Introduction
Endometrial cancer is one of the most common
gynecologic malignancies worldwide (1,2). There
are two different clinicopathological types of endometrial
carcinoma, including type I and II endometrial carcinoma (3). In clinical practice, over 80% of
endometrial cancers are type I, which commonly comprise low-grade
carcinomas. These tumors are often preceded by endometrial
premalignant disease and are always estrogen nuclear receptor (ER)
and progesterone receptor (PR)-positive, which frequently arise
during excessive estrogen exposure. Recently, much effort has been
made to confirm the involvement of aberrant estrogen metabolism in
dysregulated endometrial cancer cell growth and malignant
metastasis (4–6). We previously suggested that estrogen
promoted endometrial cancer cell proliferation and invasion by fat
mass and obesity-associated (FTO) gene (7), however, the molecular mechanism of how
FTO regulates cellular growth by estrogen remains obscure.
FTO was identified as an oncoprotein frequently
over-expressed in several types of cancer, including endometrial,
breast and pancreatic cancer (8–13). FTO
is a protein involved in energy homeostasis by controlling energy
expenditure. Depletion of FTO in mice was found to result in growth
retardation, adipose tissue reduction and lean body mass (14). Since estrogen-driven endometrial
cancer is strongly and definitively linked to obesity, two studies
previously examined the relationship between FTO gene polymorphism
and the incidence of endometrial cancer (9,13). In
our previous study, we also found that estrogen stimulation
resulted in FTO accumulation in the nucleus (7), however, the mechanism of
estrogen-driven FTO nuclear localization is not clear.
Although less than 0.1% of the total cellular
protein, kinase and phosphatase enzymes play a pivotal role in
conducting signals to control cell growth or invasion, abundant
signaling pathways have been reported to be involved in
estrogen-driven endometrial cancer. Inhibition of the PI3K/AKT
pathway was found to lead to a decrease in proliferative and
invasive activities. Blocking MAPK signaling also resulted in
similar effects. These results are consistent with our previous
study (7). Another important
signaling molecule, mTOR is an atypical serine/threonine protein
kinase that belongs to the phosphoinositide 3-kinase (PI3K)-related
kinase family and is involved in energy metabolism (15,16).
mTOR has been implicated in the development and progression of
various types of cancers including melanoma, lung and endometrial
cancer (17–27). Recent findings suggest that the mTOR
pathway may play an important role in endometrial cancer progestin
resistance (28). However, as a
molecule controlling energy expenditure, whether estrogen-driven
FTO nuclear localization is mediated by the mTOR signaling pathway
has not been studied. Therefore, the aim of the present study was
to investigate whether estrogen enhances FTO nuclear localization
and promotes endometrial cancer cell growth via the mTOR signaling
pathway.
Materials and methods
Sample collection
Forty-nine samples were obtained from the Tissue
Bank of the Department of Obstetrics and Gynecology of the Shanghai
First People's Hospital affiliated to Shanghai Jiao Tong
University, which were comprised of 18 normal endometrial tissues
and 31 cases of type I endometrial carcinoma. None of the patients
in the study had a history of prior radiotherapy or chemotherapy.
Any patient with a known history of hormone replacement was
excluded. The use of these specimens was approved by the Ethics
Committee of the Medical College of Shanghai Jiao Tong University,
China.
Immunohistochemical (IHC) staining and
analysis
IHC analysis of FTO protein expression was performed
as previously described and assessed using a semi-quantitative
method. Briefly, specimens were deparaffinized in xylene and
rehydrated in a graded series of ethanol and subsequently
endogenous peroxidase activity was blocked by a 10-min treatment
with 3.0% hydrogen peroxide. Subsequently, the sections were
subjected to antigen retrieval by boiling in citrate buffer (pH
6.0) and incubated for 30 min with 0.01% Trixon and then incubated
for 20 min with 5% bovine serum albumin (BSA). The sections were
incubated overnight with a rabbit anti-human FTO primary antibody
at 4°C in a humidity chamber, followed by a 50-min incubation with
a biotinylated secondary antibody (Dako, Carpinteria, CA, USA).
Omitted primary antibodies served as negative controls. Expression
of FTO protein was assessed using a semi-quantitative method: the
slides were evaluated for the percentage of positively stained
cells (0–4) and the intensity of the staining (0–3). The index of
FTO expression was calculated as the percentage x intensity of the
staining. Therefore, a score of 0 is negative (−), 1–4 is weakly
positive (+), 5–8 is positive (++), and 9–12 is strongly positive
(+++).
Cell lines and cell culture
To investigate the mechanism of estrogen-induced FTO
nuclear localization, the Ishikawa cell line was used in the
present study, which is an estrogen-responsive cell line derived
from a well-differentiated endometrioid carcinoma. The cells were
maintained in our laboratory after being generously provided by Dr
Masato Nishida, Tsukuba University, Tsukuba City, Japan. The cells
were maintained in Dulbecco's modified Eagle's medium (DMEM)/F-12
(1:1) medium with 10% fetal bovine serum (FBS) (both from Gibco,
Gaithersburg, MD, USA), 100 U/ml penicillin, sodium pyruvate and
L-glutamine in a humidified atmosphere of 5% CO2 at
37°C.
Immunoblot analysis
Immunblot analysis was performed as previously
described. Briefly, the harvested cells were lysed and the
supernatant was collected. Then, the protein was loaded onto
SDS-PAGE and transferred to polyvinylidene fluoride (PVDF)
membrane. The membranes were blocked with 5% skimmed milk for 1 h
and incubated overnight with the primary antibodies, followed by 1
h of incubation with the appropriate secondary antibody (1:5,000).
The anti-GAPDH or anti-lamin B1 rabbit monoclonal antibody was
diluted to 1:1,000 for use as a sample loading control. The
antibodies for FTO, GAPDH, lamin B1, p-mTOR, mTOR and ERα were
purchased from Abcam (Cambridge, UK).
Subcellular fractionation
Ishikawa cells treated with estrogen, rapamycin or
transfected with siERα were harvested and lysed with cytoplasmic
extraction buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 1.5
mM MgCl2, 1 mM dithiothreitol, 0.2% Nonidet P-40, 1 mM
NaF, 1 mM Na3VO4 and protease inhibitor
cocktail). After being centrifuged at 14,000 rpm at 4°C for 5 min,
the cytoplasmic fraction was collected. Then, the pellet was
re-suspended in nuclear extraction buffer (20 mM HEPES pH 7.9, 420
mM NaCl, 0.1 mM EDTA pH 8.0, 1.5 mM MgCl2, 1 mM
dithiothreitol, 0.2% Triton-X 100, 1 mM NaF, 1 mM
Na3VO4 and protease inhibitor cocktail), and
the nuclear fraction was collected after a 5-min
centrifugation.
Immunocytochemistry
Ishikawa cells treated with 10−9 M E2 for
48 h were cultured on coverslips before fixation with 3.7%
paraformaldehyde in phosphate-buffered saline (PBS) (10 min),
permeabilization with 0.2% Triton X-100 (10 min), blocking in 3%
BSA (1 h) and then incubation with an anti-human FTO primary
antibody (1:100, overnight). After incubation with a FITC-labeled
secondary antibody (1 h at room temperature), the cells were
photographed.
Small interfering RNA (siRNA)
transfection and hormone stimulation
The acute knockdown of siERα was performed as
previously described. Briefly, Ishikawa cells were seeded in 5 ml
of growth medium in 6-cm dishes without antibiotics, and grown to
30–50% confluency 24 h prior to transfection with 200 pmol ERα
siRNA (Shanghai GenePharma Co., Ltd.) using DharmaFECT (Thermo
Scientific). siRNA-treated and untreated Ishikawa cells were
exposed to 1 nM E2 for a further 48 h before being collected for
western blot analysis. The FTO knockdown was performed in a 96-well
with the incubated Ishikawa cells, and the cell proliferation was
determined by MTT assay.
MTT assay
To investigate the proliferative activity of
endometrial cancer cells after various treatments, the MTT assay
was performed. Briefly, Ishikawa cells were plated in a 96-well
plate (2,000 cells/well) and incubated for 24 h. The culture medium
was then changed to serum-free DMEM/F-12 (1:1) medium for 24 h. In
order to determine the effect of ICI or Rap on cellular growth,
cells were pre-treated with ICI or Rap for 1 h after stimulation
with 1 nM E2 or dimethyl sulfoxide (DMSO) for 48 h. Similarly,
prior to 1 nM E2 stimulation, the cells were transiently
transfected with FTO siRNA to investigate its role in cell
proliferation. MTT solution (20 µl of 5 mg/ml MTT in PBS)
was added to the cells. After 4 h of incubation at 37°C, the
culture medium was removed, and 150 µl of DMSO was added to
dissolve the formazan. Finally, absorbance at 490 nm was measured
with a GENios multifunction reader (Tecan, Zurich,
Switzerland).
Statistical analysis
The statistical significance of the differences in
the IHC staining in endometrial tissues was calculated using the
Chi-square test. The differences in various protein levels and cell
proliferation between groups were analyzed using the Student's
t-test. A two-sided test with P<0.05 was considered
statistically significant. All statistical analyses were performed
using SPSS 11.0 (SPSS, Inc., Chicago, IL, USA).
Results
FTO is overexpressed in endometrial
cancer and serves as a marker for poor prognosis
To understand the role of FTO in endometrial cancer
development, we first examined the expression of FTO in endometrial
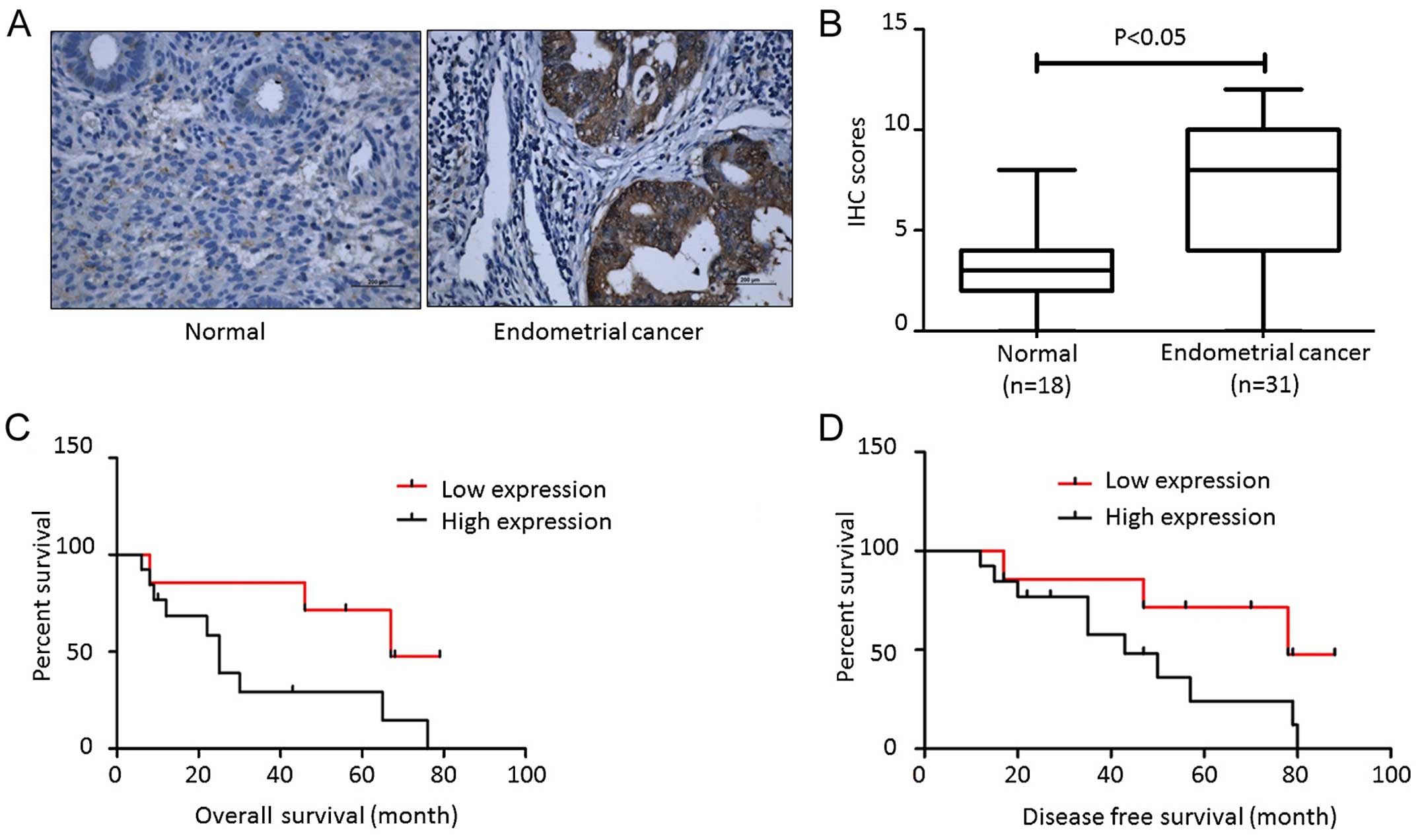
carcinoma tissues. As shown in Fig.
1A, little positive staining was detected in the normal
endometrial tissues while strong positive staining was observed in
type I endometrial carcinomas. The significant difference is
summarized in Fig. 1B. Although we
reported that FTO expression was not associated with age, stage,
grade, invasion and lymph node metastasis, we still found that
higher FTO expression correlated with poor prognosis and early
relapse (Fig. 1C and D).
Estrogen promotes FTO nuclear
localization
In our previous study, increasing nuclear expression
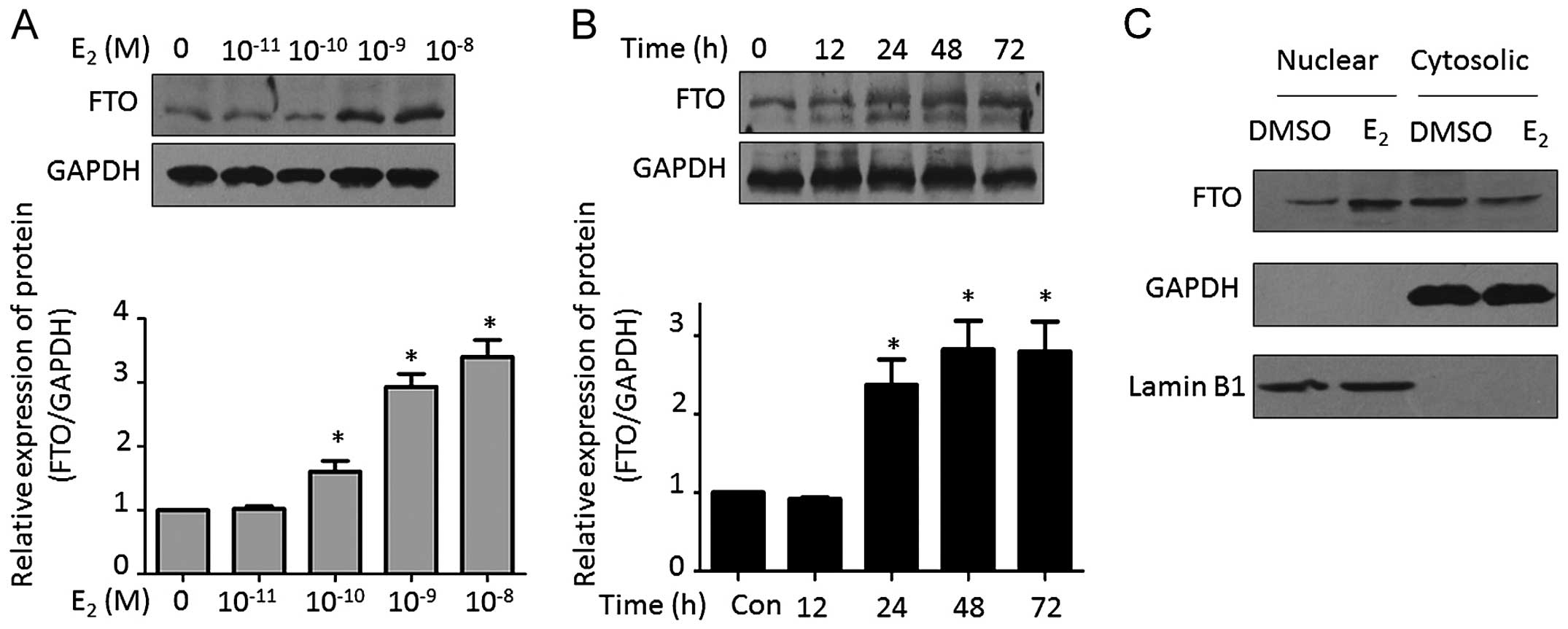
of FTO was observed after estrogen stimulation in endometrial
cancer cells (7). Consistent with
this result, we found that estrogen promoted FTO expression in a
dose-dependent manner. The major stimulating peak was observed with
1–10 nM estrogen treatment (Fig.
2A). Moreover, estrogen also induced FTO expression in a
time-dependent manner. After treatment with 1 nM estrogen for 48 h,
a significant stimulating effect was observed (Fig. 2B). We further isolated the nuclear
and cytosolic proteins. Estrogen-induced FTO was detected, however,
there was no significant alteration of FTO expression in the
cytosol following estrogen treatment (Fig. 2C).
Estrogen controls FTO nuclear
localization through the mTOR signaling pathway
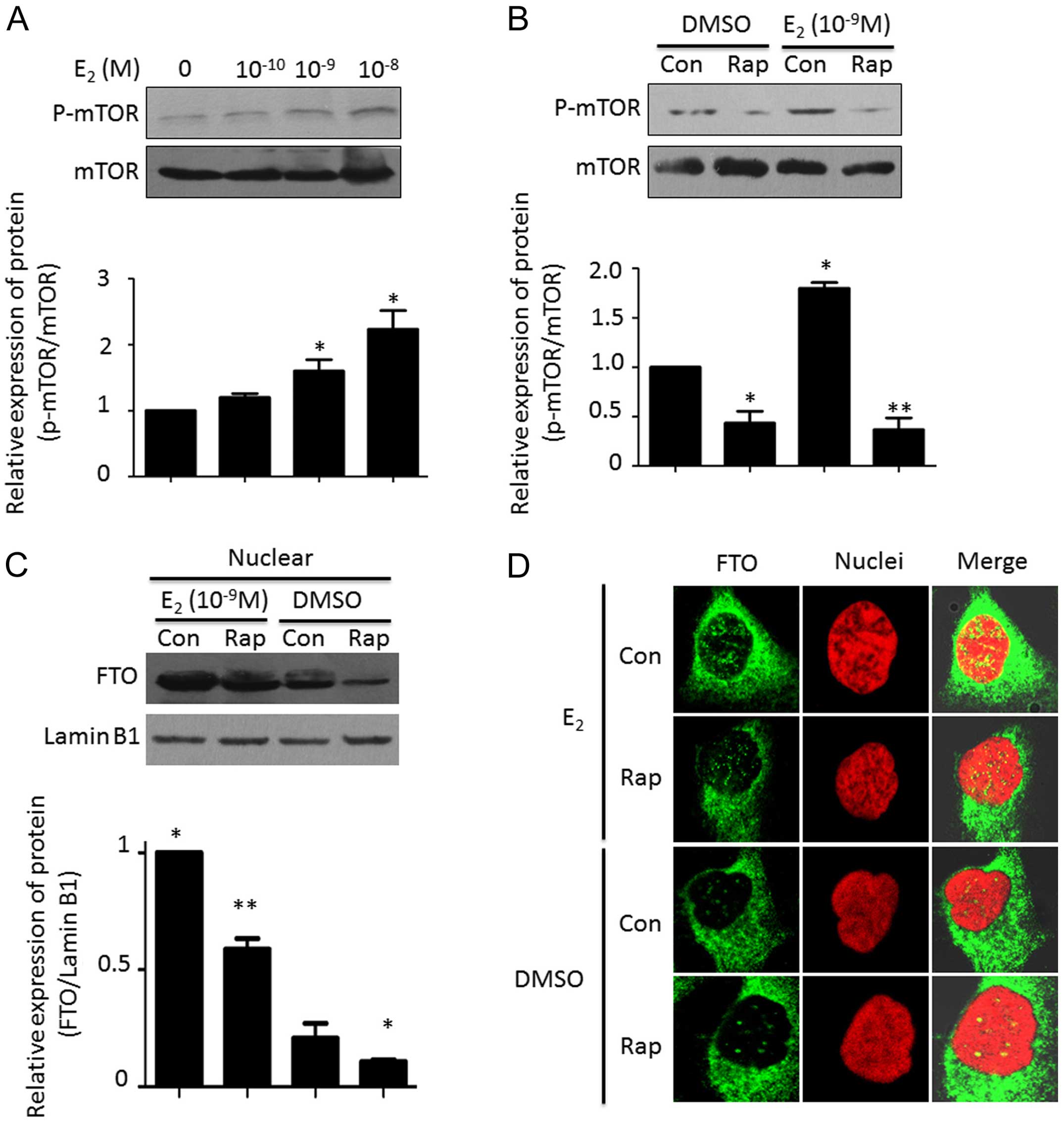
To understand the mechanism of estrogen-induced FTO
nuclear localization, we scanned the signaling pathways which may
be involve in this event. As shown in Fig. 3A, estrogen increased the
phosphorylation of mTOR in a dose-dependent manner. Pretreatment
with rapamycin potently blocked the estrogen-activated mTOR
signaling pathway (Fig. 3B). After
combined treatment with estrogen and rapamycin, we found that
estrogen-induced FTO nuclear localization was attenuated. The FTO
expression pattern in the nucleus following estrogen plus rapamycin
treatment was lower than that with estrogen stimulation alone
(Fig. 3C and D).
ERα is required for estrogen-induced FTO
nuclear localization
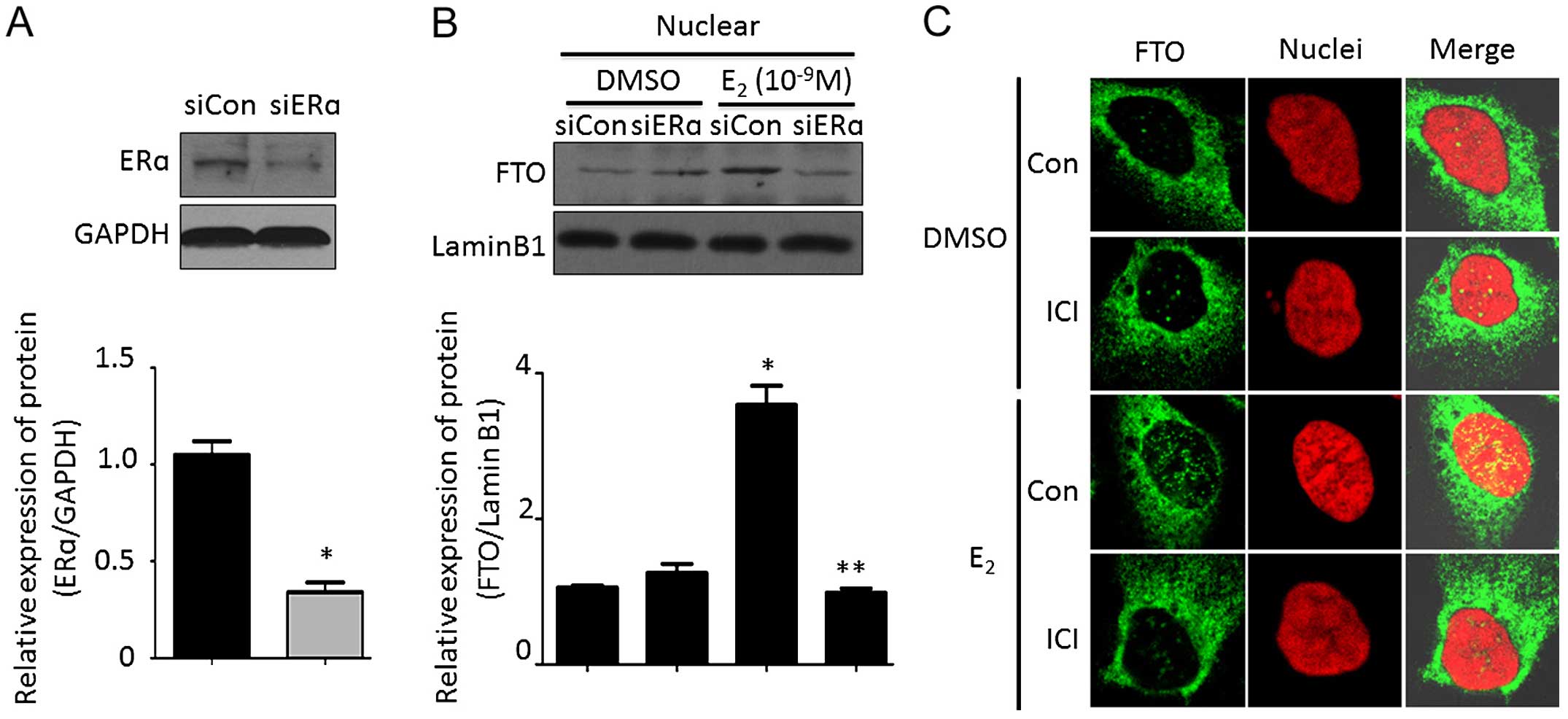
ERα plays an important role in estrogen-mediated
bio-functions. Therefore, we investigated the effect of ERα on
estrogen-induced FTO nuclear localization. As shown in Fig. 4A, acute transfection of siERα
resulted in a marked decrease in ERα protein, which had no effect
on FTO nuclear expression. However, estrogen-induced FTO expression
in the nucleus was blocked by deletion of ERα (Fig. 4B). Immunocytochemistry assay
demonstrated that estrogen stimulation accumulated FTO protein in
the nucleus, however, the increased FTO protein dot in the nucleus
was abolished by 1 µM fulvestrant (ICI 182,780; purchased
from Sigma-Aldrich) treatment, a selective estrogen receptor
antagonist (Fig. 4C). These data
suggested that ERα is required for estrogen-induced FTO nuclear
localization.
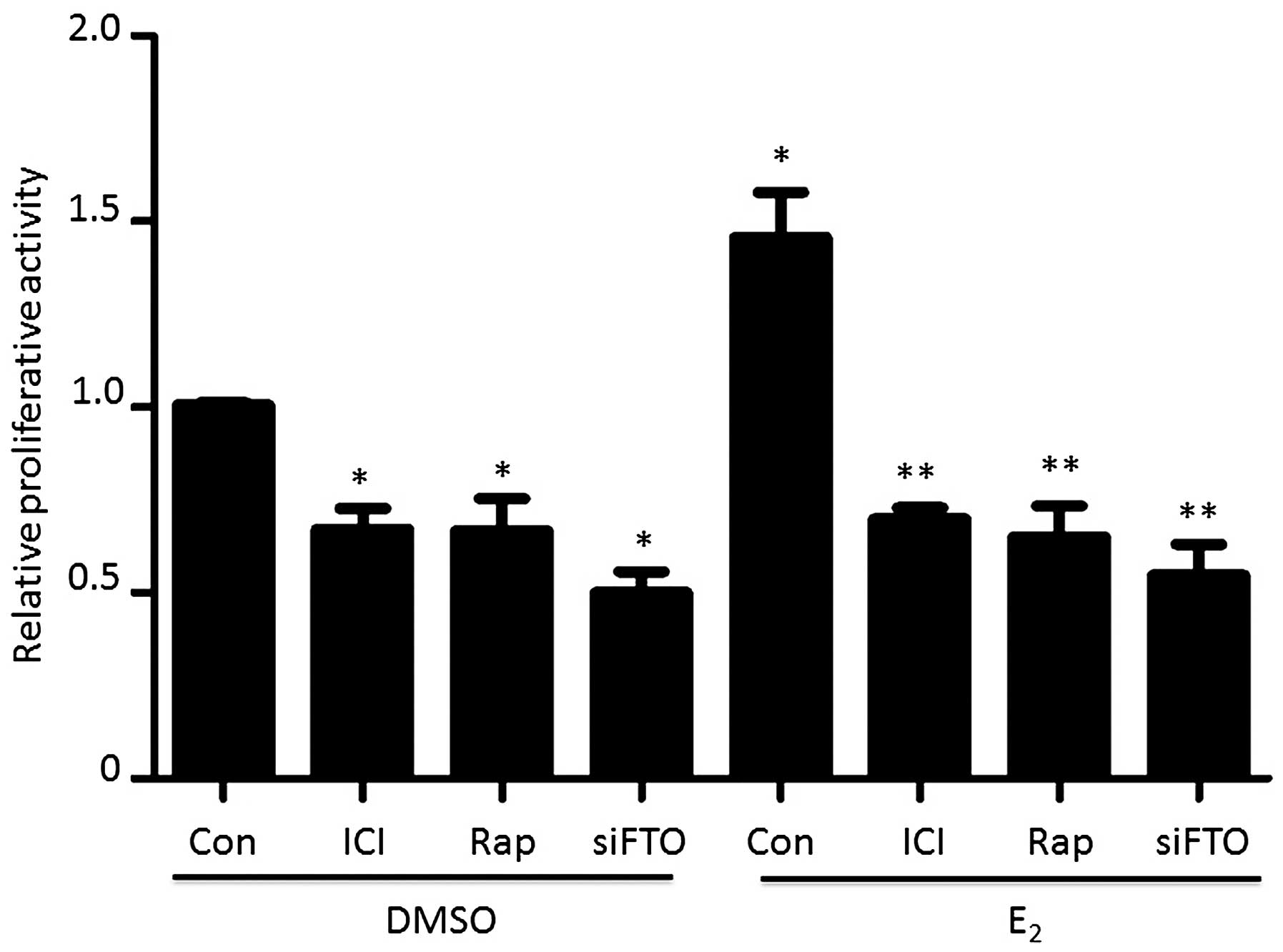
Estrogen-induced FTO nuclear localization
promotes endometrial cancer cell proliferation
To understand FTO nuclear localization in
endometrial cancer proliferation, we carried out an MTT assay. As
shown in Fig. 5, pretreatment with
ICI, to block the ERα signaling pathway, attenuated Ishikawa cell
proliferative activity. Moreover, ICI treatment also blocked
estrogen-induced cell growth. Similarly, blocking the mTOR
signaling pathway with rapamycin also abolished the
estrogen-stimulated cell proliferation. Targeting FTO by siFTO
directly resulted in decreases proliferative activity, whereas
estrogen treatment could not rescue the cellular growth.
Discussion
In the present study, we found that the fat mass and
obesity-associated (FTO) gene was overexpressed in endometrial
carcinoma tissues and estrogen induced FTO nuclear localization,
which facilitated endometrial cancer proliferation through the mTOR
signaling pathway.
Most endometrial adenocarcinomas are characterized
by positive nuclear estrogen receptor (ER) expression and
responsiveness to hormone stimulation. Increasing evidence
indicates that prolonged estrogen exposure is associated with
initiation of type 1 endometrioid cancers (29–31).
Estrogen exposure was found to result in an overall physiological
response within several hours by a genomic mechanism which depends
on estrogen binding to nuclear ER resulting in mRNA transcription
and protein synthesis of target genes. Obesity is a
well-established risk factor for endometrial cancer since obesity
in post-menopausal women has been shown to increase circulating
estrogen levels by upregulating the expression of aromatase and
enhancing aromatization of androstenedione in adipose tissue. We
documented that the FTO gene is involved in estrogen-driven
endometrial cancer development. However, the detail molecular
mechanism remains to be clarified.
An investigation with a larger sample size confirmed
that the obesity-associated polymorphism FTO rs9939609 is strongly
associated with endometrial cancer risk in non-Hispanic white women
(13). In the present study, we
re-evaluated the expression of FTO in endometrial cancer and the
association with prognosis. The higher expression in endometrial
carcinoma tissues was observed compared with the normal endometrial
tissues. In our previous study, we found that there is no
association between FTO expression and age, stage, grade, invasion
and lymph node metastasis (7).
Notably, we found that higher FTO expression was related with poor
prognosis and early relapse. These data imply that FTO plays an
important role in endometrial cancer development. It was observed
that estrogen stimulation enhanced FTO protein accumulation in
endometrial cancer cell nuclei. Yet, limited information is
available concerning its mechanism and its effect on cellular
growth. Consistent with our previous study (7), estrogen upregulated FTO protein in a
dose- and time-dependent manner (Fig.
2A and B). We further isolated the nuclear and cytosolic
proteins and found that in fact estrogen did not increase the FTO
protein level in the cytoplasm, whereas elevated FTO protein by
estrogen in the nucleus was observed. It is known that protein is
synthesized in the cytoplasm, but we did not detect any alteration
in FTO protein in the cytoplasm after estrogen treatment, which
suggests that estrogen induces FTO to transfer into the
nucleaus.
A previous study demonstrated that mTOR signaling is
involved in estrogen-driven endometrial cancer development
(32). In the present study, we
provided evidence that mTOR signaling controls estrogen-induced FTO
protein nuclear localization. As shown in Fig. 3A and B, estrogen activated mTOR
signaling and this activation was inhibited by rapamycin, an mTOR
specific inhibitor. Most importantly, rapamycin blocked the
accumulation of FTO in the nucleus with or without estrogen
stimulation (Fig. 3C and D). These
data suggest that activation of mTOR signaling is necessary for FTO
nuclear localization.
Given that ERα mediated estrogen-induced multiple
functions, we further investigated the role of ERα in FTO nuclear
localization. In the present study, we found that knockdown of ERα
had a slight effect on FTO protein nuclear localization. However,
deletion of ERα blocked estrogen-induced FTO nuclear accumulation.
We considered that ERα has no effect on FTO expression, but
estrogen-mediated function is ERα-dependent. Therefore, depletion
of ERα resulted in the inability of estrogen to bind with ERα, in
turn leading to estrogen-driven FTO nuclear accumulation failure.
These data imply that ERα is required for FTO nuclear localization.
Although we gathered significant data in the present study, to the
best of my knowledge, ERα was also located in the nucleus and
served as a transcription factor. Yet, we still raised the question
of how ERα mediates FTO nuclear accumulation. We hypothesized that
ERα may recruit some molecules and construct a protein complex,
which in turn helps FTO to transfer into the nucleus. Various
proteins have different functions when they present in different
cellular localizations, such as caveolin. In the present study, we
found that FTO nuclear localization promoted cell proliferation,
whereas blocking the nuclear accumulation by ICI or Rap
pretreatment, even by direct deletion of FTO by siRNA, resulted in
attenuated proliferative activity. The decreased proliferation
could not be rescued by estrogen stimulation.
In conclusion, the present study suggests that
overexpression of FTO in endometrial carcinoma may be a poor
prognostic marker. Importantly, FTO nuclear accumulation may be an
essential step for estrogen-driven endometrial tumor formation and
progression. Our findings may provide new insight into the
mechanisms underlying E2-induced proliferation. Additionally, the
present study further supports the possibility of using FTO as a
target for the treatment of endometrial cancer.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (NSFC no.
81372795).
References
|
1
|
Chan S: A review of selective estrogen
receptor modulators in the treatment of breast and endometrial
cancer. Semin Oncol. 29(Suppl 11): 129–133. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ito K, Utsunomiya H, Yaegashi N and Sasano
H: Biological roles of estrogen and progesterone in human
endometrial carcinoma - new developments in potential endocrine
therapy for endometrial cancer. Endocr J. 54:667–679. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bokhman JV: Two pathogenetic types of
endometrial carcinoma. Gynecol Oncol. 15:10–17. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bochkareva NV, Kolomiets LA, Kondakova IV,
Stukanov SL, Starova AB and Agarkova LA: Enzymes of estrogen
metabolism in endometrial cancer. Bull Exp Biol Med. 141:240–242.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen Y, Huang Q, Chen Q, Lin Y, Sun X,
Zhang H, Zhu M and Dong S: The inflammation and estrogen metabolism
impacts of polychlorinated biphenyls on endometrial cancer cells.
Toxicol In Vitro. 29:308–313. 2015. View Article : Google Scholar
|
|
6
|
Williams-Brown MY, Salih SM, Xu X,
Veenstra TD, Saeed M, Theiler SK, Diaz-Arrastia CR and Salama SA:
The effect of tamoxifen and raloxifene on estrogen metabolism and
endometrial cancer risk. J Steroid Biochem Mol Biol. 126:78–86.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang Z, Zhou D, Lai Y, Liu Y, Tao X, Wang
Q, Zhao G, Gu H, Liao H, Zhu Y, et al: Estrogen induces endometrial
cancer cell proliferation and invasion by regulating the fat mass
and obesity-associated gene via PI3K/AKT and MAPK signaling
pathways. Cancer Lett. 319:89–97. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
da Cunha PA, de Carlos Back LK, Sereia AF,
Kubelka C, Ribeiro MC, Fernandes BL and de Souza IR: Interaction
between obesity-related genes, FTO and MC4R, associated to an
increase of breast cancer risk. Mol Biol Rep. 40:6657–6664. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gaudet MM, Yang HP, Bosquet JG, Healey CS,
Ahmed S, Dunning AM, Easton DF, Spurdle AB, Ferguson K, O'Mara T,
et al: No association between FTO or HHEX and endometrial cancer
risk. Cancer Epidemiol Biomarkers Prev. 19:2106–2109. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hernández-Caballero ME and Sierra-Ramírez
JA: Single nucleotide polymorphisms of the FTO gene and cancer
risk: An overview. Mol Biol Rep. 42:699–704. 2015. View Article : Google Scholar
|
|
11
|
Kaklamani V, Yi N, Sadim M, Siziopikou K,
Zhang K, Xu Y, Tofilon S, Agarwal S, Pasche B and Mantzoros C: The
role of the fat mass and obesity associated gene (FTO) in breast
cancer risk. BMC Med Genet. 12:522011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lewis SJ, Murad A, Chen L, Davey Smith G,
Donovan J, Palmer T, Hamdy F, Neal D, Lane JA, Davis M, et al:
Associations between an obesity related genetic variant (FTO
rs9939609) and prostate cancer risk. PLoS One. 5:e134852010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lurie G, Gaudet MM, Spurdle AB, Carney ME,
Wilkens LR, Yang HP, Weiss NS, Webb PM, Thompson PJ, Terada K;
Australian National Endometrial Cancer Study Group; et al:
Epidemiology of Endometrial Cancer Consortium (E2C2): The
obesity-associated polymorphisms FTO rs9939609 and MC4R rs17782313
and endometrial cancer risk in non-Hispanic white women. PLoS One.
6:e167562011. View Article : Google Scholar
|
|
14
|
Fischer J, Koch L, Emmerling C, Vierkotten
J, Peters T, Brüning JC and Rüther U: Inactivation of the Fto gene
protects from obesity. Nature. 458:894–898. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wong M: A critical review of mTOR
inhibitors and epilepsy: From basic science to clinical trials.
Expert Rev Neurother. 13:657–669. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu J and Tian D: Hematologic toxicities
associated with mTOR inhibitors temsirolimus and everolimus in
cancer patients: A systematic review and meta-analysis. Curr Med
Res Opin. 30:67–74. 2014. View Article : Google Scholar
|
|
17
|
Atefi M, von Euw E, Attar N, Ng C, Chu C,
Guo D, Nazarian R, Chmielowski B, Glaspy JA, Comin-Anduix B, et al:
Reversing melanoma cross-resistance to BRAF and MEK inhibitors by
co-targeting the AKT/mTOR pathway. PLoS One. 6:e289732011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jazirehi AR, Wenn PB and Damavand M:
Therapeutic implications of targeting the PI3Kinase/AKT/mTOR
signaling module in melanoma therapy. Am J Cancer Res. 2:178–191.
2012.PubMed/NCBI
|
|
19
|
Pópulo H, Soares P and Lopes JM: Insights
into melanoma: Targeting the mTOR pathway for therapeutics. Expert
Opin Ther Targets. 16:689–705. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bae SY, Kim GD, Jeon JE, Shin J and Lee
SK: Anti-proliferative effect of (19Z)-halichondramide, a novel
marine macrolide isolated from the sponge Chondrosia corticata, is
associated with G2/M cell cycle arrest and suppression of mTOR
signaling in human lung cancer cells. Toxicol In Vitro. 27:694–699.
2013. View Article : Google Scholar
|
|
21
|
Dong LX, Sun LL, Zhang X, Pan L, Lian LJ,
Chen Z and Zhong DS: Negative regulation of mTOR activity by
LKB1-AMPK signaling in non-small cell lung cancer cells. Acta
Pharmacol Sin. 34:314–318. 2013. View Article : Google Scholar
|
|
22
|
Papadimitrakopoulou V: Development of
PI3K/AKT/mTOR pathway inhibitors and their application in
personalized therapy for non-small-cell lung cancer. J Thorac
Oncol. 7:1315–1326. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bradford LS, Rauh-Hain A, Clark RM,
Groeneweg JW, Zhang L, Borger D, Zukerberg LR, Growdon WB, Foster R
and Rueda BR: Assessing the efficacy of targeting the
phosphatidylinositol 3-kinase/AKT/mTOR signaling pathway in
endometrial cancer. Gynecol Oncol. 133:346–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen J, Zhao KN, Li R, Shao R and Chen C:
Activation of PI3K/Akt/mTOR pathway and dual inhibitors of PI3K and
mTOR in endometrial cancer. Curr Med Chem. 21:3070–3080. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cheng H, Liu P, Zhang F, Xu E, Symonds L,
Ohlson CE, Bronson RT, Maira SM, Di Tomaso E, Li J, et al: A
genetic mouse model of invasive endometrial cancer driven by
concurrent loss of Pten and Lkb1 Is highly responsive to mTOR
inhibition. Cancer Res. 74:15–23. 2014. View Article : Google Scholar :
|
|
26
|
Myers AP: New strategies in endometrial
cancer: Targeting the PI3K/mTOR pathway - the devil is in the
details. Clin Cancer Res. 19:5264–5274. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pavlidou A and Vlahos NF: Molecular
alterations of PI3K/Akt/mTOR pathway: A therapeutic target in
endometrial cancer. ScientificWorldJournal. 2014:7097362014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang Z, Dong L, Sui L, Yang Y, Liu X, Yu
Y, Zhu Y and Feng Y: Metformin reverses progestin resistance in
endometrial cancer cells by downregulating GloI expression. Int J
Gynecol Cancer. 21:213–221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chao A, Lin CY, Tsai CL, Hsueh S, Lin YY,
Lin CT, Chou HH, Wang TH, Lai CH and Wang HS: Estrogen stimulates
the proliferation of human endometrial cancer cells by stabilizing
nucleophosmin/B23 (NPM/B23). J Mol Med Berl. 91:249–259. 2013.
View Article : Google Scholar
|
|
30
|
Thorne AM, Jackson TA, Willis VC and
Bradford AP: Protein kinase Cα modulates
estrogen-receptor-dependent transcription and proliferation in
endometrial cancer cells. Obstet Gynecol Int. 2013:5374792013.
View Article : Google Scholar
|
|
31
|
Zhou C, Steplowski TA, Dickens HK, Malloy
KM, Gehrig PA, Boggess JF and Bae-Jump VL: Estrogen induction of
telomerase activity through regulation of the mitogen-activated
protein kinase (MAPK) dependent pathway in human endometrial cancer
cells. PLoS One. 8:e557302013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hou X, Zhao M, Wang T and Zhang G:
Upregulation of estrogen receptor mediates migration, invasion and
proliferation of endometrial carcinoma cells by regulating the
PI3K/AKT/mTOR pathway. Oncol Rep. 31:1175–1182. 2014.
|