Introduction
Melanoma, derived from epidermal melanocytes,
represents the most serious type of skin cancer and accounts for
80% of skin cancer-related deaths (1). Cul1, an essential scaffold of the SCF
(Skp1/Cullin/Rbx1/F-box protein) E3 ubiquitin ligase complex, has
been reported to be overexpressed in many cancer tissues and is
significantly correlated with the poor prognosis of tumors,
including hepatocellular carcinoma, colorectal cancer, glioma, lung
cancer, breast cancer and gastric cancer (2–7). In
melanoma, Cul1 expression is increased in the early stages of
melanoma (8). Cul1, combined with
BRG1, Bim and ING4, aid in the discrimination of melanoma from
dysplastic nevi (9). Cul1 enhances
melanoma cell proliferation by promoting G1-S phase transition
(10). However, the underlying
mechanisms involved in the regulation of melanoma cell
proliferation by Cul1 remain poorly understood.
The eIF4F complex plays a critical role in cancer
development by facilitating the cap-dependent translation of
oncogenic mRNAs, such as cyclin D1, c-Myc, VEGF and Mcl (11). The eIF4F complex consists of eIF4A,
eIF4G1 and eIF4E, and its assembly is largely dependent on eIF4E
availability, which is negatively regulated by 4E-BP1
phosphorylation (12). The
unphosphorylated or hypophosphorylated 4E-BP1 binds to the eIF4E
surface antagonistically with eIF4G and suppre-presses the
formation of the eIF4F complex. Phosphorylation of 4E-BP1 causes
4E-BP1 to disassociate from eIF4E and thus allows eIF4F assembly
and translation initiation. In melanoma, hyperphosphorylated 4E-BP1
was reported to be associated with worse overall and
post-recurrence survival (13).
The mammalian target of rapamycin complex 1 (mTORC1)
phosphorylates 4E-BP1 on Thr37 and Thr46, which promotes subsequent
phosphorylation of Ser65 and Thr70 and thus enhances cap-dependent
translation (14). mTORC1 consists
of mTOR, Raptor, PRAS40, GβL and DEPTOR, one of its own endogenous
inhibitors (15). DEPTOR inhibits
mTORC1 activity through binding to the FAT domain of mTOR through
its PDZ domain (16). Due to its
inhibitory effect on mTORC1 activity, DEPTOR acts, in general, as a
tumor suppressor by suppressing cap-dependent translation and cell
proliferation. DEPTOR activity is regulated largely by the control
of DEPTOR levels, which are negatively regulated by
SCFβTrCP E3 ubiquitin ligase (17–19).
By binding to DEPTOR, SCFβTrCP promotes the
ubiquitination and degradation of DEPTOR, leading to activation of
mTORC1. Given that Cul1 serves as a rigid scaffold in the SCF
complex and aberrant expression of Cul1 results in dysfunction of
SCF E3 ligases, we speculated that Cul1 may promote cap-dependent
translation and melanoma cell proliferation by promoting DEPTOR
degradation and enhancing mTORC1 activity.
In the present study, we investigated the effect of
Cul1 on DEPTOR expression, mTORC1 activity and cap-dependent
translation in melanoma cells. We found that Cul1 regulated mTORC1
activity through degradation of DEPTOR, which promoted 4E-BP1
phosphorylation and cap-dependent translation. Furthermore, we
found that suppression of mTORC1 activity or the eIF4F complex
assembly profoundly inhibited the promotive effect of Cul1 on
melanoma cell proliferation, while enhancing the eIF4F complex
activity by silencing the expression of 4E-BP1 significantly
antagonized the inhibitory effect of Cul1 depletion on melanoma
cell proliferation. Our data indicate that Cul1 promotes melanoma
cell proliferation by promoting DEPTOR degradation and enhancing
cap-dependent translation.
Materials and methods
Antibodies and reagents
Antibodies against P70S6K, pP70S6K (T389), 4EBP-1,
p4EBP-1 (T37/46), p4EBP-1 (S65), cyclin D1, eIF4E and eIF4G were
obtained from Cell Signaling Technology (Beverly, MA, USA).
Antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA)
included Cul1 and tubulin. Antibody against DEPTOR was obtained
from Millipore (Billerica, MA, USA). Anti-ubiquitin antibody was
purchased from Sigma (St. Louis, MO, USA). 4EGI-1 and PP242 was
provided by Calbiochem (Darmstadt, Germany) and Selleckchem
(Houston, TX, USA), respectively. MG132 was obtained from
Sigma.
Cells and cell culture
A375 and Mewo cells were cultured in Dulbecco's
modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA, USA)
containing 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA)
and 1% penicillin/streptomycin. All treatments with 4EGI-1 were
conducted in DMEM containing 5% FBS. Cells were maintained in a
37°C incubator at 5% CO2.
For stable overexpression of Cul1, A375 and Mewo
cells were transfected with the pCMV-2B-Cul1 vector and control
cells were transfected with the pCMV-2B backbone. Cells were
transfected with Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA,
USA) according to the manufacturer's instructions, and stable
transformants were selected using 500 µg/ml G418
(Calbiochem).
To silence the expression of Cul1, A375 and Mewo
cells were infected with appropriate amounts of lentiviral
particles carrying control shRNA or Cul1 shRNA (GeneChem Co.,
Shanghai, China). Virus-containing medium was discarded and
replaced with fresh medium after 12 h. At 48 h post-infection,
stable Cul1-knockdown cells were selected in puromycin (1
µg/ml).
Immunoblotting
Cells were lysed in RIPA buffer (150 mM NaCl, 50 mM
Tris-HCl, pH 7.5, 1% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 1 mM
EGTA, and 1% NP-40) containing protease inhibitors. Protein (40–80
µg) was electrophoresed on 10% SDS-PAGE gel after measuring
the protein concentration using the bicinchoninic acid (BCA) assay
reagent (Pierce Chemical, Rockford, IL, USA) and then transferred
to nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ,
USA). The membranes were blocked with 5% non-fat milk in 0.1%
PBS-Tween for 2 h at room temperature and then incubated with
primary antibodies overnight at 4°C, followed by incubation with
anti-rabbit/mouse/goat IgG conjugated to HRP for 2 h at room
temperature. Detection was performed using the ECL™ Advance Western
Blotting detection kit (GE Healthcare, Buckinghamshire, UK).
Ubiquitination assay
Cells were collected in lysis buffer (20 mM HEPES,
pH 7.2, 50 mM NaCl, 0.5% Triton X-100, 1 mM NaF and 1 mM DTT)
supplemented with protease inhibitors. To detect endogenous DEPTOR
ubiquitination, precleared cell lysates were incubated with the
DEPTOR antibody with gentle rotation at 4°C for 2 h, and then
protein-A beads were added for an additional 2-h incubation at 4°C
with gentle rotation. After being washed three times with lysis
buffer, the precipitated beads were analyzed by immunoblotting
using the ubiquitin antibody.
siRNA and transient transfections
siRNA for 4EBP1 and DEPTOR were purchased from
Invitrogen. A375 and Mewo cells were transfected with 4EBP1 or
DEPTOR siRNA or the negative control using Lipofectamine RNAiMAX
(Invitrogen) according to the manufacturer's instructions. At 48 h
post-transfection, the cells were lysed and subjected to assays for
immunoblotting, cap-dependent translation and apoptosis. For the
CCK-8 assay, cells were seeded into 96-well plates at 18 h
post-transfection.
m7GTP pull down assay
Cells were prepared in m7GTP lysis buffer
containing 20 mM Tris, 100 mM KCl, 20 mM β-glycerophosphate, 1 mM
EGTA, 1 mM EDTA, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl
fluoride, 0.25 mM Na3VO4, 10 mM NaF, and 1X
protease inhibitor cocktail. For the m7GTP pull down
assay, cell lysates (500 µg protein) were incubated with 30
µl of m7GTP-sepharose beads (GE Healthcare,
Chalfont St. Giles, UK) for 3 h at 4°C. Precipitates were washed
three times with 500 µl of phosphate-buffered saline
containing 0.5 mg/ml of heparin and 2 mM MgCl2, and then
analyzed by immunoblotting with the indicated antibodies.
Bicistronic luciferase assays
A375 or Mewo cells were transiently transfected with
a bicistronic luciferase reporter plasmid,
pcDNA3-rLuc-PolioIRES-fLuc, using Lipofectamine™ 2000 following the
manufacturer's instructions. This plasmid directs cap-dependent
translation of the Renilla luciferase (RL) gene and
cap-independent Polio IRES-mediated translation of the firefly (FL)
gene. At 48 h post-transfection, the luciferase activity was
measured with the Dual-Luciferase reporter assay kit (Promega,
Madison, WI, USA) according to the manufacturer's instructions.
Cap-dependent translational activity was determined by calculating
the ratio of Renilla/firefly luciferase luminescence. Assays
were performed in triplicate, and results are presented as means ±
standard deviation (SD).
Cell proliferation assays
Cells were seeded in 96-well plates (2,000
cells/well). At 18 h post-transfection, the cells were treated with
the agents as indicated for 48 h. After treatment, cell
proliferation was detected using the Cell Counting Kit-8 (CCK-8;
Dojindo Laboratories, Tokyo, Japan) assay according to the
manufacturer's instructions, and optical density (OD) was measured
at 450 nm. The OD value of the treatment group was normalized to
the values from the untreated control group. Assays were performed
in triplicate, and the results are presented as means ± standard
deviation (SD).
Cell cycle analysis
Cells were fixed with 75% ethanol overnight at
−20°C. After being washed twice with ice-cold PBS, the cells were
incubated with RNase A (100 µg/ml) for 30 min at 37°C and
then labeled with propidium iodide (50 µg/ml) for 15 min.
DNA contents were analyzed using a FACSCanto flow cytometer (BD
Biosciences, Mississauga, ON, Canada).
Statistical analysis
All data were analyzed using the unpaired Student's
t-test with GraphPad Prism 5 software. The data in this study are
presented as means ± standard deviation (SD). P<0.05 was
considered to indicate a statistically significant result.
Results
Cul1 promotes the ubiquitination and
degradation of DEPTOR
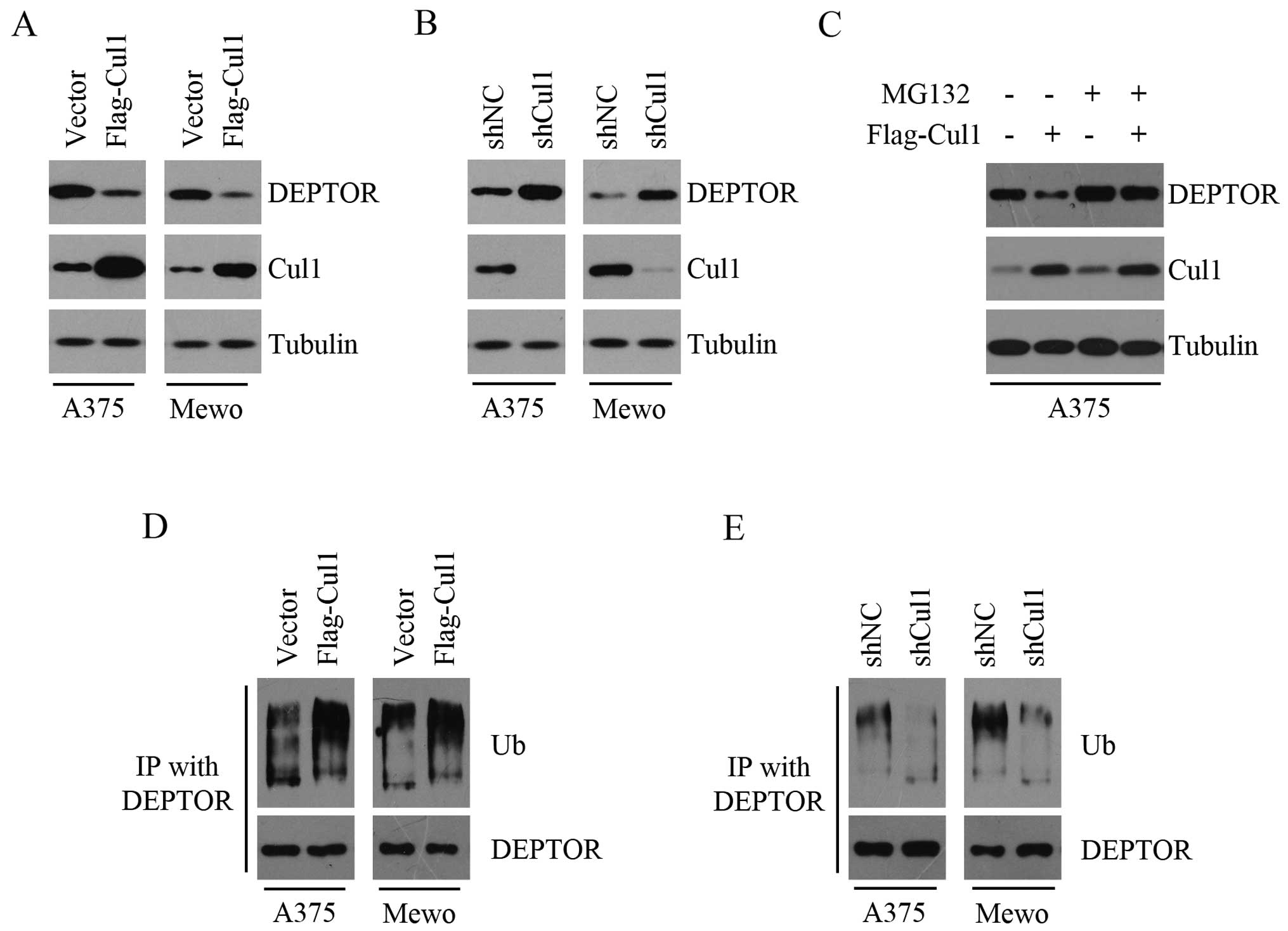
To investigate the effect of Cul1 on DEPTOR
expression, we stably overexpressed Cul1 in the A375 and Mewo cells
and found that Cul1 overexpression suppressed the expression of
DEPTOR (Fig. 1A). We next
determined the effect of Cul1 depletion on DEPTOR levels and found
that both Cul1-depleted A375 and Mewo cells had higher levels of
DEPTOR than the controls (Fig. 1B).
These results indicated an inversely correlated expression pattern
between Cul1 and DEPTOR in melanoma cells. In addition, the effect
of Cul1 on DEPTOR was suppressed in the presence of the proteasome
inhibitor MG132 (Fig. 1C), thereby
suggesting that the ubiquitin-proteasome pathway may be required
for Cul1-mediated reduction of DEPTOR protein abundance. Given that
Cul1 serves as a rigid scaffold in the SCF complex and DEPTOR is
degraded via the ubiquitin-proteasome pathway by
SCFβTrCP E3 ubiquitin ligase, we detected the effect of
Cul1 on the ubiquitination and degradation of DEPTOR. As shown in
Fig. 1D, Cul1 overexpression
promoted the ubiquitination of DEPTOR, whereas Cul1 depletion
inhibited the ubiquitination of DEPTOR (Fig. 1E). Taken together, these results
suggest that Cul1 decreases the expression of DEPTOR by promoting
the ubiquitination and degradation of DEPTOR.
Cul1 enhances mTORC1 activity by
inhibiting the expression of DEPTOR
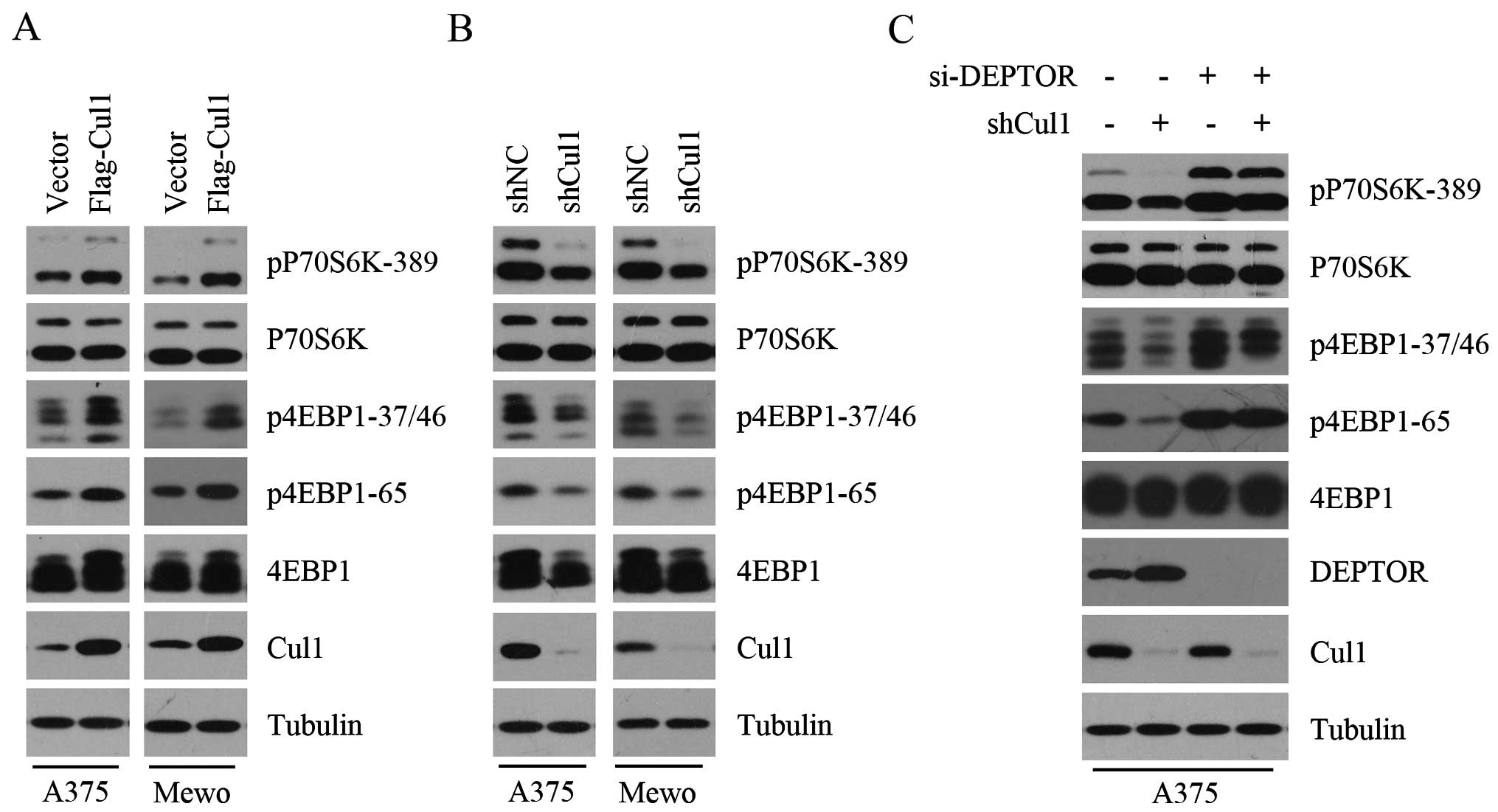
We demonstrated that Cul1 negatively regulates the
expression of DEPTOR. Since DEPTOR inhibits mTORC1 activity, we
speculated that Cul1 positively regulates mTORC1 activity. To test
this hypothesis, the phosphorylation levels of 4E-BP1 and p70S6K,
two downstream substrates of mTORC1, were detected in the control
and Cul1-overexpressing melanoma cell lines (A375 and Mewo). As
shown in Fig. 2A, Cul1
overexpression promoted the phosphorylation of 4E-BP1 and p70S6K,
indicating that Cul1 overexpression enhances mTORC1 activity. To
confirm the positive effect of Cul1 on mTORC1 activity, we further
determined the phosphorylation levels of 4E-BP1 and p70S6K in the
control and Cul1-depleted melanoma cell lines (A375 and Mewo) and
found that Cul1 knockdown profoundly attenuated the phosphorylation
of 4E-BP1 and p70S6K (Fig. 2B). To
investigate whether the negative effect of Cul1 depletion on mTORC1
activity results from DEPTOR accumulation, we silenced the
expression of DEPTOR in the control and Cul1-depleted A375 cells
and analyzed the phosphorylation of 4E-BP1 and p70S6K. As shown in
Fig. 2C, the inhibitory effect of
Cul1 depletion on the phosphorylation of 4E-BP1 and p70S6K was
rescued when DEPTOR was silenced, suggesting that Cul1 regulates
mTORC1 activity in a DEPTOR-dependent manner. Taken together, these
results suggest that Cul1 enhances mTORC1 activity by inhibiting
the expression of DEPTOR.
Cul1 activates cap-dependent
translation
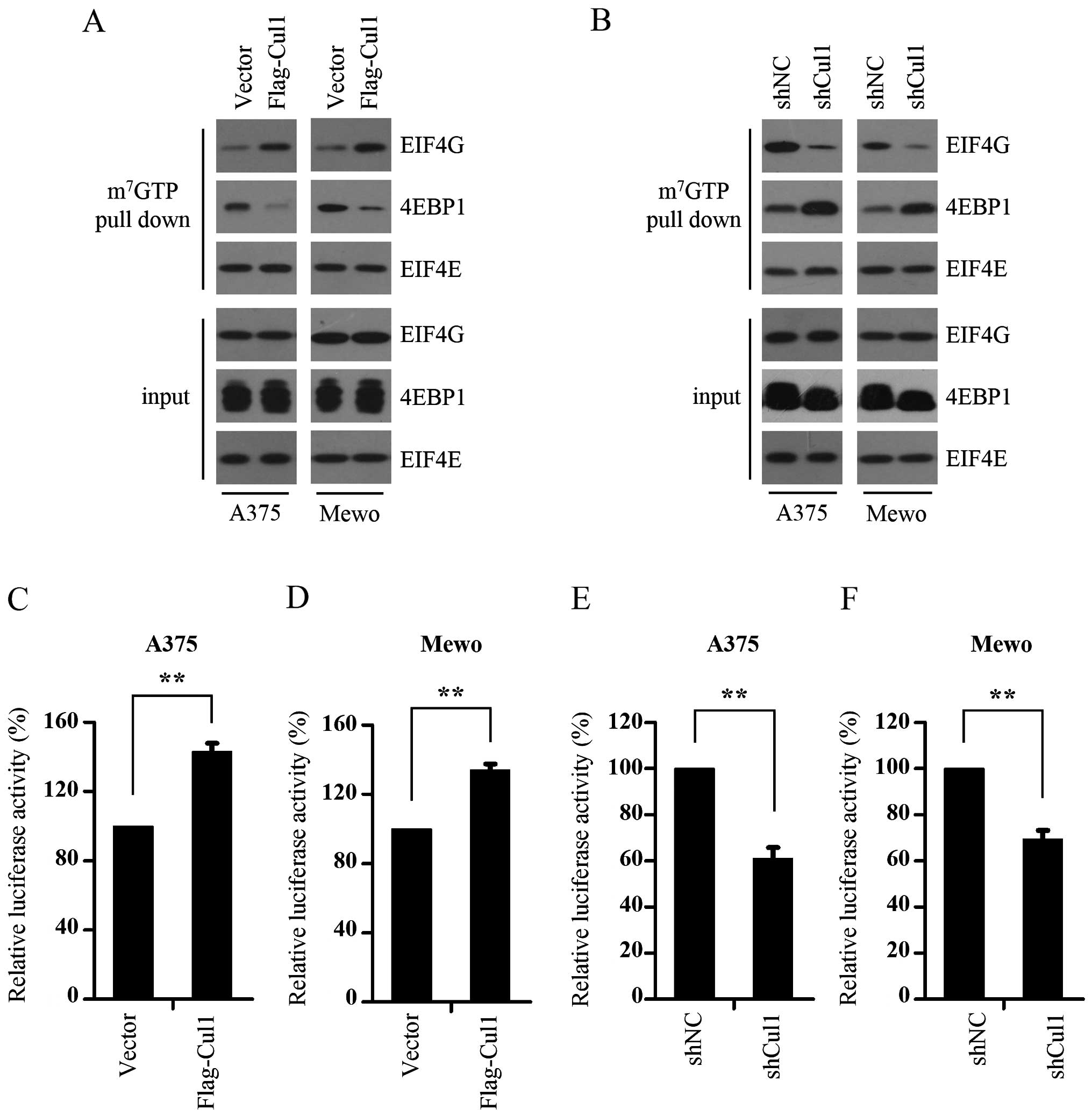
It is well-known that mTORC1 promotes the formation
of the eIF4F complex and activates cap-dependent translation by
phosphorylating 4E-BP1 and relieving its binding to eIF4E (20). As we found that Cul1 enhanced the
phosphorylation of 4E-BP1, we next investigated whether Cul1
enhances cap-dependent translation. To explore the function of Cul1
in cap-dependent translation, the effect of Cul1 on the assembly of
the eIF4F complex was determined using 7-methyl GTP sepharose bead
assay. The results show that Cul1 overexpression enhanced the
interaction of eIF4E and eIF4G, while inhibiting the interaction of
eIF4E and 4E-BP1 (Fig. 3A),
indicating that Cul1 overexpression promotes the formation of the
eIF4F complex. To confirm this result, we further detected the
assembly of the eIF4F complex in the control and Cul1-depleted
melanoma cells (A375 and Mewo) and found that Cul1 knockdown
profoundly suppressed the interaction of eIF4E and eIF4G (Fig. 3B). Given that cap-dependent
translation is dependent on the formation of the eIF4F complex, we
next detected the effect of Cul1 on cap-dependent translation in
the melanoma cells using a bicistronic luciferase reporter plasmid
that detects cap-dependent translation of the Renilla
luciferase gene and cap-independent Polio IRES-mediated translation
of the firefly luciferase gene. The results showed that Cul1
overexpression activated cap-dependent translation (Fig. 3C and D), whereas knockdown of Cul1
inhibited cap-dependent translation in both the A375 and Mewo cells
(Fig. 3E and F). To summarize,
these findings suggest that Cul1 enhances the formation of the
eIF4F complex, thus activating cap-dependent translation.
Cul1 promotes melanoma cell proliferation
by activating cap-dependent translation
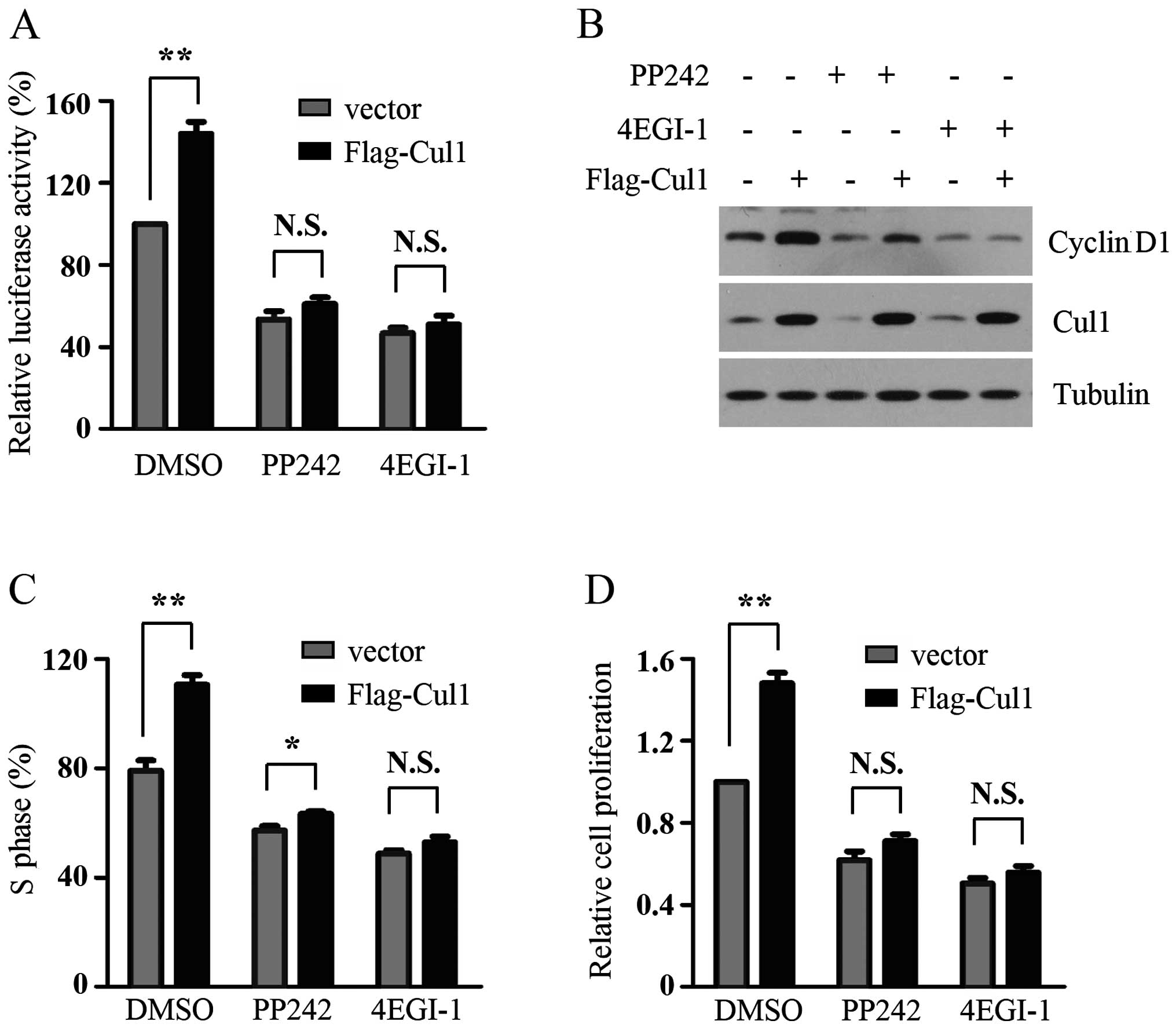
A previous study demonstrated that Cul1 enhances
melanoma cell proliferation by promoting G1-S phase transition
(10). However, the molecular
mechanism behind this is not clearly understood. Cap-dependent
translation plays a critical role in the control of cancer cell
proliferation by initiating translation of cell cycle
progression-related mRNAs, such as cyclin D1. Consistent with the
positive effect of Cul1 on cap-dependent translation and previous
results, we found that Cul1 overexpression enhanced the expression
of cyclin D1, the percentage of cells in the S phase and cell
proliferation of melanoma cells. PP242, an mTOR kinase inhibitor,
inhibited cap-dependent translation by decreasing the
phosphorylation of 4E-BP1. 4EGI-1 suppressed cap-dependent
translation initiation by disrupting the interaction of eIF4E and
eIF4G. The results showed that either PP242 or 4EGI-1 treatment
markedly reduced the promotive effect of Cul1 overexpression on
cap-dependent translation in the A375 cells (Fig. 4A). To determine whether the positive
effects of Cul1 overexpression on cell proliferation of melanoma
cells are dependent on increased cap-dependent translation, control
and Cul1-overexpressing A375 cells were treated with PP242 or
4EGI-1 for the indicated times and then the expression of cyclin
D1, the percentage of S phase cells and cell proliferation in the
melanoma cells were detected. As shown in Fig. 4B–D, the promotive effects of Cul1
overexpression on cyclin D1 expression, the percentage of S phase
cells and cell proliferation in the A375 cells were profoundly
attenuated upon PP242 or 4EGI-1 treatment. Taken together, Cul1
promotes melanoma cell proliferation by activating cap-dependent
translation.
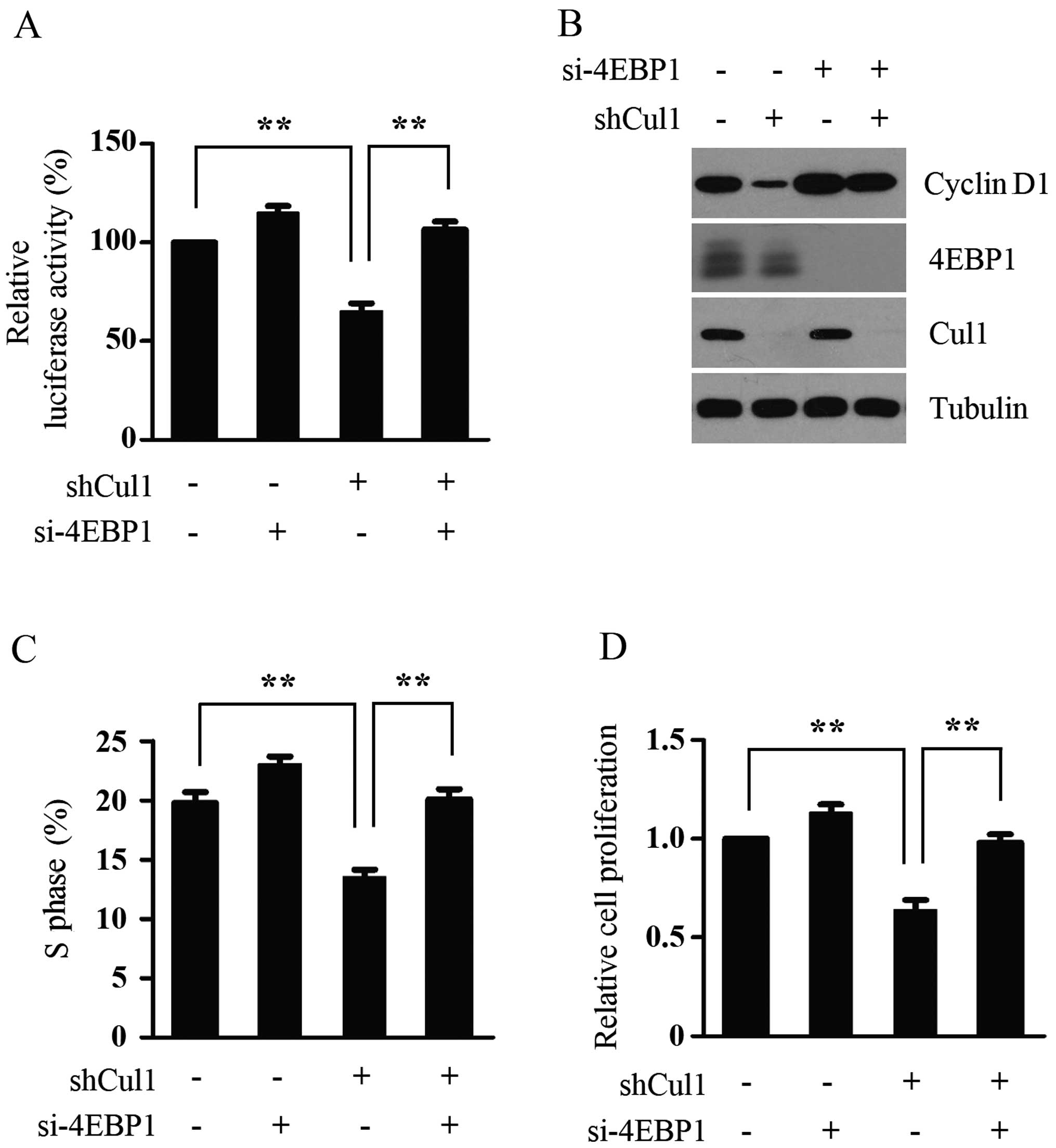
4E-BP1 mediates the effects of Cul1 on
cap-dependent translation and cell proliferation
4E-BP1 has been reported to negatively regulate cell
proliferation by selectively inhibiting the translation of
mRNA-encoding proteins involved in cell cycle progression, whereas
S6K regulates cell size in mammalian cells (21). We demonstrated that Cul1 promotes
melanoma cell proliferation by activating cap-dependent
translation. To further confirm this result and determine the
importance of 4E-BP1 dephosphorylation in mediating the effects of
Cul1 depletion on cap-dependent translation and cell proliferation
of melanoma cells, the expression of 4E-BP1 was silenced in the
control and Cul1-depleted A375 cells, and then cap-dependent
translation, the expression of cyclin D1, the percentage of S phase
cells and cell proliferation in the melanoma cells were detected.
In agreement with the inhibitory effect of Cul1 depletion on 4E-BP1
phosphorylation, Cul1 knockdown decreased cap-dependent
translation, the expression of cyclin D1, the percentage of S phase
cells and cell proliferation in the A375 cells. However, 4E-BP1
depletion significantly reversed the inhibitory effect of the
silencing of Cul1 on these processes (Fig. 5), suggesting that 4E-BP1
dephosphorylation is essential for Cul1 depletion to inhibit
cap-dependent translation and cell proliferation. Taken together,
these data suggest that 4E-BP1 mediates the effects of Cul1 on
cap-dependent translation and cell proliferation in melanoma
cells.
Discussion
Cul1 expression in melanoma tissues is profoundly
upregulated compared with that in paired normal tissues, and
increased Cul1 expression enhances melanoma cell proliferation by
promoting G1-to-S phase transition, which is consistent with its
first defined function as a regulator of the G1-to-S phase
transition in budding yeast (22).
As a scaffold protein, Cul1 binds to an adaptor protein SKP1 and an
F-box protein at the N-terminus and a RING protein RBX1 or RBX2 at
the C-terminus to constitute the functional SCF E3 ligases.
SCFβTrCP, one of the SCF E3 ligases, has been reported
to degrade DEPTOR (19). Given that
aberrant expression of Cul1 is associated with dysfunction of SCF
E3 ligases and decreased DEPTOR promotes cell proliferation via
activating mTORC1, in this study we first determined the effect of
Cul1 on DEPTOR expression levels. The results showed that the
expression level of Cul1 was conversely associated with that of
DEPTOR, suggesting the involvement of Cul1 in DEPTOR turnover. To
verify this assumption, we investigated the effect of Cul1 on
DEPTOR ubiquitination and degradation and found that Cul1 promoted
the ubiquitination of DEPTOR, while MG132, a proteasome inhibitor,
blocked its promotive effect on the degradation of DEPTOR,
suggesting that Cul1 decreases the expression of DEPTOR by
promoting the ubiquitination and degradation of DEPTOR. Whether the
expression level of Cul1 is conversely associated with that of
DEPTOR in clinical tissues remains to be addressed and is a
research direction we are currently pursuing.
DEPTOR expression negatively correlates with tumor
progression in many cancers, including colorectal cancer and
pancreatic ductal adenocarcinoma (23,24).
As a naturally occurring inhibitor of mTORC1, DEPTOR negatively
regulates cell cycle progression and cell proliferation via
suppressing mTORC1 activity. As we found that Cul1 inhibited the
expression of DEPTOR, we next analyzed the effect of Cul1 on mTORC1
activity. We found that Cul1 enhanced mTORC1 activity by inhibiting
the expression of DEPTOR. 4E-BP1 and p70S6K, two downstream
substrates of mTORC1, have been reported to regulate cell
proliferation and cell size in mammalian cells, respectively
(21). As we aimed to investigate
the underlying mechanisms involved in the regulation of melanoma
cell proliferation by Cul1, our subsequent research focused on
4E-BP1. 4E-BP1 negatively regulates the formation of the eIF4F
complex and cap-dependent translation by competing with eIF4G for
binding to eIF4E. Upon being phosphorylated by mTORC1, 4E-BP1
relieves its binding to eIF4E, permitting the assembly of the eIF4F
complex to initiate cap-dependent translation. Activation of mTORC1
was reported to be strongly associated with malignant melanocytic
lesions in vivo and inhibition of mTORC1 activity using
rapamycin suppressed the proliferation of melanoma-derived cell
lines (25,26). Hyperphosphorylated 4E-BP1 was
reported to be associated with worse overall and post-recurrence
survival of metastatic melanoma patients (13). The expression of eIF4E is strongly
elevated in melanoma and positively correlated with that of VEGF
and cyclin D1 (27), the mRNAs of
which are translated in a cap-dependent translation manner. These
studies indicate that cap-dependent translation plays a key role in
melanoma development. Since we found that Cul1 activated mTORC1 and
enhanced the phosphorylation of 4E-BP1 in melanoma cells, we
speculated that Cul1 may activate cap-dependent translation. Using
7-methyl GTP sepharose bead and bicistronic luciferase reporter
assays, we validated this hypothesis.
Cap-dependent translation promotes cell
proliferation through initiating the translation of mRNA encoding
proteins involved in cell cycle progression, such as cyclin D1 and
c-myc. Therefore, it is reasonable to speculate that Cul1 promotes
melanoma cell proliferation by activating cap-dependent
translation. Our results showed that blocking cap-dependent
translation using PP242 or 4EGI-1 profoundly attenuated the
promotive effects of Cul1 overexpression on the expression of
cyclin D1, the percentage of S phase cells and cell proliferation
in melanoma cells. Subsequently, we aimed to ascertain the role of
4E-BP1 phosphorylation in mediating the effects of Cul1 on
cap-dependent translation and cell proliferation of melanoma cells.
Cul1 depletion dephosphorylates 4E-BP1 and promotes 4E-BP1 to
compete with eIF4G for binding to eIF4E, leading to the inhibition
of cap-dependent translation. We relieved the sequestered eIF4E by
hypophosphorylated 4E-BP1 by silencing the expression of 4E-BP1 in
control and Cul1-depleted A375 cells and found that silencing of
4E-BP1 significantly reversed the suppressive effect of the
silencing of Cul1 on cap-dependent translation and cell
proliferation. However, 4E-BP1 knockdown did not completely restore
cap-dependent translation and cell proliferation suppressed by Cul1
depletion, suggesting that Cul1 may regulate other molecules
involved in cell proliferation in melanoma cells. In support of
this, Cul1 has been reported to regulate cell proliferation by
decreasing the expression of p27 in many types of cancer cells,
including melanoma cells (6,7,10).
In summary, we found that Cul1 promoted
cap-dependent translation and melanoma cell proliferation by
promoting DEPTOR degradation and activating mTORC1. Cul1 acts as a
scaffold to constitute the intact SCF E3 ligases, consisting of 4
functional components: a substrate-recognizing F-box protein, an
adaptor protein SKP1, a scaffold protein Cul1, a RING protein RBX1
or RBX2 (28). In addition to Cul1,
other components of SCF E3 ligases have been reported to be
implicated in skin cancers. RBX2, a RING protein, was reported to
promote the development of skin cancer by promoting inhibitor of
IκBα degradation to activate NF-κB (29). The expression of SKP2, an F-box
protein, is gradually increased during melanomagenesis from
melanocytic nevi to metastatic melanoma and is associated with a
poorer 5-year survival of melanoma patients (30). SKP2 promotes cell proliferation by
targeting the degradation of p27 and ING3 (31,32).
βTrCP1 and βTrCP2, two F-box proteins, are overexpressed in
DMBA/TPA-induced mouse skin papillomas and contribute to
skin papillomagenesis by accelerating degradation of IκBα (29,33).
These studies demonstrate that SCF E3 ubiquitin ligases play a
critical role in skin carcinogenesis and represent a potential
therapeutic target for the treatment of human skin cancer. In fact,
MLN4924, an indirect inhibitor of SCF E3 ligases, was shown to be
well tolerated and effective in patients with metastatic melanoma
in phase I clinical trials (34).
In addition, our results indicate that Cul1 promotes melanoma cell
proliferation by activating cap-dependent translation,
demonstrating that targeting mTORC1 or the eIF4F complex may be
effective for the treatment of melanoma patients with elevated Cul1
expression.
Acknowledgments
The present study was supported by the Guizhou
Province Chinese Native Medicine Modernization Special Project
(20125018 to Y.C.) and Guiyang Science and Technology Bureau
Science and Technology Innovation Platform Project (2012303 to
Y.C.).
Abbreviations:
|
Cul1
|
Cullin1
|
|
SCF
|
Skp1/Cullin/Rbx1/F-box protein
|
|
eIF4E
|
eukaryotic translation initiation
factor 4E
|
|
4E-BP1
|
eIF4E-binding protein 1
|
|
p70S6K
|
ribosomal p70 S6 kinase
|
|
mTORC1
|
mammalian target of rapamycin complex
1
|
References
|
1
|
Sosman JA, Kim KB, Schuchter L, Gonzalez
R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ,
Flaherty KT, et al: Survival in BRAF V600-mutant advanced melanoma
treated with vemurafenib. N Engl J Med. 366:707–714. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu W, Wang Y, Zhang C, Huang B, Bai J and
Tian L: Cullin1 is up-regulated and associated with poor patients'
survival in hepatocellular carcinoma. Int J Clin Exp Pathol.
8:4001–4007. 2015.PubMed/NCBI
|
|
3
|
Wang W, Chen Y, Deng J, Zhou J, Gu X, Tang
Y, Zhang G, Tan Y, Ge Z, Huang Y, et al: Cullin1 is a novel
prognostic marker and regulates the cell proliferation and
metastasis in colorectal cancer. J Cancer Res Clin Oncol.
141:1603–1612. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fan YC, Zhu YS, Mei PJ, Sun SG, Zhang H,
Chen HF, Chen C and Miao FA: Cullin1 regulates proliferation,
migration and invasion of glioma cells. Med Oncol. 31:2272014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xu M, Yang X, Zhao J, Zhang J, Zhang S,
Huang H, Liu Y and Liu J: High expression of Cullin1 indicates poor
prognosis for NSCLC patients. Pathol Res Pract. 210:397–401. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bai J, Yong HM, Chen FF, Mei PJ, Liu H, Li
C, Pan ZQ, Wu YP and Zheng JN: Cullin1 is a novel marker of poor
prognosis and a potential therapeutic target in human breast
cancer. Ann Oncol. 24:2016–2022. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bai J, Zhou Y, Chen G, Zeng J, Ding J, Tan
Y, Zhou J and Li G: Overexpression of Cullin1 is associated with
poor prognosis of patients with gastric cancer. Hum Pathol.
42:375–383. 2011. View Article : Google Scholar
|
|
8
|
Chen G, Cheng Y, Martinka M and Li G: Cul1
expression is increased in early stages of human melanoma. Pigment
Cell Melanoma Res. 23:572–574. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang G and Li G: Novel multiple markers
to distinguish melanoma from dysplastic nevi. PLoS One.
7:e450372012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen G and Li G: Increased Cul1 expression
promotes melanoma cell proliferation through regulating p27
expression. Int J Oncol. 37:1339–1344. 2010.PubMed/NCBI
|
|
11
|
Spilka R, Ernst C, Mehta AK and Haybaeck
J: Eukaryotic translation initiation factors in cancer development
and progression. Cancer Lett. 340:9–21. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kong J and Lasko P: Translational control
in cellular and developmental processes. Nat Rev Genet. 13:383–394.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
O'Reilly KE, Warycha M, Davies MA, Rodrik
V, Zhou XK, Yee H, Polsky D, Pavlick AC, Rosen N, Bhardwaj N, et
al: Phosphorylated 4E-BP1 is associated with poor survival in
melanoma. Clin Cancer Res. 15:2872–2878. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chiarini F, Evangelisti C, McCubrey JA and
Martelli AM: Current treatment strategies for inhibiting mTOR in
cancer. Trends Pharmacol Sci. 36:124–135. 2015. View Article : Google Scholar
|
|
15
|
Zoncu R, Efeyan A and Sabatini DM: mTOR:
From growth signal integration to cancer, diabetes and ageing. Nat
Rev Mol Cell Biol. 12:21–35. 2011. View
Article : Google Scholar
|
|
16
|
Wang Z, Zhong J, Inuzuka H, Gao D, Shaik
S, Sarkar FH and Wei W: An evolving role for DEPTOR in tumor
development and progression. Neoplasia. 14:368–375. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Duan S, Skaar JR, Kuchay S, Toschi A,
Kanarek N, Ben-Neriah Y and Pagano M: mTOR generates an
auto-amplification loop by triggering the βTrCP- and CK1α-dependent
degradation of DEPTOR. Mol Cell. 44:317–324. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gao D, Inuzuka H, Tan MK, Fukushima H,
Locasale JW, Liu P, Wan L, Zhai B, Chin YR, Shaik S, et al: mTOR
drives its own activation via SCF(βTrCP)-dependent degradation of
the mTOR inhibitor DEPTOR. Mol Cell. 44:290–303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao Y, Xiong X and Sun Y: DEPTOR, an mTOR
inhibitor, is a physiological substrate of SCF(βTrCP) E3 ubiquitin
ligase and regulates survival and autophagy. Mol Cell. 44:304–316.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Silvera D, Formenti SC and Schneider RJ:
Translational control in cancer. Nat Rev Cancer. 10:254–266. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dowling RJ, Topisirovic I, Alain T,
Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj
A, Liu Y, et al: mTORC1-mediated cell proliferation, but not cell
growth, controlled by the 4E-BPs. Science. 328:1172–1176. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kipreos ET, Lander LE, Wing JP, He WW and
Hedgecock EM: cul-1 is required for cell cycle exit in C. elegans
and identifies a novel gene family. Cell. 85:829–839. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lai EY, Chen ZG, Zhou X, Fan XR, Wang H,
Lai PL, Su YC, Zhang BY, Bai XC and Li YF: DEPTOR expression
negatively correlates with mTORC1 activity and tumor progression in
colorectal cancer. Asian Pac J Cancer Prev. 15:4589–4594. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li H, Sun GY, Zhao Y, Thomas D, Greenson
JK, Zalupski MM, Ben-Josef E and Sun Y: DEPTOR has growth
suppression activity against pancreatic cancer cells. Oncotarget.
5:12811–12819. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pópulo H, Soares P and Lopes JM: Insights
into melanoma: Targeting the mTOR pathway for therapeutics. Expert
Opin Ther Targets. 16:689–705. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Marone R, Erhart D, Mertz AC, Bohnacker T,
Schnell C, Cmiljanovic V, Stauffer F, Garcia-Echeverria C, Giese B,
Maira SM, et al: Targeting melanoma with dual phosphoinositide
3-kinase/mammalian target of rapamycin inhibitors. Mol Cancer Res.
7:601–613. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang SX, Hewitt SM, Steinberg SM, Liewehr
DJ and Swain SM: Expression levels of eIF4E, VEGF, and cyclin D1,
and correlation of eIF4E with VEGF and cyclin D1 in multi-tumor
tissue microarray. Oncol Rep. 17:281–287. 2007.PubMed/NCBI
|
|
28
|
Jia L and Sun Y: SCF E3 ubiquitin ligases
as anticancer targets. Curr Cancer Drug Targets. 11:347–356. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gu Q, Bowden GT, Normolle D and Sun Y:
SAG/ROC2 E3 ligase regulates skin carcinogenesis by stage-dependent
targeting of c-Jun/AP1 and IkappaB-alpha/NF-kappaB. J Cell Biol.
178:1009–1023. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen G, Cheng Y, Zhang Z, Martinka M and
Li G: Cytoplasmic Skp2 expression is increased in human melanoma
and correlated with patient survival. PLoS One. 6:e175782011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li Q, Murphy M, Ross J, Sheehan C and
Carlson JA: Skp2 and p27kip1 expression in melanocytic
nevi and melanoma: An inverse relationship. J Cutan Pathol.
31:633–642. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen G, Wang Y, Garate M, Zhou J and Li G:
The tumor suppressor ING3 is degraded by SCF(Skp2)-mediated
ubiquitin-proteasome system. Oncogene. 29:1498–1508. 2010.
View Article : Google Scholar
|
|
33
|
Bhatia N, Herter JR, Slaga TJ, Fuchs SY
and Spiegelman VS: Mouse homologue of HOS (mHOS) is overexpressed
in skin tumors and implicated in constitutive activation of
NF-kappaB. Oncogene. 21:1501–1509. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nawrocki ST, Griffin P, Kelly KR and Carew
JS: MLN4924: A novel first-in-class inhibitor of NEDD8-activating
enzyme for cancer therapy. Expert Opin Investig Drugs.
21:1563–1573. 2012. View Article : Google Scholar : PubMed/NCBI
|