Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignant tumors worldwide, especially in Asian countries.
It accounts for ~70% of liver cancers and may originate from both
liver cirrhosis and non-cirrhotic liver, indicating the existence
of different hepatocarcinogenesis pathways (1–3).
Although the precise mechanisms may be different according to the
different risk factors, the multi-step process of
hepatocarcinogenesis may be divided into several phases: chronic
liver injury, cell death, cirrhosis and regeneration, DNA damage,
dysplasia, and finally HCC (4–6).
Through surgical resection and liver transplantation, HCC is
potentially curable at early stage. Unfortunately, most of the HCC
patients at diagnosis are in the advanced-stage with serious
background liver disease which is not suitable for such therapy
(7). Moreover, HCC is highly
resistant to traditional medical treatment such as radiation and
chemotherapy (8). Therefore, the
need for the development of effective prevention, and therapy of
the disease is urgent.
G protein-coupled receptor kinase 2 (GRK2) is a
serine/threonine protein kinase that is ubiquitously expressed. It
was originally found to desensitize G protein coupled receptor
(GPCR) signaling by phosphorylating the agonist-occupied receptor
(9,10). β-arrestin then binds to the
phosphorylated receptor to form a protein complex which prevents
the coupling of the receptor from its G-protein, leading to
signaling attenuation (11).
Despite of its conventional role as a kinase in receptor
desensitization, an increasing number of evidence shows that GRK2
can interact with other transmembrane receptors to participate in
various cellular processes, such as receptor tyrosine kinases
(RTKs) (12,13). GRK2 translocates to the
ligand-activated PDGFR (platelet-derived growth factor receptor)
and EGFR (epidermal growth factor receptor), and phosphorylates
their intracellular domains. Although the relationship between GRK2
and some human diseases including heart failure, hypertension,
rheumatoid arthritis has been intensely characterized (14,15),
its role in cancer, particularly in the proliferation of HCC,
remains largely unknown.
EGR1, known as early growth response 1, is a member
of the immediate-early gene family. It can be induced by growth
factors, cytokines and stress signals such as radiation, damage
(16). MAPK families, known as
ERK1/2, JNK and p38 MAPK, are most commonly involved in EGR1
activation (17). EGR1 is also a
member of zinc-finger transcription factors with an amino terminal
activation domain near the carboxyl terminal end of the protein
sequence (18,19), so it can bind to the promoter of the
target gene to regulate the gene expression and be involved in many
biological functions, including wound repair, cell growth,
migration and invasion (20).
We selected two HCC cell lines, HepG2, with a high
expression of GRK2, and HCCLM3, with a low expression of GRK2. To
directly study the effect of GRK2 on EGR1 expression, HepG2 cell
line with a functional GRK2 siRNA expression was created, and
remarkable elevation in EGR1 expression was observed in the
presence of IGF1. HCCLM3 with GRK2 overexpression was created, and
remarkable decrease in EGR1 expression was observed in the presence
of IGF1. Furthermore, we found that GRK2 negatively regulates
IGF1-induced activation of IGF1R signal pathway though binding to
IGF1R. This regulation of EGR1 by GRK2 may be essential for HCC
cell growth as evidenced by the in vitro cell growth and
migration studies. Thus, our data suggest, for the first time, that
the regulation of EGR1 by GRK2 is critical for HCC growth and
enforced GRK2 may offer a potential therapeutic approach against
HCC.
Materials and methods
Reagent and antibodies
All chemicals and HRP-conjugated anti-rabbit, mouse,
goat IgG were purchased from Sigma-Aldrich. Antibodies against
EGR1, phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), p44/42 MAPK
(Erk1/2), phospho-Akt (Ser473), Akt, phospho-IGF1R β (Tyr1135),
IGF1R β, and human recombinant IGF-1 were from Cell Signaling
Technology; antibodies against GRK2, actin and protein A/G
plus-agarose were purchased from Santa Cruz Biotechnology.
Cell culture and transfection
Human HCC cell lines, HepG2, SMCC7721, HCCLM3 were
maintained in DMEM (Hyclone) supplemented with 10% FBS (Hyclone)
and 2 nmol/l L-glutamine and penicillin-streptomycin. Cells were
cultured in an incubator with humidified air at 37°C with 5%
CO2. Lipofectamine-3000 (Invitrogen) was employed for
transfection according to the manufacturer's instruction. The
stably transfected cell lines were obtained after being selected by
800 µg/ml G418 for 3–4 weeks. Double-strand siRNAs targeting
GRK2, EGR1 and scrambled control was synthesized using the
following sequences, siRNA-GRK2; forward, 5′-GCA UCA UGC AUG GCU
ACA UdTdT, reverse, 5′-AUG UAG CCA UGC AUG AUG CdTdT; siRNA-EGR1;
forward, 5′-CCU GGA GCC UGC ACC CAA CdTdT, reverse, 5′-GUU GGG UGC
AGG CTC CAG GdTdT, siRNA-scramble: forward 5′-AUG AAC GUG AAU UGC
UCA AdTdT, reverse 5′-UUG AGC AAU UCA CGU UCA UdTdT. SiRNA duplexes
were purchased from GenePharma.
RT-PCR and real-time PCR
Total RNA was isolated by TRIzol (Invitrogen)
according to the manufacturer's instructions, and 1 µg RNA
was converted to cDNA using the high capacity cDNA reverse
transcription kits (Invitrogen). The following primer sets were
used for RT-PCR and real-time PCR: ACTIN-Fw: 5′-GAC CTG ACT GAC TAC
CTC ATG AAG AT-3′, ACTIN-Re: 5′-GTC ACA CTT CAT GAT GGA GTT GAA
GG-3′; EGR1-Fw: 5′-CTG ACC GCA GAG TCT TTT CCT G-3′, EGR1-Re:
5′-TGG GTG CCG CTG AGT AAA TG-3′; GRK2-Fw: 5′-GTT GCT GCA GAG GGA
TGT CAA CCG-3′, GRK2-Re: 5′-GTC AGG AAG GGG TTG CCC ATC TTG
G-3′.
Immunoprecipitation
Cells were washed with ice-cold PBS and lysed in 800
µl NP-40 solubilization buffer for 0.5 h. The lysate was
centrifuged and the supernatant was incubated with 1 µg of
anti-IGF1R antibody and 15 µl of 50% slurry of protein A/G
plus-agarose beads at 4°C overnight. The beads were subsequently
washed, and the proteins bound to the beads were separated by
SDS-PAGE. The samples were detected in the subsequent western blot
with anti-GRK2 and anti-IGF1R antibody.
Western blot analysis
Cells were harvested and lysed in the RIPA lysis
buffer to prepare the whole cell extract. Protein concentration was
determined using Bradford assay (Bio-Rad, Hercules, CA, USA).
Lysates were subjected to SDS-PAGE and immunoblot analysis.
Horseradish peroxidase-conjugated secondary antibodies and enhanced
chemiluminescence detection (ImageQuant) was used to detect
specific immunoreactive proteins.
Cell proliferation assays
The proliferation of HCC cells were examined using
Cell Counting Kit 8 (Dojindo) according to the manufacturer's
instructions. Equal numbers of cells in a volume of 100 µl
were seeded in a 96-well plates. The cells were transduced with
GRK2 expression plasmid, GRK2 siRNA duplex and their corresponding
controls with and without IGF1 stimulation. The plates were
incubated for 3 days. Cell proliferation was determined every 24 h.
For each measurement, 10 µl CCK was added into each well and
incubated at 37°C with 5% CO2 for 1 h. The absorbance of
the plate was taken at 450 nm in an ELISA plate reader. All assays
were done in triplicate.
Tumor colony formation assay
After the stable transfected HCCLM3/pc-GRK2 and its
control cells were transfected with EGR1 siRNA or scrambled siRNA
for 48 h, 5000 cells were suspended in 1.5 ml DMEM with 0.35% agar,
50 ng/ml IGF1 and 5% fetal bovine serum and seeded in 6-well plates
which were pre-coated with the same medium except containing 0.5%
agar. The cells were allowed to form colonies for 2 weeks. The
colonies were stained with 0.1% crystal violet for 10 min at 37°C
and counted.
Wound healing assay
HepG2 and HCCLM3 cells transfected with
corresponding plasmids or siRNA were seeded into 24-well plates, at
80–90% confluency, the cell monolayer was disrupted with a cell
scraper. After washing with PBS three times to remove cell debris,
the remaining cells were treated with 50 ng/ml IGF1 and then images
were captured by microscope at 0 and 24 h after treatment. Cell
motility was evaluated according to the following formula: Cell
motility = (distance 24 h - distance 0 h)/distance 0 h.
Statistical analysis
All experiments were performed in triplicates. The
data represent the mean ± SD. Statistical analysis was performed
with SPSS 17.0 software. Student's T-test was used to analyze the
significance between two groups. Statistical significance was set
at P<0.05, P<0.01.
Results
Regulation of IGF1-induced human HCC cell
proliferation and migration by GRK2
A recent study indicates that GRK2 has an inhibitory
role in regulating IGF1-stimulated signaling pathway in HEK293T
cells (21). This observation
promoted us to investigate whether GRK2 is critical for
IGF1-induced human HCC cell proliferation and migration. To gain a
profile of basal GRK2 levels in HCC cells, we examined GRK2
expression in three HCC cell lines by western blot and RT-PCR
analysis, with the highest level in the HepG2 cell line and the
lowest level in the HCCLM3 cell line (Fig. 1A and B). We thus selected HepG2 for
GRK2 knockdown experiments (HepG2/si-GRK2) and HCCLM3 cell lines
for GRK2 overexpression (HCCLM3/pc-GRK2) experiments. As shown in
Fig. 1C, E and G, suppression of
GRK2 in HepG2 led to an increase in IGF1-induced cell proliferation
and migration compared with control cells. While in HCCLM3, as
shown in Fig. 1D, F and H,
overexpression of GRK2 led to a decrease in IGF1-induced cell
proliferation and migration. These results indicate that GRK2 can
negatively regulate IGF1-induced tumor cell growth. However, GRK2
alone can not affect HCC cell growth without IGF1 stimulation
(Fig. 1C–H).
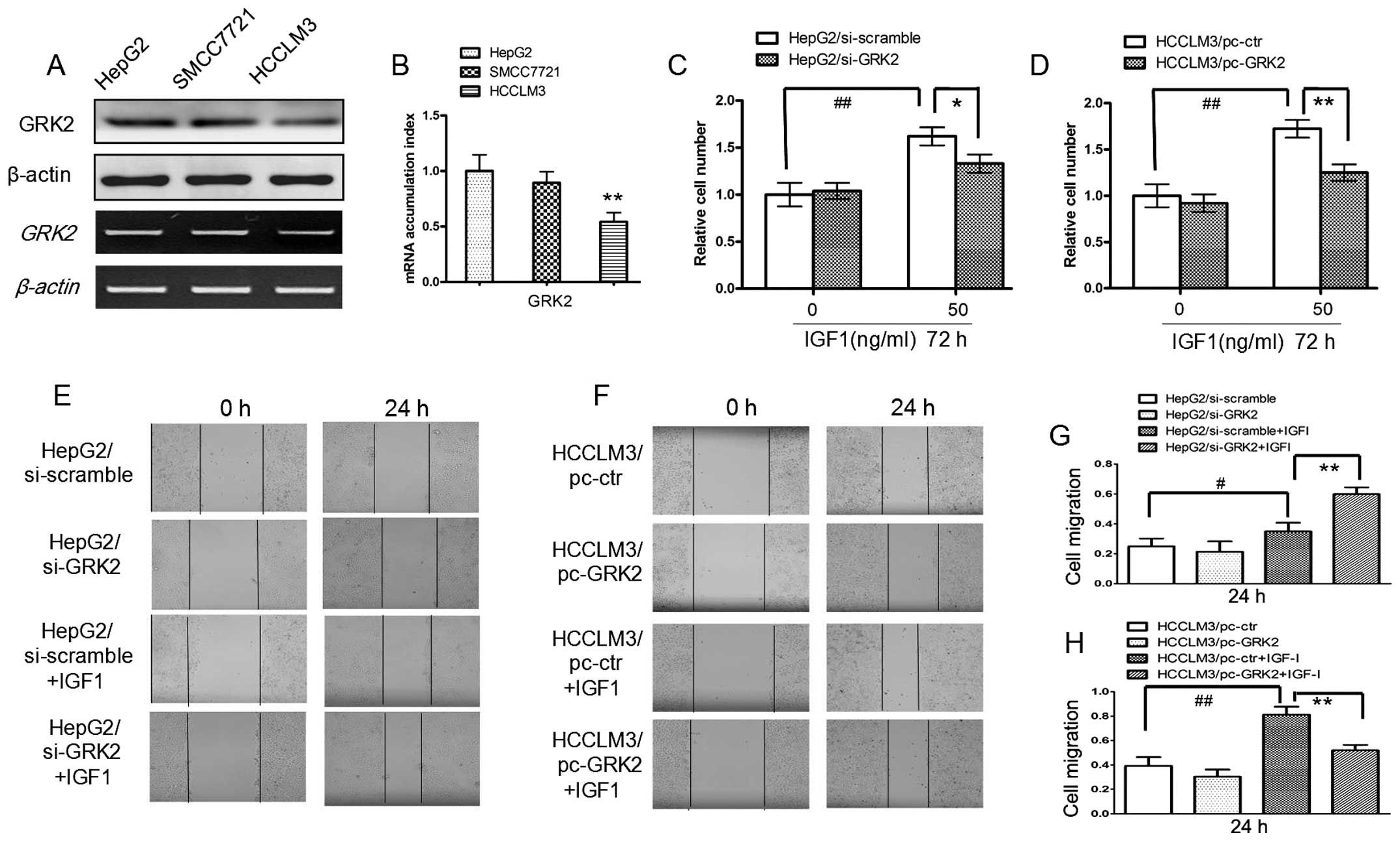 | Figure 1The regulation of IGF1-induced human
HCC cell proliferation and migration by GRK2. (A and B) GRK2 levels
were evaluated on three HCC cell lines, HepG2, SMCC7721, HCCLM3 by
western blotting, RT-PCR and real-time PCR (**P<0.01
compared with HepG2). (C and D) Growth of HepG2/si-scramble,
HepG2/si-GRK2 and HCCLM3/pc-ctr, HCCLM3/pc-GRK2 cells treated with
50 ng/ml IGF1 or the vehicle (0.1% DMSO) for 72 h. (E-H)
Wound-healing analysis of HCC cell migration for 0 and 24 h, images
and quantitative analysis of HepG2/si-scramble, HepG2/si-GRK2 and
HCCLM3/pc-ctr, HCCLM3/pc-GRK2 cells treated with 50 ng/ml IGF1 or
the vehicle at 0 and 24 h; (##P<0.01,
#P<0.05 compared with ctr group,
**P<0.01, *P<0.05 compared with
ctr+IGF1 group). |
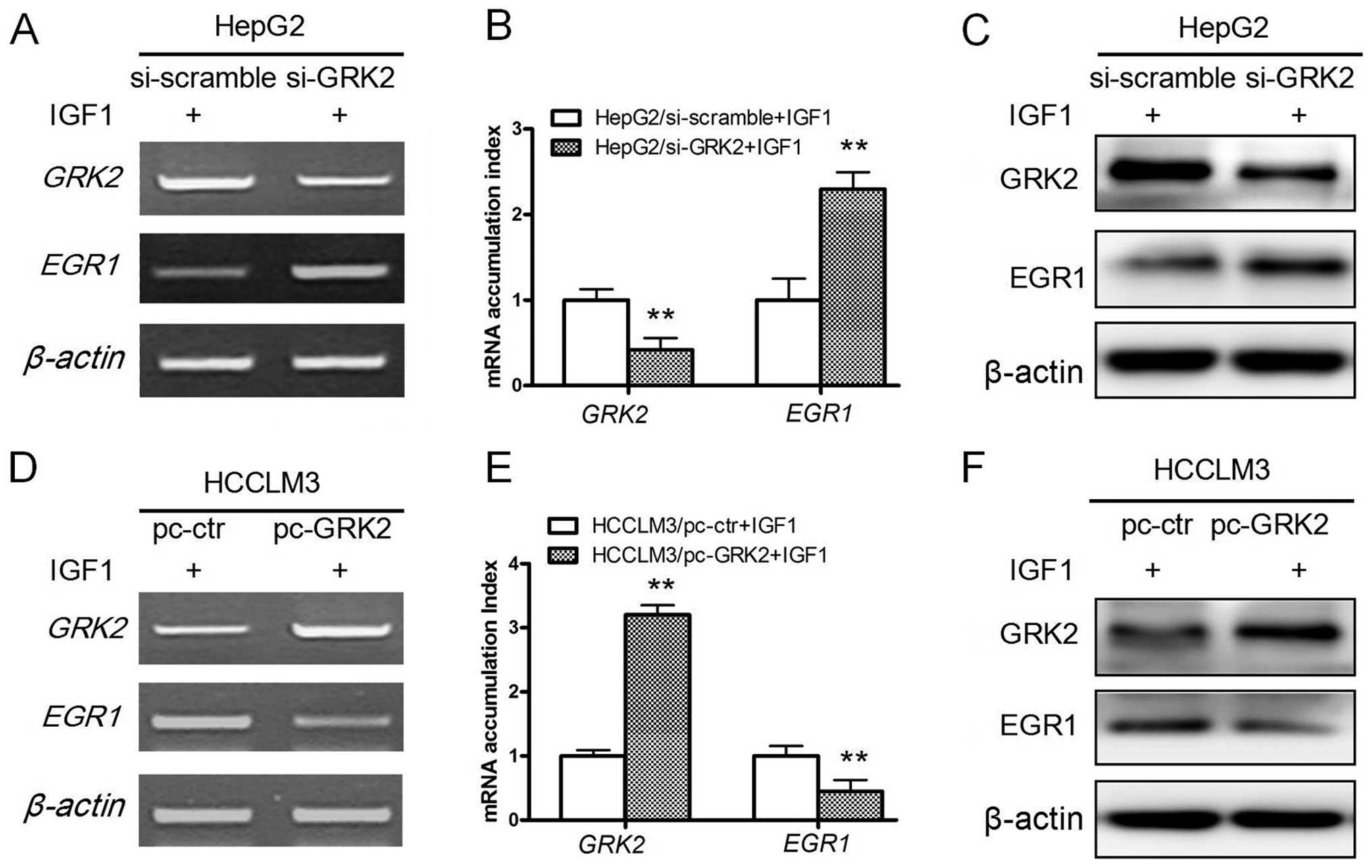
GRK2 induces downregulation of EGR1
expression in the IGF1 treatment
To determine the mechanisms of proliferation
inhibition role of GRK2, we next investigated GRK2 regulated genes
in HCC cells. From unpublished microarray results, we found that
EGR1 expression can be inhibited by GRK2 over-expression in the
presence of IGF1. As shown in Fig.
2A–C, significant increases of EGR1 levels were observed in
GRK2 siRNA cells compared with the control cell as evidenced by
RT-PCR, real-time PCR and western blot experiments. In contrast to
GRK2 downregulation, overexpression of GRK2 in HCCLM3 led to a
decrease in EGR1 protein and mRNA levels (Fig. 2D–F). These results revealed that
GRK2 induced downregulation of the EGR1 expression in the IGF1
treatment of HCC cell lines.
GRK2 negatively regulates the
IGF1-induced IGF1R signaling pathway through interacting with
IGF1R
Previous studies indicated that activated IGF-1R can
stimulate the downstream Erk signaling to ultimately transactivate
the EGR1 transcription factor (22,25).
In order to explore the mechanisms by which GRK2 regulates the EGR1
expression, we investigated the influence of ectopic expression of
GRK2 on IGF1 signaling pathways, and analyzed the IGF1R, ERK and
AKT activation in HCC cells expressing high levels of GRK2, or a
siRNA directed against GRK2. As shown in Fig. 3A and B, silencing of GRK2 led to an
increase in IGF1-induced activation of IGF1R, ERK and AKT in HepG2
cells. Conversely, in Fig. 3C and
D, overexpression of GRK2 caused a decrease in IGF1-induced
IGF1R, ERK and AKT activation in HCCLM3 cells.
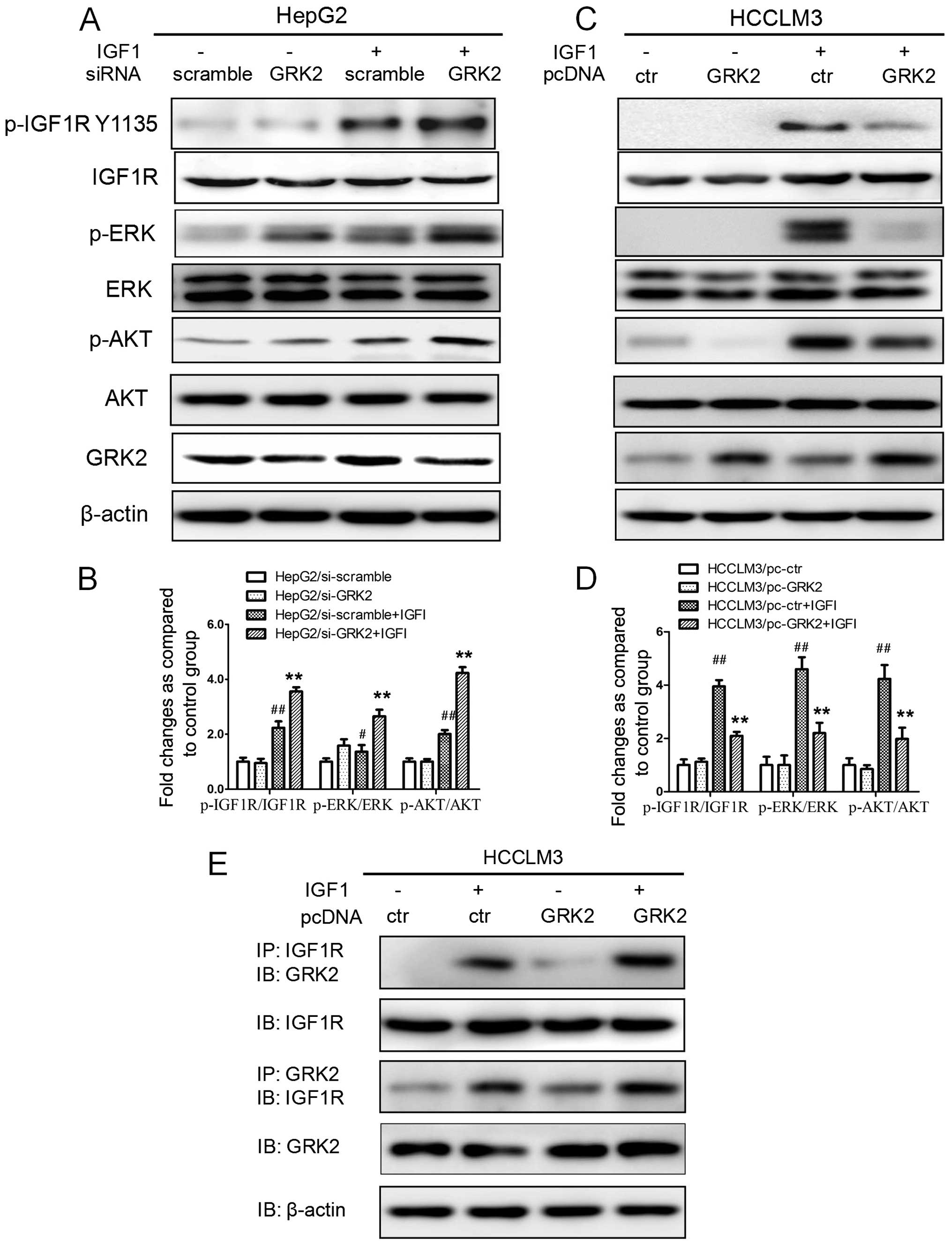 | Figure 3GRK2 negatively regulates the
IGF1-induced IGF1R signaling pathway through interacting with
IGF1R. (A and B) Whole cell lysates from HepG2/si-GRK2,
HCCLM3/pc-GRK2 cells and the control cells which were treated with
50 ng/ml IGF1 or the vehicle were subjected to SDS-PAGE analyses
for the expression of GRK2, IGF1R, ERK and AKT. Activation of
IGF1R, Erk and AKT was examined by immunoblot assays using
anti-phosphorylated antibody. (C and D) The histogram corresponding
to the quantitative analysis of IGF1R, Erk and AKT activation are
shown. The band intensities of phosphorylation of IGF1R, Erk and
AKT were normalized to that of the corresponding
non-phosphorylation. The data from three independent experiments
are shown as the mean ± SD. Densitometry values in the histograms
were expressed as fold-change relative to the normal control,
##P<0.01 compared with normal control group,
**P<0.01 compared with control plus IGF1 group. (E)
HCCLM3 cells were either transfected with control or GRK2
expressing plasmid, serum starved for 12 h, and stimulated or not
with IGF1 (50 ng/ml) for 10 min. The cell lysates were
immunoprecipitated with anti-IGF1R or GRK2 antibody, followed by
immunoblotting for GRK2 or IGF1R. β-actin levels in the total
protein lysates before IP were used as loading controls. |
GRK2 can desensitize GPCR signaling and some
tyrosine kinase receptors by binding to the agonist-occupied
receptor (9,10). Then we analyzed the relationship
between GRK2 and IGF1R in an HCC cell line. The association of GRK2
and IGF1R was observed in the IGF1 treatment of HCCLM3 cells, and
after GRK2 overexpression, this association was increased in the
stimulated conditions (Fig. 3E),
suggesting that GRK2 overexpression increased the formation of
GRK2-IGF1R complex in a ligand-dependent manner. These results
demonstrated that overexpression of the GRK2 suppressed IGF1R
signaling pathway through interacting with IGF1R, and this
regulation may lead to decreased EGR1 expression.
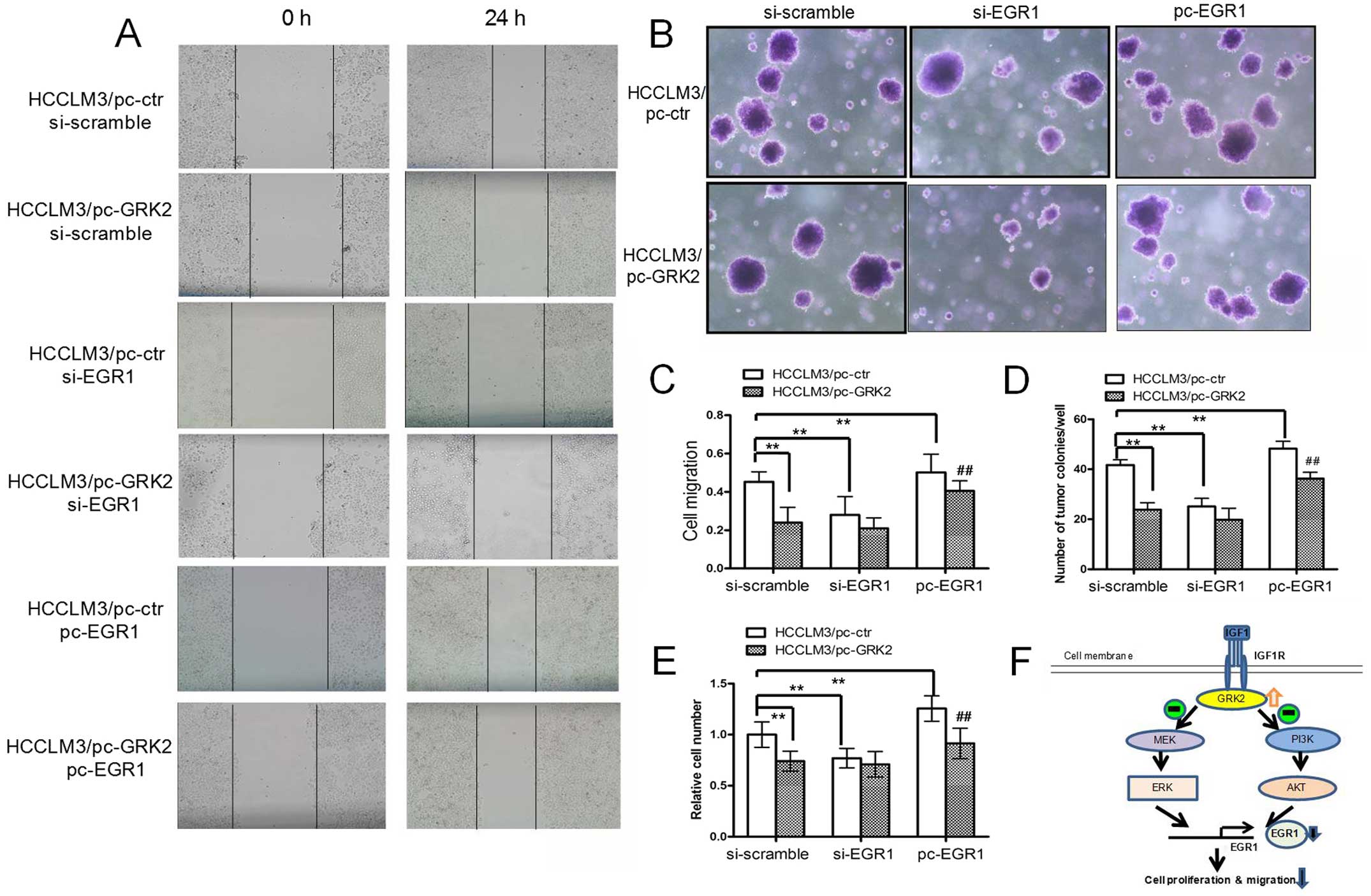
Anti-proliferative and migratory effect
by overexpression of GRK2 is EGR1-mediated
Since GRK2 has been involved in HCC cell
proliferation and migration, we investigated whether EGR1 is
critical for GRK2-regulation of these functions. Stably transfected
HCCLM3/pc-GRK2 and HCCLM3/pc-ctr cells were transiently transfected
with either siRNA-EGR1 or EGR1 expression plasmid, the cell scratch
migration (Fig. 4A and C), tumor
colony-forming (Fig. 4B and D) and
proliferation assays (Fig. 4E) were
performed with the cells in the presence of IGF1. Overexpression of
GRK2 or suppression of EGR1 in HCCLM3/pc-ctr cells led to
significant decrease in cell proliferation, colony growth and
migration compared to control cells. Of note, treatment with EGR1
siRNA minimized the difference in cell growth and migration between
HCCLM3/pc-GRK2 cells and control cells, while overexpression of
EGR1 restored the anti-proliferative and migratory effect by GRK2
overexpression in HCCLM3 cells. These results indicated that GRK2
inhibits IGF1-induced HCC cell growth and migration through
downregulating EGR1.
Discussion
GRK2 is known to have antiproliferative, antitumor
activities in some tumors. Enforced overexpression of GRK2
inhibited cell proliferation in human thyroid cancer cells and in
human smooth muscle cells (23,24);
however, the mechanisms by which the proliferation activity was
mediated by GRK2 is not clearly understood. In this study, we
illustrated a novel mechanism that enforced GRK2 blocks HCC cell
proliferation and migration induced by IGF1, including repressing
EGR1 expression and blocking the IGF1R signaling pathway by
interacting with IGF1R. This is the first report of GRK2′s
involvement in the regulation of EGR1 expression.
In this study, GRK2 overexpression alone was not
able to suppress the proliferation of HCCLM3 cells in the absence
of IGF1, which seems to contradict the published data that enforced
GRK2 by adenovirus transduction inhibited HCC cell growth (24). One explanation may be the
differences of cell types, Mahlavu and Huh7 cells were chosen in
the previous study, whereas HCCLM3 and HepG2 were used in the
current study. Another possible explanation is that the time
lengths in the proliferation assay are different, in the previous
study the test time was four days, and GRK2 inhibited the growth of
Mahlavu and Huh7 cells on the third and fourth day, but there is no
difference of cell growth in the first two days. In the current
study, we tested three days of cell proliferation, and did not
detect the difference during this period.
While EGR1 is crucial for the proliferation of
hepatocytes and plays an important role in liver regeneration
(26), the expression level of EGR1
and its impact in HCC are controversial. One study found that EGR1
is overexpressed in HCC (27),
while another study found downregulation of EGR1 expression
(28). Also studies have indicated
that increased HGF/c-Met signaling could lead to growth-promoting
functions of EGR1 in HCC (29,30).
In our study, the growth-promoting effect of EGR1 may arise due to
increased MEK/ERK and PI3K/AKT signaling by IGF1. Many studies show
that transcription factor EGR1 is a key regulator in mediating
HGF-induced genes that encode regulators of cell proliferation,
migration, invasion, and metastasis (29–31).
Taken together, these observations presented in this
study indicated that GRK2 plays regulatory functions in HCC cell
growth. They also suggested that enforced GRK2 may offer a
potential therapeutic approach in the treatment of HCC.
Acknowledgments
This study was supported by National Natural Science
Foundation of China (no. 81502123; 81330081; 81202596), Natural
Science Foundation of Anhui Province (no. 1308085QH130), Anhui
Province Nature Science Foundation in University (no. KJ2014A119),
Grants for Scientific Research of BSKY from Anhui Medical
University (no. XJ201212), Specialized Research Fund for the
Doctoral Program of Higher Education (no. 20113420120006;
20123420110003), Program for tackling key problems in science and
technology by Anhui Province (no. 1301042098).
References
|
1
|
Geisler F and Strazzabosco M: Emerging
roles of Notch signaling in liver disease. Hepatology. 61:382–392.
2015. View Article : Google Scholar
|
|
2
|
Ding J and Wang H: Multiple interactive
factors in hepatocarcinogenesis. Cancer Lett. 346:17–23. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huang J, Xu L, Luo Y, He F, Zhang Y and
Chen M: The inflammation-based scores to predict prognosis of
patients with HCC after hepatectomy. Med Oncol. 31:8832014.
View Article : Google Scholar
|
|
4
|
Coulouarn C and Clément B: Stellate cells
and the development of liver cancer: Therapeutic potential of
targeting the stroma. J Hepatol. 60:1306–1309. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cervello M, McCubrey JA, Cusimano A,
Lampiasi N, Azzolina A and Montalto G: Targeted therapy for HCC:
Novel agents on the horizon. Oncotarget. 3:236–260. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stauffer JK, Scarzello AJ, Jiang Q and
Wiltrout RH: Chronic inflammation, immune escape, and oncogenesis
in the liver: A unique neighborhood for novel intersections.
Hepatology. 56:1567–1574. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bruix J and Sherman M; Practice Guidelines
Committee, American Association for the Study of Liver Diseases:
Management of hepatocellular carcinoma. Hepatology. 42:1208–1236.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cheng AL, Kang YK, He AR, Lim HY, Ryoo BY,
Hung CH, Sheen IS, Izumi N, Austin T, Wang Q, et al: Safety and
efficacy of tigatuzumab plus sorafenib as first-line therapy in
subjects with advanced hepatocellular carcinoma: a phase 2
randomized study. J Hepatol. 63:896–904. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tutunea-Fatan E, Caetano FA, Gros R and
Ferguson SS: GRK2 targeted knock-down results in spontaneous
hypertension, and altered vascular GPCR signaling. J Biol Chem.
290:5141–5155. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Penela P, Ribas C, Aymerich I, Eijkelkamp
N, Barreiro O, Heijnen CJ, Kavelaars A, Sánchez-Madrid F and Mayor
F Jr: G protein-coupled receptor kinase 2 positively regulates
epithelial cell migration. EMBO J. 27:1206–1218. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma L and Pei G: β-arrestin signaling and
regulation of transcription. J Cell Sci. 120:213–218. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo J, Chen H, Ho J, Mancini J, Sontag T,
Laporte SA, Richard DE and Lebrun JJ: TGFbeta-induced GRK2
expression attenuates AngII-regulated vascular smooth muscle cell
proliferation and migration. Cell Signal. 21:899–905. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang X, Yang P and Ma L: Kinase
activity-independent regulation of cyclin pathway by GRK2 is
essential for zebrafish early development. Proc Natl Acad Sci USA.
106:10183–10188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Avendaño MS, Lucas E, Jurado-Pueyo M,
Martínez-Revelles S, Vila-Bedmar R, Mayor F Jr, Salaices M, Briones
AM and Murga C: Increased nitric oxide bioavailability in adult
GRK2 hemizygous mice protects against angiotensin II-induced
hypertension. Hypertension. 63:369–375. 2014. View Article : Google Scholar
|
|
15
|
Lymperopoulos A, Rengo G, Funakoshi H,
Eckhart AD and Koch WJ: Adrenal GRK2 upregulation mediates
sympathetic overdrive in heart failure. Nat Med. 13:315–323. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liang XH, Deng WB, Li M, Zhao ZA, Wang TS,
Feng XH, Cao YJ, Duan EK and Yang ZM: Egr1 protein acts downstream
of estrogen-leukemia inhibitory factor (LIF)-STAT3 pathway and
plays a role during implantation through targeting Wnt4. J Biol
Chem. 289:23534–23545. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chu L, Wang T, Hu Y, Gu Y, Su Z and Jiang
H: Activation of Egr-1 in human lung epithelial cells exposed to
silica through MAPKs signaling pathways. PLoS One. 8:e689432013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Grotegut S, von Schweinitz D, Christofori
G and Lehembre F: Hepatocyte growth factor induces cell scattering
through MAPK/Egr-1-mediated upregulation of Snail. EMBO J.
25:3534–3545. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma Y, Cheng Q, Ren Z, Xu L, Zhao Y, Sun J,
Hu S and Xiao W: Induction of IGF-1R expression by EGR-1
facilitates the growth of prostate cancer cells. Cancer Lett.
317:150–156. 2012. View Article : Google Scholar
|
|
20
|
Ma J, Ren Z, Ma Y, Xu L, Zhao Y, Zheng C,
Fang Y, Xue T, Sun B and Xiao W: Targeted knockdown of EGR-1
inhibits IL-8 production and IL-8-mediated invasion of prostate
cancer cells through suppressing EGR-1/NF-kappaB synergy. J Biol
Chem. 284:34600–34606. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zheng H, Worrall C, Shen H, Issad T,
Seregard S, Girnita A and Girnita L: Selective recruitment of G
protein-coupled receptor kinases (GRKs) controls signaling of the
insulin-like growth factor 1 receptor. Proc Natl Acad Sci USA.
109:7055–7060. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Peppel K, Jacobson A, Huang X, Murray JP,
Oppermann M and Freedman NJ: Overexpression of G protein-coupled
receptor kinase-2 in smooth muscle cells attenuates mitogenic
signaling via G protein-coupled and platelet-derived growth factor
receptors. Circulation. 102:793–799. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Métayé T, Levillain P, Kraimps JL and
Perdrisot R: Immunohistochemical detection, regulation and
antiproliferative function of G-protein-coupled receptor kinase 2
in thyroid carcinomas. J Endocrinol. 198:101–110. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wei Z, Hurtt R, Gu T, Bodzin AS, Koch WJ
and Doria C: GRK2 negatively regulates IGF-1R signaling pathway and
cyclins' expression in HepG2 cells. J Cell Physiol. 228:1897–1901.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Criswell T, Beman M, Araki S, Leskov K,
Cataldo E, Mayo LD and Boothman DA: Delayed activation of
insulin-like growth factor-1 receptor/Src/MAPK/Egr-1 signaling
regulates clusterin expression, a pro-survival factor. J Biol Chem.
280:14212–14221. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mueller L, Broering DC, Meyer J, Vashist
Y, Goettsche J, Wilms C and Rogiers X: The induction of the
immediate-early-genes Egr-1, PAI-1 and PRL-1 during liver
regeneration in surgical models is related to increased portal
flow. J Hepatol. 37:606–612. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kawashıta Y, Ohtsuru A, Kaneda Y, Nagayama
Y, Kawazoe Y, Eguchi S, Kuroda H, Fujioka H, Ito M, Kanematsu T, et
al: Regression of HCC in vitro and in vivo by radiosensitizing
suicide gene therapy under the inducible and spatial control of
radiation. Hum Gene Ther. 10:1509–1519. 1999. View Article : Google Scholar
|
|
28
|
Hao MW, Liang YR, Liu YF, Liu L, Wu MY and
Yang HX: Transcription factor EGR1 inhibits growth of HCC and
esophageal carcinoma cell lines. World J Gastroenterol. 2:203–207.
2002.
|
|
29
|
Lee KH and Kim JR: Hepatocyte growth
factor induced up-regulations of VEGF through Egr-1 in
hepatocellular carcinoma cells. Clin Exp Metastasis. 26:685–692.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ozen E, Gozukizil A, Erdal E, Uren A,
Bottaro DP and Atabey N: Heparin inhibits Hepatocyte Growth Factor
induced motility and invasion of hepatocellular carcinoma cells
through early growth response protein 1. PLoS One. 7:e427172012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shin SY, Kim JH, Baker A, Lim Y and Lee
YH: Transcription factor Egr-1 is essential for maximal matrix
metalloproteinase-9 transcription by tumor necrosis factor alpha.
Mol Cancer Res. 8:507–519. 2010. View Article : Google Scholar : PubMed/NCBI
|