Introduction
Worldwide, bladder cancer (BCa) is the 9th most
common malignant tumor and the 13th leading cause of death due to
cancer (1). Clinicopathologically,
BCa is divided into superficial and invasive types (2–4). The
instillation of chemodrug or Bacillus Calmette-Guérin (BCG) plus
transurethral resection of BCa (TUR-bt) procedure is considered to
be the most effective treatment for T1 and T2a superficial BCa
(5). For Ta, Tis superficial BCa
and invasive BCa, surgery (including radical cystectomy)
accompanied by cisplatin-based chemotherapy is recommended and
proved to be effective for promoting the progress-free survival
(PFS) of patients (6–9). However, in instillation of chemodrug
for superficial BCa or systematic chemotherapy for invasive BCa,
chemoresistance is the vital obstacle, leading to treatment
failure, whereas, the mechanism of how chemoresistance develops is
still unclear (7).
The ABC superfamily is the most abundant
trans-membrane protein family encoded in the human genome, which
plays important roles in pumping xenobiotics and anti-neoplastic
drugs (e.g. chemodrugs) out of cells against a concentration
gradient to maintenance the balance of the microenvironment, thus
resulting in a low drug concentration in the cells and leading to
the failure of chemotherapy (10).
To date, 49 members of ABC superfamily has been discovered and
divided into 7 subfamilies according to their structure (ABCA to
ABCG) (11), among of them, ʻABCB1ʼ
(MDR1/P-gp) is regarded as the main one due to its special role in
chemoresistance. MDR1 is a 170 kDa protein and consists of two
nucleotide-binding domains (NBDs) and two transmembrane domains
(TMDs), which localize in apical membrane of kidney, placenta,
liver, adrenal glands, intestine and blood-brain barrier cells
(12). Overexpression of MDR1 has
been associated with various types of cancers, such as acute
myeloid leukemia, childhood tumors, breast cancers and
hematological malignancies, which can be regulated by tumor-related
signaling, such as PI3K/Akt signaling (13–16).
Widely, chemodrug is reported to be one of the key inducers of MDR1
(17–20).
In the present study, cisplatin-resistant BCa cell
lines were generated to study the mechanism of chemoresistance in
BCa, and further signaling pathway dissections demonstrated that
HIF-1α→MDR1 pathway played critical role in the development of
resistance to cisplatin in BCa, providing an avenue for BCa
chemotherapeutics.
Materials and methods
Cell culture
Human BCa cell lines T24, and J82 were obtained from
American Type Culture Collection (ATCC; Manassas, VA, USA) and were
cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented
by 10% fetal bovine serum (FBS), (Invitrogen, Carlsbad, CA, USA).
Cells were cultured in an atmosphere with 5% CO2 at 37°C
(incubators, Thermo Scientific, Germany).
In order to get cisplatin-resistant cell lines, the
parental T24 and J82 cells were supplemented by 20 µM
cisplatin. Medium was refreshed every two days to remove the dead
cells and washed thrice using sterile phosphate-buffered saline
(PBS) (pH 7.2). The cisplatin-treatment for parental T24/J82 cells
for >3 months and MTT was used to verify the cisplatin
sensitivity in the end of treatment (the cisplatin-resistant cell
was tagged as T24Cis-R and J82Cis-R).
Western blotting
Pretreated cells were harvested at 80% confluency
and washed with cold PBS three times. Total cellular protein
lysates were prepared with RIPA buffer [50 mM Tris (pH 8.0), 150 mM
NaCl, 0.1% SDS, 1% NP40 and 0.5% sodium deoxycholate] containing
proteinase inhibitors [1% inhibitor cocktail and 1 mM PMSF, both
from Sigma (St. Louis, MO, USA)]. Protein (30 µg) was
separated on 10% SDS-PAGE gels and transferred to nitrocellulose
membranes. The membranes were blocked at room temperature for 1 h
with 5% skim milk in Tris-buffered saline (pH 7.6, TBS). Primary
antibodies were applied at different dilutions (GAPDH, 1:15,000;
HIF-1α, 1:300; MDR1, 1:400) in 5% skim milk in TBS at 4°C
overnight, followed by TBST (with Tween-20) washes. Membranes were
incubated with fluorescent secondary antibodies coupled to the
first antibody at room temperature in the dark for 1 h, followed by
TBST washes and signaling detection using Odyssey detection system
(both from Licor, Rockford, IL, USA). MG-132 (Sigma-Aldrich, USA)
was used to inhibit the proteasome-dependent degradation when
necessary (10 µM, 4 h before the protein harvest). GAPDH was
used as loading control (for total cell fraction).
Real-time PCR
Cellular total RNA was isolated using TRIzol reagent
(Invitrogen) and quantitated by absorbance at 260 nm. RNA (2
µg) was reverse transcribed using RevertAid™ First Strand
cDNA Synthesis kit (MBI Fermentas, St. Leon-Rot, Germany) according
to the manufacturer's protocol. For real-time PCR, we used the SYBR
Premix Ex Taq™ II system (Takara Biotechnology, Co., Ltd, Dalian,
China) and the Bio-Rad CFX96™ Real-Time system (Bio-Rad, Hercules,
CA, USA). Briefly, 12.5 µl SSYBR Premix Ex Taq™ II, 1
µl primer (F and R, respectively), 200 ng cDNA and 9.5
µl distilled and deionized water were mixed together,
followed by two stage, pre-degeneration for 95°C, 30 sec, one
repeat; and PCR reaction, 95°C 5 sec followed by 60°C, 30 sec, 30
repeats; and the third stage as dissociation, 95°C, 15 sec followed
by 60°C, 30 sec and another 95°C, 15 sec. GAPDH was used as the
loading control. Primers used are as follows: MDR1
(NM_000927) F, 5′-CAG GAA CCT GTA TTG TTT GCC ACC AC-3′ and R,
5′-TGC TTC TGC CCA CCA CTC AAC TG-3′; HIF-1α (NM_001530.3)
F, 5′-TTG CTC ATC AGT TGC CAC TTC C-3′ and R, 5′-AGC AAT TCA TCT
GTG CTT TCA TGT C-3′; GAPDH (NM_002046.4) F, 5′-AAC AGC GAC
ACC CAT CCT C-3′ and R, 5′-CAT ACC AGG AAA TGA GCT TGA CAA-3′.
Cell viability assay
Cell viability was assessed using a
tetrazolium-based assay (MTT). Pretreated cells were incubated in
the absence or presence of cisplatin/doxorubicin for the indicated
times, and then washed once with PBS and incubated with 0.5 mg/ml
of MTT at 37°C for 1 h. The reagent was reduced by living cells to
form an insoluble blue formazan product. After incubation, cells
were lysed with DMSO. Colorimetric analysis using a 96-well
microplate reader was performed at a wavelength of 490 nm. The
experiments were performed in triplicate.
Boyden chamber assay
Cell ability of migration/invasion was determined by
the Boyden chamber assay. Chambers with pores of 8-µm
diameter were obtained from Millipore (Switzerland). For migration
assay, 0.2 ml FBS-free DMEM suspension with 10,000 cells was added
to the upper chamber in 24-well plates, and 0.8 ml FBS-free DMEM
was added to the lower chamber. After 12 h incubation, the chambers
were washed with PBS (pH 7.4) three times to remove the cells in
the upper chamber and fixed with 4% formalin for 15 min, then
stained with crystal violet (0.01% in the ethanol) for 25 min
followed by washing three times with PBS. The cells were counted
using an inverted microscope, and five visions were randomly taken
in the ×200 magnification, and the average number of cells was
analyzed. For the invasive assay, the cell suspension (10,000
cells/well) in the upper chamber contained 0.2 ml mixture of
FBS-free DMEM/Matrigel at a ratio 8/1 (Matrigel; Sigma). Cells were
incubated for 36 h and the rest of procedure was conducted
according to the protocol of the migration assay.
RNAi and plasmid transfections
RNAi transfection: (siRNA-HIF-1α sense, CGT TGT GAG
TGG TAT TAT TTT and antisense, AAT AAT ACC ACT CAC AAC GTA;
siRNA-MDR1 sense, GGA AAA GAA ACC AAC UGU CdT dT and antisense, dT
dTC CUU UUC UUU GGU UGA CAG) were used to silence the expression of
HIF-1α and MDR1 in
T24Cis-R/J82Cis-R cells, Lipofectamine 2000
was used according to its protocol. Forced expression of HIF-1α or
MDR1 in parental T24/J82 cells completed by
HA-HIF1αP402A/P564A-pc-DNA3 and pHa-MDRwt
plasmids, were obtained from Addgene (Addgene plasmid, #18955 and
#10957, http://www.addgene.org). Lipofectamine
2000 was used to transfect plasmid into target cells, G418 was used
to select HIF-1α high-expression stable clone. Both
T24HIF-1α/J82HIF-1α and
T24MDR1/J82MDR1 were monitored using
real-time PCR for their HIF-1α or MDR1 expression efficiency.
Proliferative assay
5-Bromo-2′-deoxyuridine (BrdU) incorporation assay
was used to monitor the proliferative ability of tumor cells.
Pretreated cells were seeded on 8-well glass (Millipore) until
50–70% confluent, and BrdU was added into the medium (3
µg/ml), followed by 4 h incubation and then rinsed with PBS
for 10 min to remove residual free BrdU. Cells were then fixed with
4% paraformaldehyde for 45 min, followed by rinsing with PBS for 20
min. 0.1% Triton X-100 was used to permeabilize the cell membrane
for 15 min and 2N HCl added for 25 min to unspool DNA into single
strands to allow primary antibody access to the incorporated BrdU.
Cells were then rinsed with PBS for 10 min and non-specific
epitopes were blocked by 10% BSA for 20 min. Anti-BrdU antibody
(1:200) in 10% BSA was added to cells overnight at 4°C. Cells were
rinsed with PBS, followed by incubation with TRTIC-labeled
secondary antibody for 1 h at room temperature, and finally rinsed
with PBS to remove the free antibody. The fluorescence intensity of
TRITC was monitored by SuperMicro Orifice Plate Spectrophotometer
(BioTek, USA) in 547 nm.
Animal experiments
In order to demonstrate the ability of
tumorigenesis, parental BCa cells, T24/J82 and chemore-sistant BCa
cells, T24Cis-R/J82Cis-R,
T24HIF-1α/J82HIF-1α and
T24MDR1/J82MDR1 were implanted subcutaneously
in both flanks of the mice. In brief, 106 BCa cells
mixed with Matrigel (V/V=1:2) were injected into subcutaneous of
the two flanks of mice, 5 weeks later, tumor mass was harvested,
weighed, fixed with 4% formalin and prepared for pathological
analysis.
Hematoxylin and eosin (H&E) and
immunohistochemistry (IHC) staining
For H&E staining, the tissue sections were
de-waxed and rehydrated routinely. The sections were stained in
hematoxylin for 5 min, and washed in running tap water for 5 min.
Then the sections were stained in eosin for 30 sec, dehydrated and
mounted by routine methods. The representative fields were chosen
for presentation in the figures.
IHC staining was conducted using the Image-Pro Plus
System (Olympus, Japan). Tissues were deparaffinized, rehydrated
and subjected to 5-min pressure-cooking antigen retrieval, 15-min
endogenous enzyme block, 60-min primary antibody incubation and
30-min DakoCytomation EnVision-HRP reagent incubation for rabbit
antibodies. Signals were detected by adding substrate hydrogen
peroxide using diaminobenzidine (DAB) as a chromogen followed by
hematoxylin counterstaining. Negative control slices were prepared
by omitting the primary antibody. Stained (brown) cells are
indicated in the figures.
Statistical analysis
ANOVA test was used to analyze the statistical
discrepancy in >3 groups. Student's t-test was used to detect
any statistically significant difference between two groups.
P-values <0.05 were considered to indicate a statistically
significant result.
Results
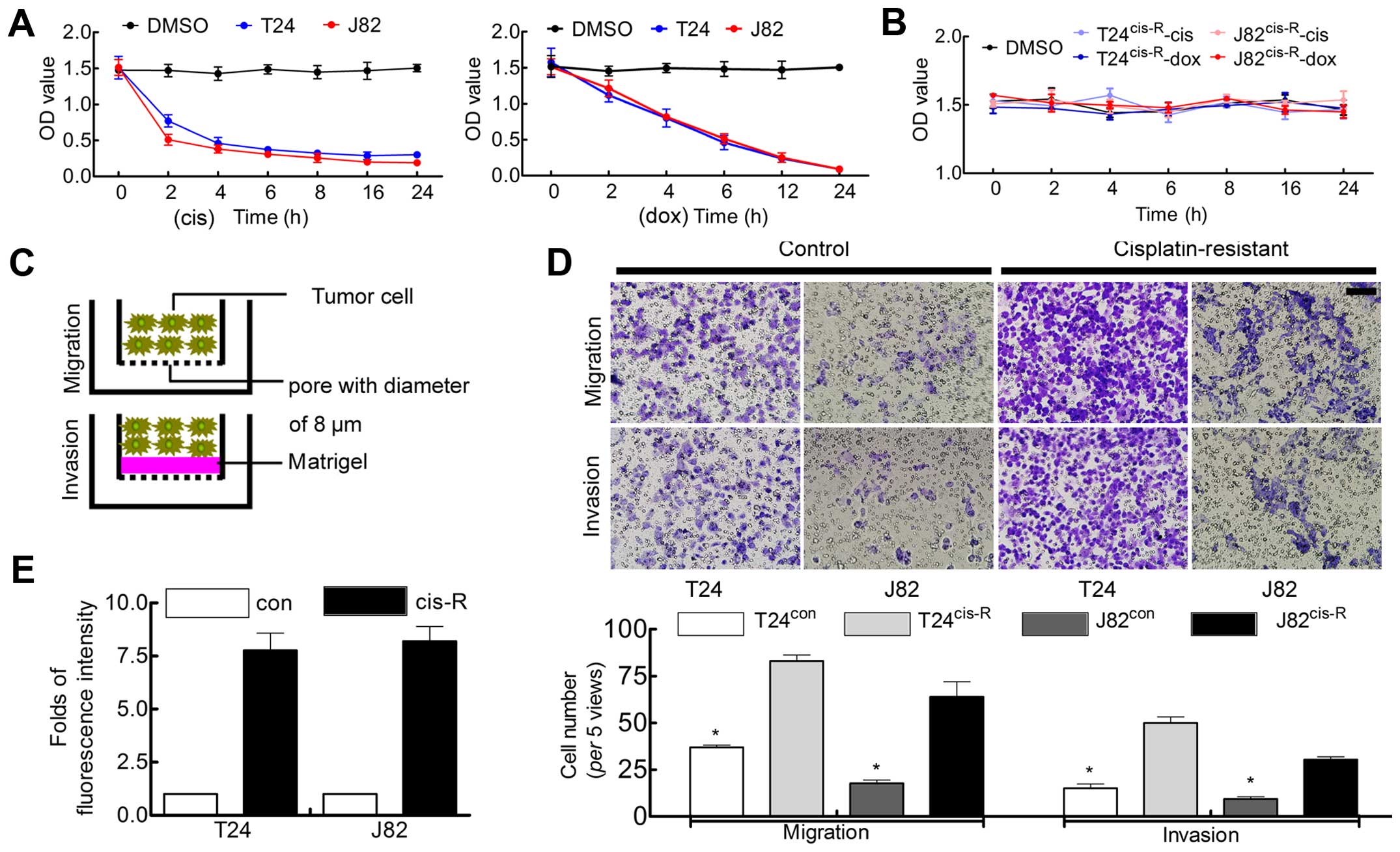
Decreased sensitivity of BCa cells to
cisplatin in prolonged treatment
Acquired drug resistance of cancer cells leads to
the failure of chemotherapy (12).
In BCa, cisplatin is regarded as the most effective components of
classical chemoregimen, such as M-AVC regimen; however, BCa cell
acquired resistance to the treatment leads to inevitable tumor
progression (21). Thus, in the
present study, cisplatin (20 µM) was used to treat BCa
cells, in addition, doxorubicin (40 µM) was used as parallel
experiment. Our results indicated that, along with the prolonged
exposure to chemodrug, the slope of cell viability gradually
decreased, for cisplatin (Fig. 1A),
indicating the decreased sensitivity of cells induced by the
chemodrug.
Chemoresistant BCa cells show enhanced
ability of proliferation and malignant behavior
Previous experiment suggested that cisplatin
sensitivity gradually decreased by the prolonged time of treatment,
therefore, 20 µM of cisplatin was used to treat T24/J82
cells as described in Materials and methods. More than three months
later, we observed that cisplatin-treated BCa cell lines displayed
chemoresistance to cisplatin, even non-sensitivity to doxorubicin
(Fig. 1B). These cell lines were
tagged with T24Cis-R and J82Cis-R in the
following investigation.
In order to clarify whether cisplatin-induced
chemo-resistance affected tumor malignancy and proliferation,
Boyden chamber assay and BrdU incorporation were applied. As
expected, T24Cis-R and J82Cis-R cells showed
enhanced malignant behavior (Fig. 1C
and D) and ability of proliferation (Fig. 1E).
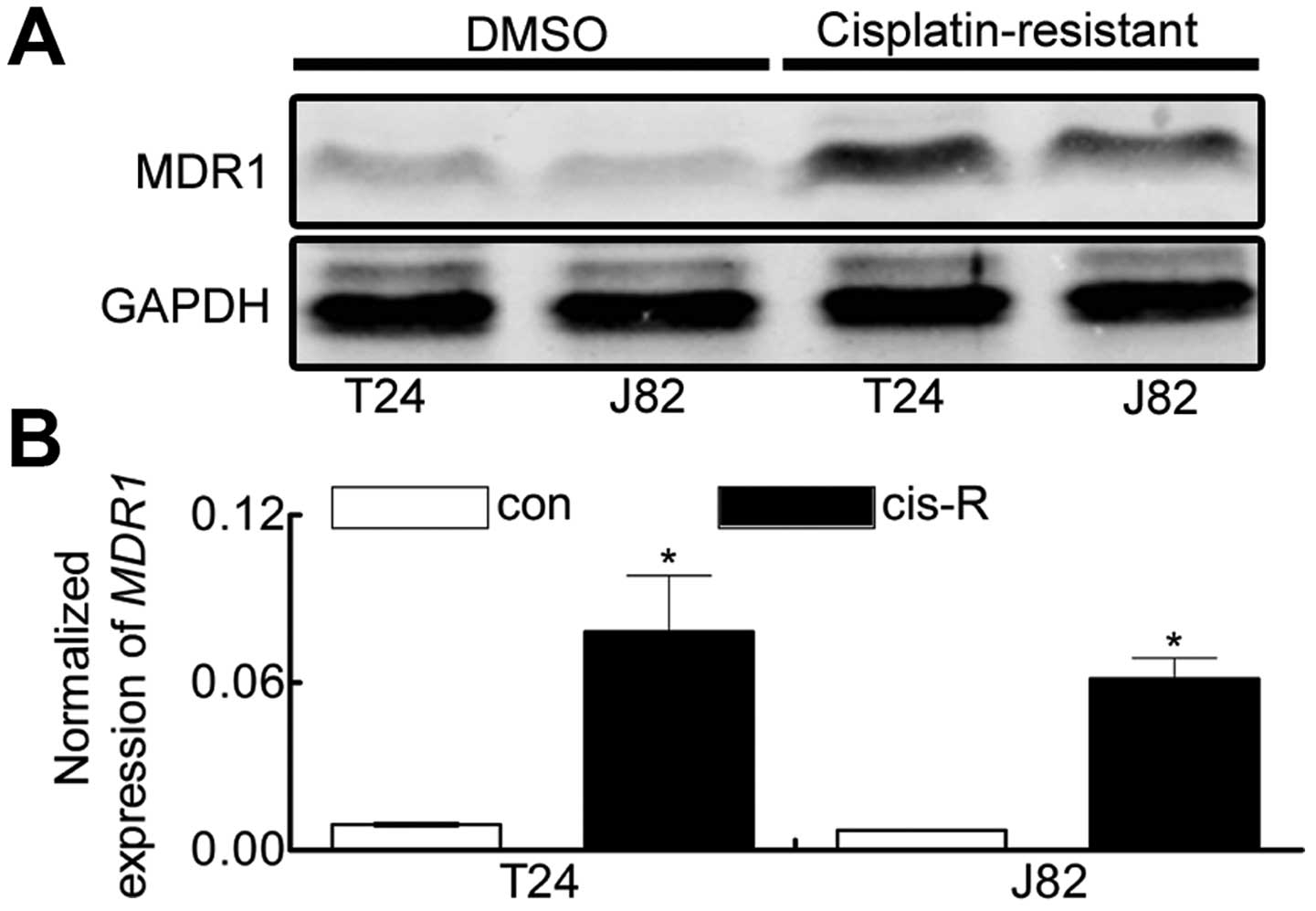
Cisplatin treatment induces elevated
expression of MDR1 (p-gp) in T24 and J82 cell lines
Increasing body of evidence indicates that the ABC
transporter family is irreplaceable in acquired chemoresistance,
and the most important one is the ABCB1 family, encoding MDR1
(P-gp) protein (22–24). Therefore we assessed whether MDR1
was involved in cisplatin-induced chemoresistance in our study. The
expression of MDR1 in T24Cis-R and J82Cis-R
cells were demonstrated using western blotting (Fig. 2A) and real-time PCR (Fig. 2B). Our results suggested that,
comparing with control cells, T24Cis-R and
J82Cis-R cells exhibited increased expression of MDR1,
giving evidence that cisplatin-induced chemoresistance is possibly
involved in upregulation of MDR1.
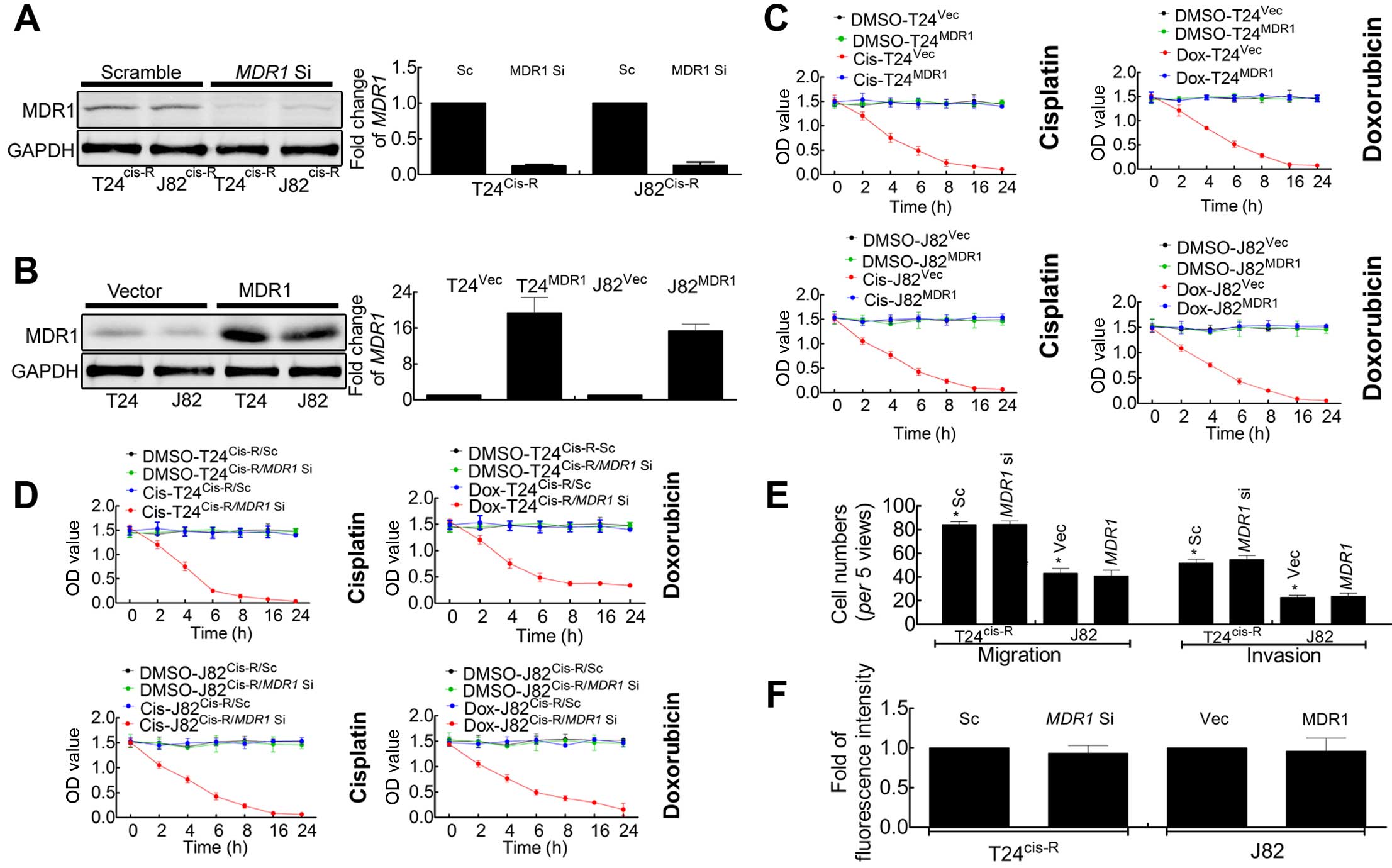
Knockdown of MDR1 in T24Cis-R
and J82Cis-R cells attenuates cisplatin-induced
chemoresistance
Large number of reports have pointed out the
important roles of MDR1 in chemoresistance (25,26),
in order to demonstrate this point, the expression of MDR1 in
T24Cis-R and J82Cis-R were knocked down by
RNAi, as indicated in Fig. 3A. The
MTT analysis suggested that chemodrug sensitivity significantly
increased in T24Cis-R/MDR1
Si and J82Cis-R/MDR1
Si compared with T24Cis-R/Sc and
J82Cis-R/Sc, respectively (Fig. 3C).
In addition to chemoresistance, we postulated that
the decreased expression of MDR1 may affect the malignancy of
T24Cis-R and J82Cis-R. Notably, the Boyden
chamber assay (Fig. 3E) and BrdU
incorporation (Fig. 3F) suggested
that there was no significant difference in cell proliferation and
malignant behaviors between
T24Cis-R/MDR1 Si vs.
T24Cis-R/Sc, or the
J82Cis-R/MDR1 Si vs.
J82Cis-R/Sc (data not shown).
Forced expression of MDR1 decreases drug
sensitivity in parental T24 and J82 cells
The above results suggested that knockdown of MDR1
in T24Cis-R and J82Cis-R cells led to the
increased chemodrug sensitivity, but had non-effect on tumor cell
malignancy, necessitating re-direction of our research. MDR1
plasmid was used to force expression of MDR1 in parental T24 and
J82 cells (Fig. 3B), followed by
MTT, Boyden chamber assay and BrdU incorporation to monitor the
cell viability, malignancy and proliferation. Our results suggested
that, consistent with the above findings, forced expression of MDR1
in parental T24 and J82 cells resulted in decreased chemodrug
sensitivity (Fig. 3D) and still had
non effect on tumor cell malignancy (Fig. 3E) and proliferation (Fig. 3F) (T24 data not shown).
Taken together, our results indicated that in T24
and J82 cells, cisplatin-induced chemodrug resistance was mediated
by upregulation of MDR1, the process of which was proved to have
non-effect on tumor cell malignancy and proliferation. However,
T24Cis-R and J82Cis-R cells manifested
enhanced ability of malignancy and proliferation, giving us new
questions and need for further investigation.
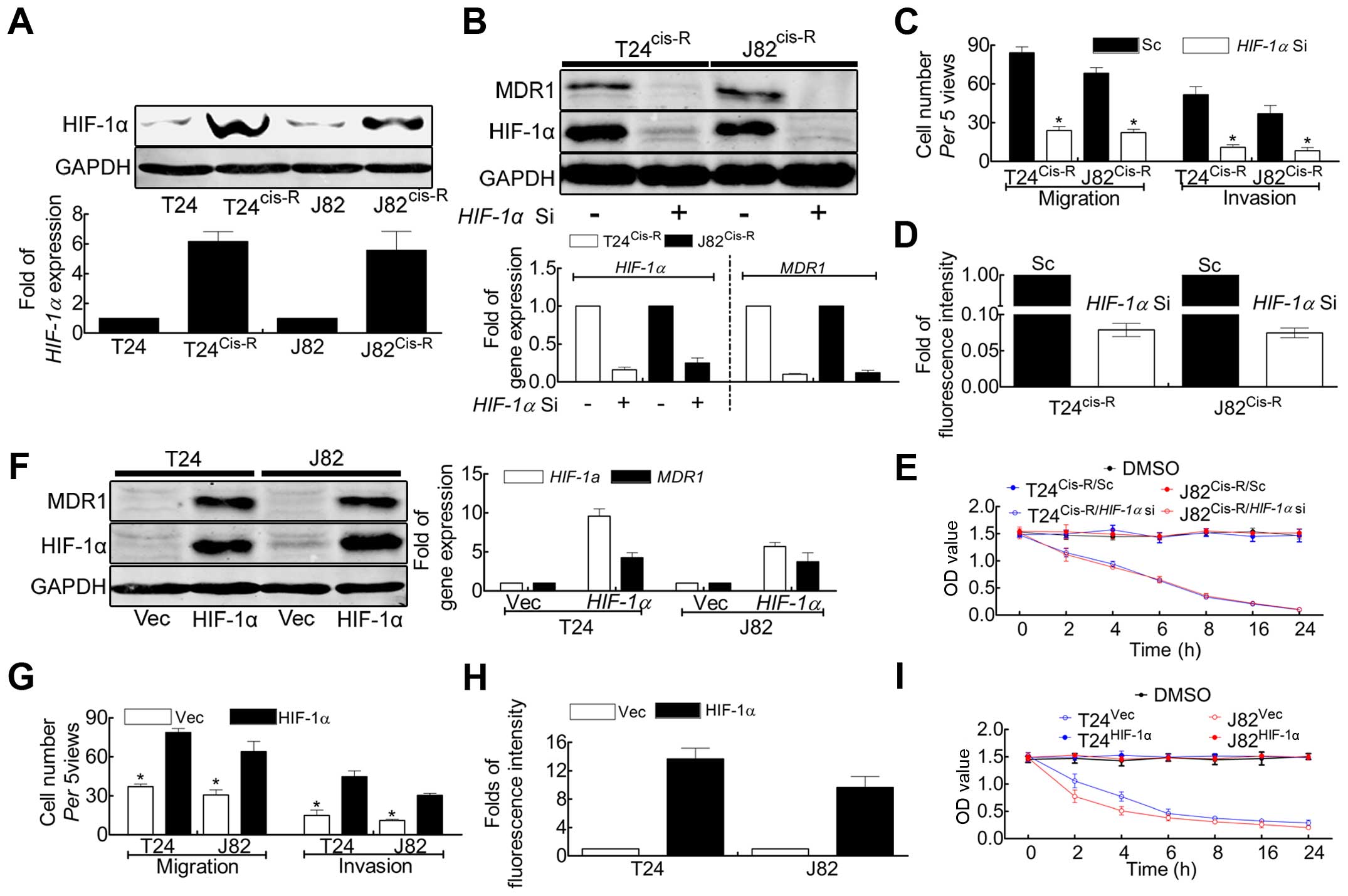
HIF-1α is involved in cisplatin-induced
upregulation of MDR1 in T24Cis-R and J82Cis-R
cells
Our previous investigation proved that
hypoxia-induced factor-1α (HIF-1α) played key roles in promoting
BCa cell migration/invasion and proliferation (27). Yet, MDR1 was reported to be one of
the target genes of HIF-1α (28),
evoking us to postulate that cisplatin-induced tumor malignancy
maybe medicated by HIF-1α. Expectedly, the expression of HIF-1α was
strongly elevated in T24Cis-R and J82Cis-R
cells comparing with parental T24 and J82 (Fig. 4A), leading us to investigate the
mechanism. Thus, HIF-1α was knocked down in T24Cis-R and
J82Cis-R cells (Fig.
4B), followed by Boyden chamber assay and BrdU incorporation.
Our results were consistence with the postulation that knockdown of
HIF-1α expression in T24Cis-R and J82Cis-R
cells resulted in deceased expression of MDR1, attenuated ability
of migration/invasion (Fig. 4C) and
proliferation (Fig. 4D),
accompanied by increased chemodrug sensitivity (Fig. 4E).
To further confirm this mechanism, forced expression
of HIF-1α/Vec was monitored in parental T24 and J82 cells as shown
in Fig. 4F, indicating that forced
expression of HIF-1α upregulated MDR1 in both protein and mRNA
levels. The followed results suggested that forced expression of
HIF-1α in parental T24 and J82 cells gave rise to tumor cell
migration/invasion (Fig. 4G) and
proliferation (Fig. 4H),
accompanied by decreased chemodrug sensitivity (Fig. 4I).
Collectively, we provided evidence that HIF-1α was
elevated in T24Cis-R and J82Cis-R cells,
through which cisplatin induced upregulation of MDR1, leading to
chemoresistance.
Demonstration the enhanced ability of
tumorigenesis in vivo and validation of our conclusion in human BCa
tissue
To demonstrate the enhanced ability of tumorigenesis
in T24Cis-R and J82Cis-R, both cell lines, in
line with their parental cells, were mixed with Matrigel and
injected into both flanks of the nude mice (Fig. 5A). The visible tumor mass was
harvested and validated by H&E staining (Fig. 5B).
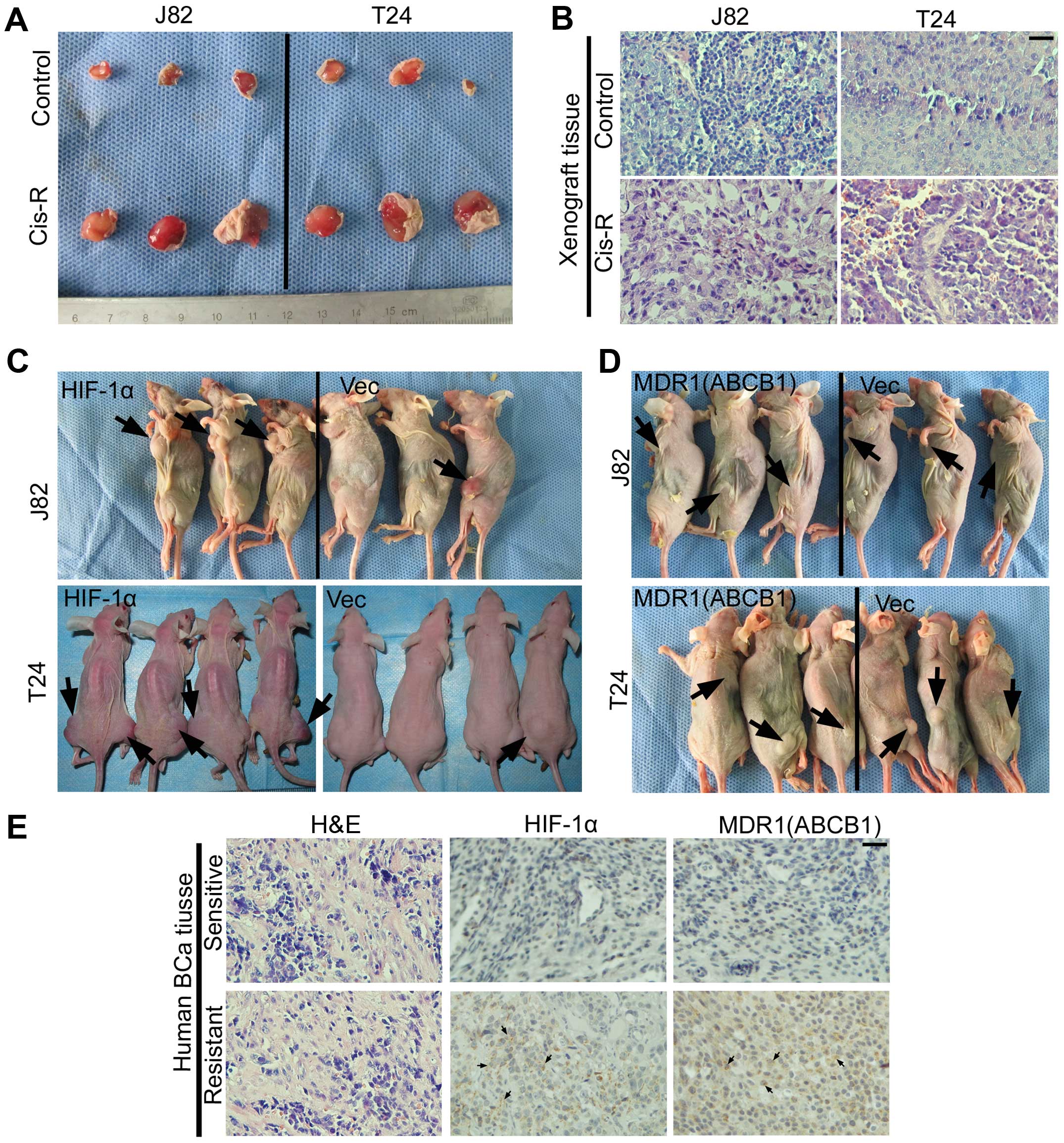 | Figure 5Validation of our conclusion in
vivo and human BCa tissue. (A) The harvested xenograft tumor
mass generated from J82con/J82Cis-R and
T24con/J82Cis-R indicating the enhanced
tumorigenetic ability of cisplatin-resistant cell lines. (B)
H&E staining for validation of the xenograft tumor mass,
indicating the cancerous structure, bar, 100 µm. (C)
Xenograft of T24HIF-1α/T24Vec and
J82HIF-1α/J82Vec in mice to monitor the
discrepancy of tumorigenesis, suggesting enhanced tumorigenic
ability of higher-HIF-1α-expression cell lines vs. the
corresponding Vector, black arrow, the tumor mass. (D) Xenografts
of T24MDR1/T24Vecand J82MDR1/J82Vec in mice
to monitor the discrepancy of tumorigenesis, suggesting that there
is no visible difference between the enhanced tumorigenic ability
of higher-MDR1-expression cell lines vs. the corresponding vector,
black arrow, and the tumor mass. (E) H&E and IHC staining for
the human BCa tissue, indicating the elevated expression of HIF-1α
and MDR1 in chemoresistant BCa tissues vs. chemosensitive BCa
tissues. Black arrow, positive cell; bar, 100 µm. |
To clarify whether there is a difference in
tumorigenic ability between high-HIF-1α expressing cells and
parental cells, both the T24Vec/T24HIF-1α and
J82Vec/J82HIF-1α cells were used. As
indicated in Fig. 5C, comparing
with vector, T24HIF-1α/J82HIF-1α showed
enhanced ability of tumorigenesis. In addition, in agreement with
our mechanistic conclusion, there is no significant discrepancy of
tumorigenesis between T24MDR1/J82MDR1 and
T24Vec/J82Vec (Fig. 5D).
Finally, to further confirm our conclusion in human
BCa tissue, 8 chemoresistant BCa tissues and 5 chemosensitive BCa
tissues were used to monitor the expression of HIF-1α and MDR1. As
expected, and in line with mechanistic investigation, IHC staining
for HIF-1α and MDR1 in human BCa tissues (Fig. 5D) suggested that, comparing with
chemosensitive BCa, chemoresistant BCa tissue showed higher
expression of HIF-1α and MDR1.
Discussion
Despite the improvement of surgery,
chemotherapy/radiotherapy is still the final regimen for invasive
BCa patient or the first choice for patient who cannot be helped by
surgery. Cisplatin-based chemodrug is recommended for most BCa
patients due to its high efficiency, such as M-VAC regimen
(3); however, due to multiple
chemoresistance, treatment failure still exists, and its mechanism
is unknown.
Theoretically, chemoresistance of tumor cells is
divided into initial and acquired types (29). The former suggests that there is a
fraction of chemoresistant cells in the tumor mass, which initiates
tumorigenesis after chemotherapy; the latter emphasizes that the
ability of chemoresistance is induced by chemodrugs, manifesting an
inevitable result for chemotherapy. Mechanistically,
chemoresistance can be ascribed to various reasons including drug
inactivation, off-target, cell death inhibition, epigenetics,
decreased drug uptake/increased drug efflux and EMT (30,31).
The drug efflux system attracts more and more attentions for its
irreplaceable roles in chemoresistance, which is mainly mediated by
ABC superfamily. Consistent with other studies, our data suggested
that prolonged time of cisplatin-treatment obviously led to the
elevated expression of MDR1, accompanied by attenuation of
cisplatin sensitivity and enhanced tumor cell malignant behavior
(Figs. 1 and 5A).
In our reversal experiment, we found that forced
expression of MDR1 in T24 and J82 (T24MDR1 and
J82MDR1) resulted in decreased sensitivity to chemodrug,
however, with no effect on tumor malignancy and proliferation
(Figs. 3 and 5D). These results created a dilemma since
in T24Cis-R and J82Cis-R, high expression of
MDR1 was accompanied by enhanced ability of malignancy and
proliferation (Figs. 3 and 5A). In BCa cell lines, our previous
investigation had proved that HIF-1α played irreplaceable roles in
monitoring the tumor cell malignancy (27). Furthermore, HIF-1α was proved to
target the expression of MDR1 directly (32). Previous investigations indicated
that activation of HIF-1α could be induced by hypoxia (33), VHL mutation, and PHD mutation; in
addition, reactive oxygen species (ROS) was regarded as the key
(34). Mechanistical investigation
indicated that ROS was elevated in cisplatin-treated tumor cells
(35). Therefore, we postulate that
in our study, elevated expression of HIF-1α was possibly induced by
ROS, which was the production of cisplatin-treatment, leading to
the expression of MDR1, accompanied by enhanced tumor cell
malignancy (Fig. 4) and
proliferation (Fig. 5C). However,
more studies are needed to support our postulation. In other
aspects, we found that, both cisplatin-induced MDR1 and forced
elevation of MDR1 led to doxorubicin resistance, indicating a
ubiquitous role in chemo-resistance for MDR1.
Taken together, in the present investigation, we
provide evidence that elevated expression of HIF-1α induced by
cisplatin monitor the upregulation of MDR1, resulting in
chemoresistance, in other aspects, elevated expression of HIF-1α
initiates enhanced tumor cell malignant behavior and ability of
proliferation. Thus, HIF-1α plays vital roles in cisplatin-induced
chemoresistance in BCa therapeutics, which provides us another view
to understand the acquired chemoresistance in BCa.
Acknowledgments
The present study was partly supported by the
National Natural Science Foundation of China (NSFC; no. 81172436 to
Y.S.), and Overall Innovation Projects of Scientific and
Technological Resources of Shaanxi Province (No. 2013KTCL03-04 to
Y.X.).
References
|
1
|
Skeldon SC and Goldenberg SL: Bladder
cancer: A portal into men's health. Urol Oncol. 33:40–44. 2015.
View Article : Google Scholar
|
|
2
|
Bellmunt J, Orsola A, Leow JJ, Wiegel T
and De Santis M: Bladder cancer: ESMO Practice Guidelines for
diagnosis, treatment and follow-up. Ann Oncol. 25(Suppl 3):
iii40–iii48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Witjes JA, Compérat E, Cowan NC, De Santis
M, Gakis G, Lebret T, Ribal MJ, Van der Heijden AG and Sherif A;
European Association of Urology: EAU guidelines on muscle-invasive
and metastatic bladder cancer: Summary of the 2013 guidelines. Eur
Urol. 65:778–792. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pang KH and Catto JWF: Bladder cancer.
Surgery. 31:523–529. 2013.
|
|
5
|
Svatek RS, Hollenbeck BK, Holmäng S, Lee
R, Kim SP, Stenzl A and Lotan Y: The economics of bladder cancer:
costs and considerations of caring for this disease. Eur Urol.
66:253–262. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sio TT, Ko J, Gudena VK, Verma N and
Chaudhary UB: Chemotherapeutic and targeted biological agents for
metastatic bladder cancer: A comprehensive review. Int J Urol.
21:630–637. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nadal R and Bellmunt J: New treatments for
bladder cancer: When will we make progress? Curr Treat Options
Oncol. 15:99–114. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li H, Yu G, Shi R, Lang B, Chen X, Xia D,
Xiao H, Guo X, Guan W, Ye Z, et al: Cisplatin-induced epigenetic
activation of miR-34a sensitizes bladder cancer cells to
chemotherapy. Mol Cancer. 13:82014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Petrelli F, Coinu A, Cabiddu M, Ghilardi
M, Vavassori I and Barni S: Correlation of pathologic complete
response with survival after neoadjuvant chemotherapy in bladder
cancer treated with cystectomy: A meta-analysis. Eur Urol.
65:350–357. 2014. View Article : Google Scholar
|
|
10
|
Jones PM and George AM: A reciprocating
twin-channel model for ABC transporters. Q Rev Biophys. 47:189–220.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chang G and Roth CB: Structure of MsbA
from E. coli: A homolog of the multidrug resistance ATP binding
cassette (ABC) transporters. Science. 293:1793–1800. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Anreddy N, Gupta P, Kathawala RJ, Patel A,
Wurpel JN and Chen ZS: Tyrosine kinase inhibitors as reversal
agents for ABC transporter mediated drug resistance. Molecules.
19:13848–13877. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Misra S, Ghatak S and Toole BP: Regulation
of MDR1 expression and drug resistance by a positive feedback loop
involving hyaluronan, phosphoinositide 3-kinase, and ErbB2. J Biol
Chem. 280:20310–20315. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kuo MT, Liu Z, Wei Y, Lin-Lee YC, Tatebe
S, Mills GB and Unate H: Induction of human MDR1 gene expression by
2-acety-laminofluorene is mediated by effectors of the
phosphoinositide 3-kinase pathway that activate NF-kappaB
signaling. Oncogene. 21:1945–1954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Burris HA III: Overcoming acquired
resistance to anticancer therapy: Focus on the PI3K/AKT/mTOR
pathway. Cancer Chemother Pharmacol. 71:829–842. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tazzari PL, Cappellini A, Ricci F,
Evangelisti C, Papa V, Grafone T, Martinelli G, Conte R, Cocco L,
McCubrey JA, et al: Multidrug resistance-associated protein 1
expression is under the control of the phosphoinositide 3
kinase/Akt signal transduction network in human acute myelogenous
leukemia blasts. Leukemia. 21:427–438. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hu XF, Slater A, Wall DM, Kantharidis P,
Parkin JD, Cowman A and Zalcberg JR: Rapid up-regulation of mdr1
expression by anthracyclines in a classical multidrug-resistant
cell line. Br J Cancer. 71:931–936. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schöndorf T, Neumann R, Benz C, Becker M,
Riffelmann M, Göhring UJ, Sartorius J, von König CH, Breidenbach M,
Valter MM, et al: Cisplatin, doxorubicin and paclitaxel induce mdr1
gene transcription in ovarian cancer cell lines. Recent Results
Cancer Res. 161:111–116. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou Y and Ling XL: Establishment of a
cisplatin-induced multidrug resistance cell line SK-Hep1/DDP. Chin
J Cancer. 29:167–171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Housman G, Byler S, Heerboth S, Lapinska
K, Longacre M, Snyder N and Sarkar S: Drug resistance in cancer: An
overview. Cancers. 6:1769–1792. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Galluzzi L, Vitale I, Michels J, Brenner
C, Szabadkai G, Harel-Bellan A, Castedo M and Kroemer G: Systems
biology of cisplatin resistance: Past, present and future. Cell
Death Dis. 5:e12572014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
He T, Mo A, Zhang K and Liu L: ABCB1/MDR1
gene polymorphism and colorectal cancer risk: A meta-analysis of
case-control studies. Colorectal Dis. 15:12–18. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Holland IB: ABC transporters, mechanisms
and biology: An overview. Essays Biochem. 50:1–17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Al-Shawi MK: Catalytic and transport
cycles of ABC exporters. Essays Biochem. 50:63–83. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Koenderink JB, Kavishe RA, Rijpma SR and
Russel FG: The ABCs of multidrug resistance in malaria. Trends
Parasitol. 26:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dean M and Annilo T: Evolution of the
ATP-binding cassette (ABC) transporter superfamily in vertebrates.
Annu Rev Genomics Hum Genet. 6:123–142. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guan Z, Ding C, Du Y, Zhang K, Zhu JN,
Zhang T, He D, Xu S, Wang X and Fan J: HAF drives the switch of
HIF-1α to HIF-2α by activating the NF-κB pathway, leading to
malignant behavior of T24 bladder cancer cells. Int J Oncol.
44:393–402. 2014.
|
|
28
|
Tsai YP and Wu KJ: Hypoxia-regulated
target genes implicated in tumor metastasis. J Biomed Sci.
19:1022012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–726. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Marín-Aguilera M, Codony-Servat J, Reig Ò,
Lozano JJ, Fernández PL, Pereira MV, Jiménez N, Donovan M, Puig P,
Mengual L, et al: Epithelial-to-mesenchymal transition mediates
docetaxel resistance and high risk of relapse in prostate cancer.
Mol Cancer Ther. 13:1270–1284. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sui H, Zhu L, Deng W and Li Q:
Epithelial-mesenchymal transition and drug resistance: Role,
molecular mechanisms, and therapeutic strategies. Oncol Res Treat.
37:584–589. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xie J, Li DW, Chen XW, Wang F and Dong P:
Expression and significance of hypoxia-inducible factor-1α and
MDR1/P-glycoprotein in laryngeal carcinoma tissue and hypoxic Hep-2
cells. Oncol Lett. 6:232–238. 2013.PubMed/NCBI
|
|
33
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Movafagh S, Crook S and Vo K: Regulation
of hypoxia-inducible factor-1α by reactive oxygen species: New
developments in an old debate. J Cell Biochem. 116:696–703. 2015.
View Article : Google Scholar
|
|
35
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|