folkman J and D'Amato RJ:
2-methoxyestradiol inhibits hypoxia-inducible factor-1{alpha} and
suppresses growth of lesions in a mouse model of endometriosis. Am
J Pathol. 172:534–544. 2008.
|
|
10
|
Escuin D, Kline ER and Giannakakou P: Both
microtubule-stabilizing and microtubule-destabilizing drugs inhibit
hypoxia-inducible factor-1alpha accumulation and activity by
disrupting microtubule function. Cancer Res. 65:9021–9028.
2005.
|
|
11
|
Chua YS, Chua YL and Hagen T: Structure
activity analysis of 2-methoxyestradiol analogues reveals targeting
of microtubules as the major mechanism of antiproliferative and
proapoptotic activity. Mol Cancer Ther. 9:224–235. 2010.
|
|
12
|
D'Amato RJ, Lin CM, Flynn E, Folkman J and
Hamel E: 2-Methoxyestradiol, an endogenous mammalian metabolite,
inhibits tubulin polymerization by interacting at the colchicine
site. Proc Natl Acad Sci USA. 91:3964–3968. 1994.
|
|
13
|
Benedikt MB, Mahlum EW, Shogren KL,
Subramaniam M, Spelsberg TC, Yaszemski MJ and Maran A:
2-methoxyestradiol-mediated anti-tumor effect increases
osteoprotegerin expression in osteosarcoma cells. J Cell Biochem.
109:950–956. 2010.
|
|
14
|
Bu S, Blaukat A, Fu X, Heldin NE and
Landström M: Mechanisms for 2-methoxyestradiol-induced apoptosis of
prostate cancer cells. FEBS Lett. 531:141–151. 2002.
|
|
15
|
Rankin EB and Giaccia AJ: The role of
hypoxia-inducible factors in tumorigenesis. Cell Death Differ.
15:678–685. 2008.
|
|
16
|
Kilic M, Kasperczyk H, Fulda S and Debatin
KM: Role of hypoxia inducible factor-1 alpha in modulation of
apoptosis resistance. Oncogene. 26:2027–2038. 2007.
|
|
17
|
Sermeus A, Cosse JP, Crespin M, Mainfroid
V, de Longueville F, Ninane N, Raes M, Remacle J and Michiels C:
Hypoxia induces protection against etoposide-induced apoptosis:
Molecular profiling of changes in gene expression and transcription
factor activity. Mol Cancer. 7:272008.
|
|
18
|
Yu EZ, Li YY, Liu XH, Kagan E and McCarron
RM: Antiapoptotic action of hypoxia-inducible factor-1 alpha in
human endothelial cells. Lab Invest. 84:553–561. 2004.
|
|
19
|
Mabjeesh NJ, Escuin D, LaVallee TM,
Pribluda VS, Swartz GM, Johnson MS, Willard MT, Zhong H, Simons JW
and Giannakakou P: 2ME2 inhibits tumor growth and angiogenesis by
disrupting microtubules and dysregulating HIF. Cancer Cell.
3:363–375. 2003.
|
|
20
|
Brown JM and Wilson WR: Exploiting tumour
hypoxia in cancer treatment. Nat Rev Cancer. 4:437–447. 2004.
|
|
21
|
Flamant L, Notte A, Ninane N, Raes M and
Michiels C: Anti-apoptotic role of HIF-1 and AP-1 in paclitaxel
exposed breast cancer cells under hypoxia. Mol Cancer.
9:1912010.
|
|
22
|
Sun HC, Qiu ZJ, Liu J, Sun J, Jiang T,
Huang KJ, Yao M and Huang C: Expression of hypoxia-inducible
factor-1 alpha and associated proteins in pancreatic ductal
adenocarcinoma and their impact on prognosis. Int J Oncol.
30:1359–1367. 2007.
|
|
23
|
Swinson DE, Jones JL, Cox G, Richardson D,
Harris AL and O'Byrne KJ: Hypoxia-inducible factor-1 alpha in non
small cell lung cancer: Relation to growth factor, protease and
apoptosis pathways. Int J Cancer. 111:43–50. 2004.
|
|
24
|
Kuo KL, Lin WC, Ho IL, Chang HC, Lee PY,
Chung YT, Hsieh JT, Pu YS, Shi CS and Huang KH: 2-methoxyestradiol
induces mitotic arrest, apoptosis, and synergistic cytotoxicity
with arsenic trioxide in human urothelial carcinoma cells. PLoS
One. 8:e687032013.
|
|
25
|
Zhou NN, Zhu XF, Zhou JM, Li MZ, Zhang XS,
Huang P and Jiang WQ: 2-Methoxyestradiol induces cell cycle arrest
and apoptosis of nasopharyngeal carcinoma cells. Acta Pharmacol
Sin. 25:1515–1520. 2004.
|
|
26
|
Rajkumar SV, Richardson PG, Lacy MQ,
Dispenzieri A, Greipp PR, Witzig TE, Schlossman R, Sidor CF,
Anderson KC and Gertz MA: Novel therapy with 2-methoxyestradiol for
the treatment of relapsed and plateau phase multiple myeloma. Clin
Cancer Res. 13:6162–6167. 2007.
|
|
27
|
Sweeney C, Liu G, Yiannoutsos C, Kolesar
J, Horvath D, Staab MJ, Fife K, Armstrong V, Treston A, Sidor C, et
al: A phase II multicenter, randomized, double-blind, safety trial
assessing the pharmacokinetics, pharmacodynamics, and efficacy of
oral 2-methoxyestradiol capsules in hormone-refractory prostate
cancer. Clin Cancer Res. 11:6625–6633. 2005.
|
|
28
|
James J, Murry DJ, Treston AM, Storniolo
AM, Sledge GW, Sidor C and Miller KD: Phase I safety,
pharmacokinetic and pharmacodynamic studies of 2-methoxyestradiol
alone or in combination with docetaxel in patients with locally
recurrent or metastatic breast cancer. Invest New Drugs. 25:41–48.
2007.
|
|
29
|
Matei D, Schilder J, Sutton G, Perkins S,
Breen T, Quon C and Sidor C: Activity of 2 methoxyestradiol (Panzem
NCD) in advanced, platinum-resistant ovarian cancer and primary
peritoneal carcinomatosis: A Hoosier Oncology Group trial. Gynecol
Oncol. 115:90–96. 2009.
|
|
30
|
Bottsford-Miller JN, Coleman RL and Sood
AK: Resistance and escape from antiangiogenesis therapy: Clinical
implications and future strategies. J Clin Oncol. 30:4026–4034.
2012.
|
|
31
|
Ma L, Li G, Zhu H, Dong X, Zhao D, Jiang
X, Li J, Qiao H, Ni S and Sun X: 2-Methoxyestradiol synergizes with
sorafenib to suppress hepatocellular carcinoma by simultaneously
dysregulating hypoxia-inducible factor-1 and -2. Cancer Lett.
355:96–105. 2014.
|
|
32
|
Semenza GL: Evaluation of HIF-1 inhibitors
as anticancer agents. Drug Discov Today. 12:853–859. 2007.
|
|
33
|
Harris AL: Hypoxia - a key regulatory
factor in tumour growth. Nat Rev Cancer. 2:38–47. 2002.
|
|
34
|
Teicher BA, Holden SA, al-Achi A and
Herman TS: Classification of antineoplastic treatments by their
differential toxicity toward putative oxygenated and hypoxic tumor
subpopulations in vivo in the FSaIIC murine fibrosarcoma. Cancer
Res. 50:3339–3344. 1990.
|
|
35
|
Mueck AO and Seeger H: 2-Methoxyestradiol
- biology and mechanism of action. Steroids. 75:625–631. 2010.
|
|
36
|
Piret JP, Mottet D, Raes M and Michiels C:
Is HIF-1alpha a pro-or an anti-apoptotic protein? Biochem
Pharmacol. 64:889–892. 2002.
|
|
37
|
Greijer AE and van der Wall E: The role of
hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J
Clin Pathol. 57:1009–1014. 2004.
|
|
38
|
Shen H, Yang Y, Xia S, Rao B, Zhang J and
Wang J: Blockage of Nrf2 suppresses the migration and invasion of
esophageal squamous cell carcinoma cells in hypoxic
microenvironment. Dis Esophagus. 27:685–692. 2014.
|
|
39
|
Visagie MH and Joubert AM: In vitro
effects of 2-methoxyestra-diol-bis-sulphamate on reactive oxygen
species and possible apoptosis induction in a breast adenocarcinoma
cell line. Cancer Cell Int. 11:432011.
|
|
40
|
Stander BA, Marais S, Vorster CJ and
Joubert AM: In vitro effects of 2-methoxyestradiol on morphology,
cell cycle progression, cell death and gene expression changes in
the tumorigenic MCF-7 breast epithelial cell line. J Steroid
Biochem Mol Biol. 119:149–160. 2010.
|
|
41
|
Vorster C and Joubert A: In vitro effects
of 2-methoxyestradiol-bis-sulphamate on cell growth, morphology and
cell cycle dynamics in the MCF-7 breast adenocarcinoma cell line.
Biocell. 34:71–79. 2010.
|
|
42
|
Chander SK, Foster PA, Leese MP, Newman
SP, Potter BV, Purohit A and Reed MJ: In vivo inhibition of
angiogenesis by sulphamoylated derivatives of 2-methoxyoestradiol.
Br J Cancer. 96:1368–1376. 2007.
|
|
43
|
Tevaarwerk AJ, Holen KD, Alberti DB, Sidor
C, Arnott J, Quon C, Wilding G and Liu G: Phase I trial of
2-methoxyestra-diol NanoCrystal dispersion in advanced solid
malignancies. Clin Cancer Res. 15:1460–1465. 2009.
|
|
44
|
Visagie M, Theron A, Mqoco T, Vieira W,
Prudent R, Martinez A, Lafanechère L and Joubert A: Sulphamoylated
2-methoxyestra-diol analogues induce apoptosis in adenocarcinoma
cell lines. PLoS One. 8:e719352013.
|
|
45
|
Ricker JL, Chen Z, Yang XP, Pribluda VS,
Swartz GM and Van Waes C: 2-methoxyestradiol inhibits
hypoxia-inducible factor 1alpha, tumor growth, and angiogenesis and
augments paclitaxel efficacy in head and neck squamous cell
carcinoma. Clin Cancer Res. 10:8665–8673. 2004.
|
|
46
|
Sato M, Tanaka T, Maeno T, Sando Y, Suga
T, Maeno Y, Sato H, Nagai R and Kurabayashi M: Inducible expression
of endothelial PAS domain protein-1 by hypoxia in human lung
adenocarcinoma A549 cells. Role of Src family kinases-dependent
pathway. Am J Respir Cell Mol Biol. 26:127–134. 2002.
|
|
47
|
Uchida T, Rossignol F, Matthay MA, Mounier
R, Couette S, Clottes E and Clerici C: Prolonged hypoxia
differentially regulates hypoxia-inducible factor (HIF)-1alpha and
HIF-2alpha expression in lung epithelial cells: Implication of
natural antisense HIF-1alpha. J Biol Chem. 279:14871–14878.
2004.
|
|
48
|
Wu XH, Qian C and Yuan K: Correlations of
hypoxia-inducible factor-1α/hypoxia-inducible factor-2α expression
with angio-genesis factors expression and prognosis in non-small
cell lung cancer. Chin Med J. 124:11–18. 2011.
|
|
49
|
Kim WY, Perera S, Zhou B, Carretero J, Yeh
JJ, Heathcote SA, Jackson AL, Nikolinakos P, Ospina B, Naumov G, et
al: HIF2α cooperates with RAS to promote lung tumorigenesis in
mice. J Clin Invest. 119:2160–2170. 2009.
|