Introduction
The mortality of ovarian cancer is the highest
(1,2) of all cancers in women. Due to the
difficulty of detection at an early stage, most patients with
ovarian cancer are diagnosed at a late stage, usually with
metastases (3), resulting in poor
prognoses. Therefore, any inhibition of metastasis will improve the
therapeutic outcome.
Of the 11 membranes in membrane-associated RING-CH
(MARCH) family proteins, some molecules play an important part in
immune response (4). The RING
domain of MARCH1, localized in the cytoplasmic N-terminal region
(5,6) participates in the ubiquitin transfer
from E2 to its substrate (5).
MARCH1 regulates the antigen presentation (7) and T cell costimulatory functions of
dendritic cells by attenuating the cell-surface expression of its
substrates MHC class II and CD86 molecules (8–10).
MARCH1 is also capable of autoregulating its expression through
dimerization and autoubiquitination (11). MARCH8, a close homolog of MARCH1
(12), has been identified as a
suppressor of the IL-1β-induced NF-κB pathway (13). MARCH8-mediated polyubiquitination
(13) and degradation of IL1RAP
(14) is an important mechanism for
negative regulation of IL-1β-induced signaling pathways.
Previous studies of MARCH1 focus on its function in
the immune system. However, the role of MARCH1 in tumors has not
been clarified. In the present study, we explored the role of
MARCH1 in ovarian cancer cells. The results show that MARCH1 is
overexpressed in ovarian cancer tissues. Silencing MARCH1 inhibits
proliferation, migration and invasion of ovarian cancer cell SKOV3,
and downregulates the NF-κB and the Wnt/β-catenin pathways.
Materials and methods
Tissue specimens and
immunohistochemistry
A tissue microarray (TMA) slide containing malignant
and non-neoplastic ovarian tissues (n=72) was provided by US Biomax
Inc. Cancer Tissue Bank Collection (US Biomax Inc., Rockville, MD,
USA). Another 4 normal ovarian tissues were supplied by the Second
Affiliated Hospital of Chongqing Medical University. The use of
archived cancer samples was approved by the relevant Ethics
Commission. The TMA slide and sample sections were deparaffinized
and rehydrated. Antigen was retrieved using 0.01 M sodium-citrate
buffer (pH 6.0) at a sub-boiling temperature for 20 min after
boiling in a microwave oven. The slide and sections were incubated
with 3% hydrogen peroxide for 10 min to block endogenous
peroxidase. After 15 min of pre-incubation in 5% normal goat serum
to prevent non-specific staining, the samples were incubated with
the antibody to MARCH1 (1:100l; bs-9335R; Bioss, Beijing, China) at
4°C overnight. Secondary (Bioss Biotechnology) antibody was added
and incubated for 30 min. The section was incubated in horseradish
enzyme-labeled chain avidin solution (Bioss Biotechnology) for 30
min at room temperature. Color was developed using a
diaminobenzidine (DAB) substrate kit. Counterstaining was carried
out with hematoxylin.
MARCH1 immunoreactivity was graded as follows: 0
(absence of staining), 1 (weakly stained), 2 (moderately stained)
and 3 (strongly stained). The percentage of positive tumor cells
was scored as follows: 0 (absence of positive cells), 1 (≤33%
positive tumor cells), 2 (33–66% positive tumor cells) and 3 (≥66%
positive tumor cells). The staining score was calculated (staining
intensity score x the percentage score), and the criteria was as
absence: IHC=0, weak; >0 IHC ≤4; and strong, ≥5 IHC ≤9). The
Mann-Whitney U test was used to assess the associations between
MARCH1 overexpression and clinicopathological variables of
epithelial ovarian cancer (EOC) (n=45) samples.
Cell culture and transfection
Human ovarian cancer SKOV3 cells were cultured in
RPMI-1640 medium (Thermo Scientific, Waltham, MA, USA) supplemented
with 10% fetal bovine serum (FBS) (Kang Yuan Biology, China) and 1%
antibiotics (Beyotime, Tianjin, China) at 37°C and 5%
CO2.
Small interfering RNAs (siRNAs) for MARCH1 and
negative control (NC) siRNAs were synthesized by GenePharma Co.,
Ltd. (Shanghai, China). MARCH1 or NC siRNAs were transfected into
SKOV3 cell using a transfection kit from GenePharma Co., Ltd.,
according to the manufacturer's protocol. The sequences for MARCH1
siRNA were: 444, 5′-GCAAGUAUGACUUCAUAAUTT-3′ and
5′-AUUAUGAAGUCAUACUUGCTT-3′; 540, 5′-CUGUCACAUUCCACGUAAUTT-3′ and
5′-AUUACGUGGAAUGUGACAGTT-3′; 693, 5′-CAGGAGGUCUUGUCUUCAUTT-3′ and
5′-AUGAAGACAAGACCUCCUGTT-3′.
Scratch assay
Cells were plated into a 6-well plate with complete
medium and grown to 50% confluence. The medium was then replaced
with antibiotic-free medium. Cells were transfected, and the medium
was replaced with complete medium 24 h after transfection. When
cells were grown to 100% confluence, a wound was created by
scraping the confluent monolayer cells with a p200 pipette tip.
Cells were then grown in medium supplemented with 1% FBS. The
distance between the two sides of cell-free area was photographed
using 10x objective in an Olympus photomicroscope, and was measured
by Matrigel invasion assays at 0, 24 and 48 h. All experiments were
performed in triplicate.
Transwell invasion assay
Cellular migration was determined using a Transwell
assay. Cells were transfected for 48 h and subsequently
dissociated. Cells (1.0×105) were re-suspended in
medium-free of serum and growth factors, and placed in the upper
chamber with an 8-µm pore, 6.5-mm polycarbonate filter
(Corning, New York, NY, USA) coated with Matrigel basement membrane
matrix (BD Biosciences, Bedford, MA, USA) for 2 h at 37°C before
cells were added. The insert was placed in a well with complete
medium. Cells were incubated for 48 h, and cells that did not
migrate through the pores were removed with a cotton swab. Cells on
the lower surface of the membrane were fixed in 4%
paraformaldehyde, stained with 0.5% crystal violet (Beyotime), and
counted under a microscope (magnification, ×200).
Cell viability
Cell growth was analyzed using a WST-8 assay
(Dojindo Laboratories, Kumamoto, Japan). Cells were plated in a
96-well plate with a density of 5×103 cells/well and
siRNA was transfected after 24 h. Cell viability was determined at
24, 48, 72, 96 and 120 h.
5-Ethynyl-2′-deoxyuridine (EdU)-based
proliferation assay
Cell proliferation was measured by the EdU DNA Cell
Proliferation kit (RiboBio) according to the manufacturer's
protocol.
RNA isolation and real-time RT-PCR with
reverse transcription
Total RNA was isolated using the High-purity Total
RNA Rapid Extraction kit (RP1201; BioTeke, Beijing, China).
Real-time RT-PCR was performed using iScript cDNA Synthesis and
SYBR-Green Gene Expression Assay kits (Bio-Rad, Philadelphia, PA,
USA). Real-time RT-PCR and data collection were performed on a
CFX96 instrument (Bio-Rad).
Western blot analysis
Total protein was extracted using cell lysis buffer,
and the protein concentration was determined using the BCA assay
(both from Beyotime). Protein (100 µg) was subjected to
SDS-PAGE, and then transferred to polyvinylidene fluoride membranes
(Millipore, Bedford, MA, USA). The membranes were blocked for 2 h
in 5% skimmed milk (Difco Laboratories, Detroit, MI, USA). Then, a
membrane was incubated with the primary antibodies, including
polyclonal rabbit anti-MARCH1 antibody (1:200; bs-9335R; Bioss),
polyclonal rabbit anti-NF-κB p65 (1:1,000; ab7970), polyclonal
rabbit anti-NF-κB p50 (1:1,000; ab7971) (both from Abcam Inc.,
Cambridge, MA, USA), polyclonal rabbit anti-β-catenin antibody
(1:500; bs-1165R; Bioss), polyclonal rabbit anti-Histone H1, and
polyclonal rabbit anti-E-cadherin antibody, overnight at 4°C. The
membrane was incubated with the HRP-conjugated secondary antibody
for 2 h. GAPDH was detected with a polyclonal antibody and served
as the reference (1:1,000; AB10016; Sangon Biotech, Shanghai,
China). Proteins were visualized with the ECL system (Beyotime)
using the ChemiDoc XRS system (Bio-Rad).
Immunofluorescence microscopy
Cells were transfected with siRNAs. After 48 h,
cells were fixed with 4% paraformaldehyde and stained with
immunofluorescence. Hoechst was used to label the nucleus.
Luciferase reporter assay
Cells were seeded on a 24-well plate, and
transfected with siRNAs. After 48 h, cells were transfected
(GeneCopoeia) with 500 ng TOP flash or NF-κB reporter and 10 ng
pRL-TK (Promega, Madison, WI, USA) plasmids using EndoFectin™-Plus.
Assays were performed in accordance with the dual-luciferase assay
specifications (Promega) using the Mithras LB 940 luminometer
(Berthold, Bad Wildbad, Germany). The activity of firefly
luciferase was normalized to measure the transfection efficiency.
All experiments were performed at least 3 times.
Statistical analysis
All statistical analyses were performed using SPSS
17.0 software (SPSS, Inc., Chicago, IL, USA). Data are presented as
mean ± standard deviation and Student's t-test or ANOVA was used.
Statistical significance was set at p<0.05.
Results
Immunohistochemistry profile of MARCH1 in
normal/benign ovary and ovarian cancer tissue samples
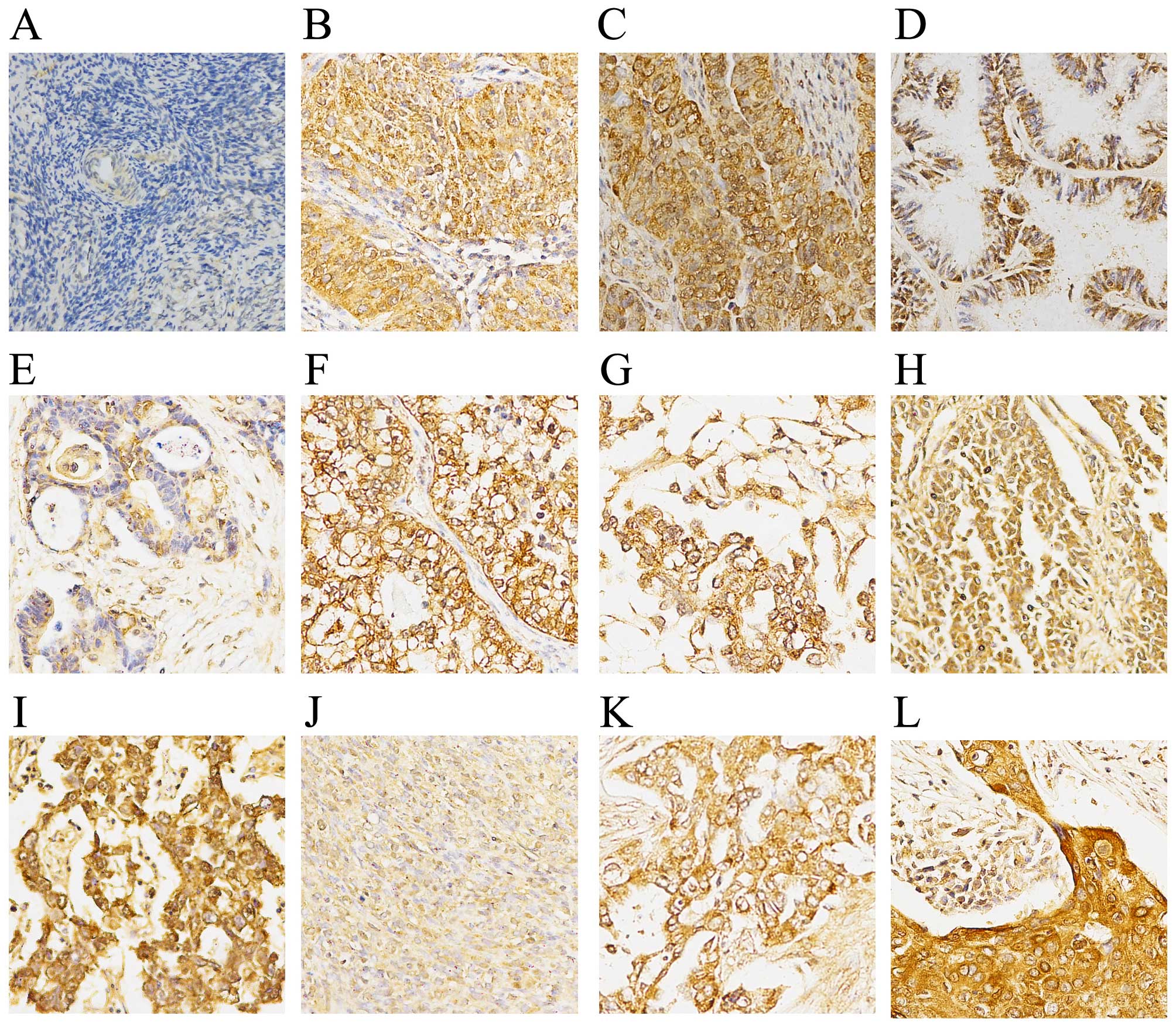
The immunohistochemistry results demonstrated a
negative expression of MARCH1 in normal and para-cancerous normal
ovary tissue (Fig. 1). Of the
primary ovarian cancer samples (n=63), 34 (54.97%) had high
expression and 29 (46.03%) had low expression. In order to
determine whether MARCH1 expression correlates to
clinicopathological type, epithelial ovarian cancer (EOC) tissues
(n=45) were grouped into serous and non-serous (mucinous,
clear-cell and endometrioid) cancers. No correlation between MARCH1
expression and clinicopathological variables are noted (Table I).
 | Table IAssociation of MARCH1 expression with
clinicopathological characteristics in 45 patients with EOC. |
Table I
Association of MARCH1 expression with
clinicopathological characteristics in 45 patients with EOC.
|
Characteristics | No. of
pts.
(n=45) | MARCH1 expression
| P-value |
|---|
Low no.
(%) | High no.
(%) |
|---|
| Age (years) | | | | 0.913 |
| <51 | 28 | 12 (42.86) | 16 (57.14) | |
| ≥51 | 17 | 7 (41.18) | 10 (58.82) | |
| FIGO stage | | | | 0.625 |
| I–II | 36 | 16 (44.44) | 20 (55.56) | |
| III–IV | 9 | 3 (33.33) | 6 (66.67) | |
| Grade | | | | 0.202 |
| 1–2 | 24 | 8 (33.33) | 16 (66.67) | |
| 3 | 21 | 11 (52.38) | 10 (47.62) | |
| Tumor type | | | | 0.971 |
| Serous | 38 | 16 (42.11) | 22 (57.89) | |
| Non-serousa | 7 | 3 (42.86) | 4 (57.14) | |
MARCH1 expression in ovarian cancer cell
lines
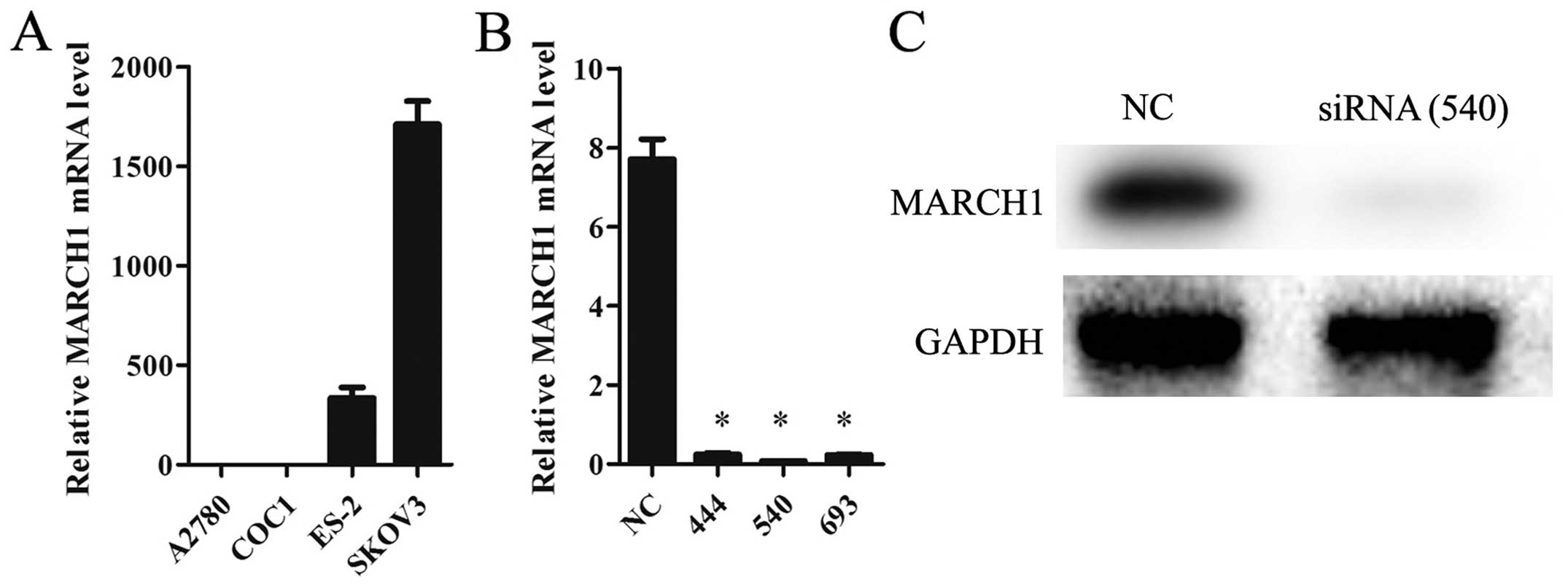
The mRNA level of MARCH1 in ovarian cancer cell
lines A2780, COC1, ES-2 and SKOV3 was analyzed. The highest level
was detected in SKOV3 cells (Fig.
2A), so these were chosen for a knockdown trial. Of the 3 siRNA
sequences, 540 was found to be the most suitable one for our
purposes (Fig. 2B), and was thus
used in all subsequent experiments.
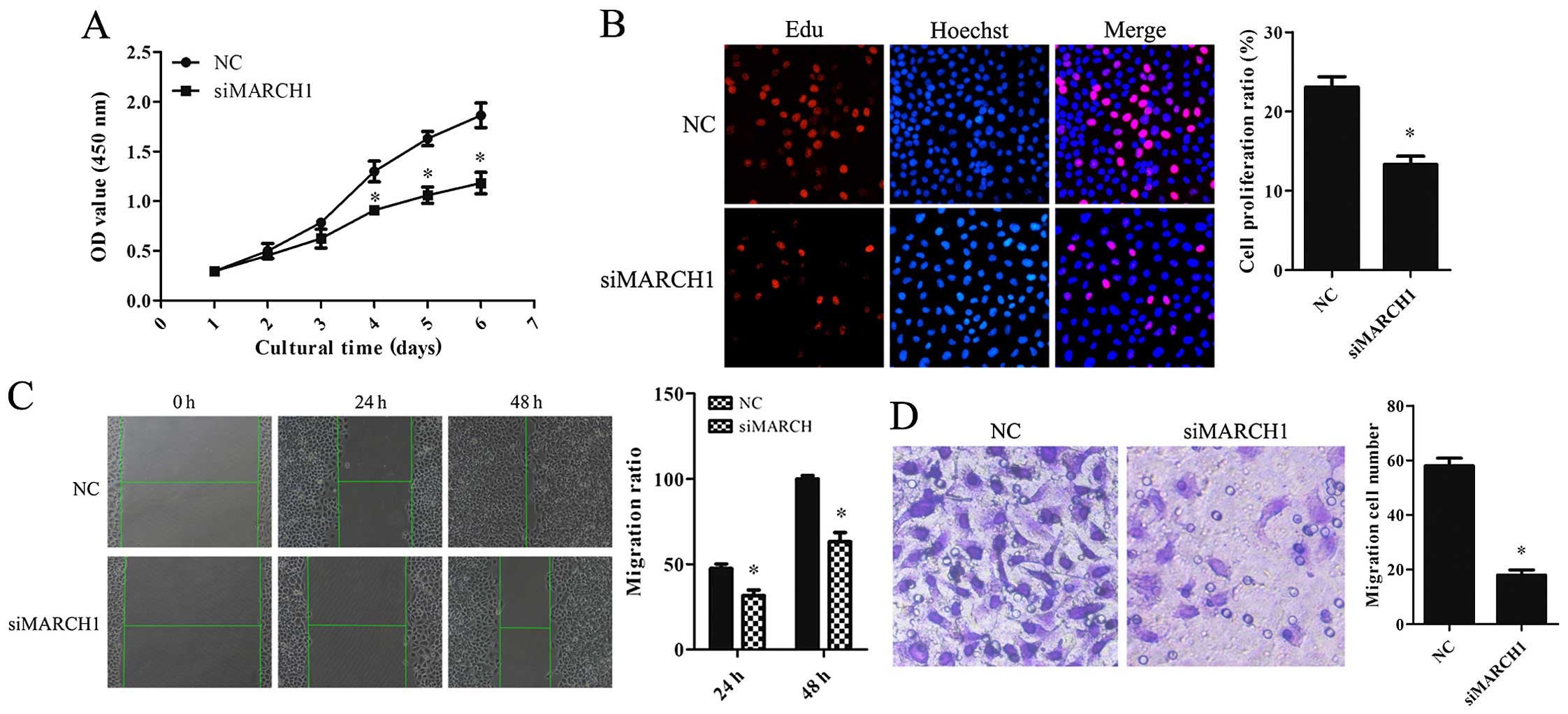
Knockdown of MARCH1 inhibits cell
proliferation, migration and invasion
The effect of MARCH1 on the cell biological behavior
was analyzed. Cell Counting Kit-8 (CCK-8) and EdU assays showed
that the proportion of proliferating cells was reduced in
MARCH1-silenced cells (Fig. 3A and
B). The scratch assay showed that migration ability was
impaired in MARCH1-silenced cells (Fig.
3C). The invasion assay showed a decrease in invaded cells
(Fig. 3D).
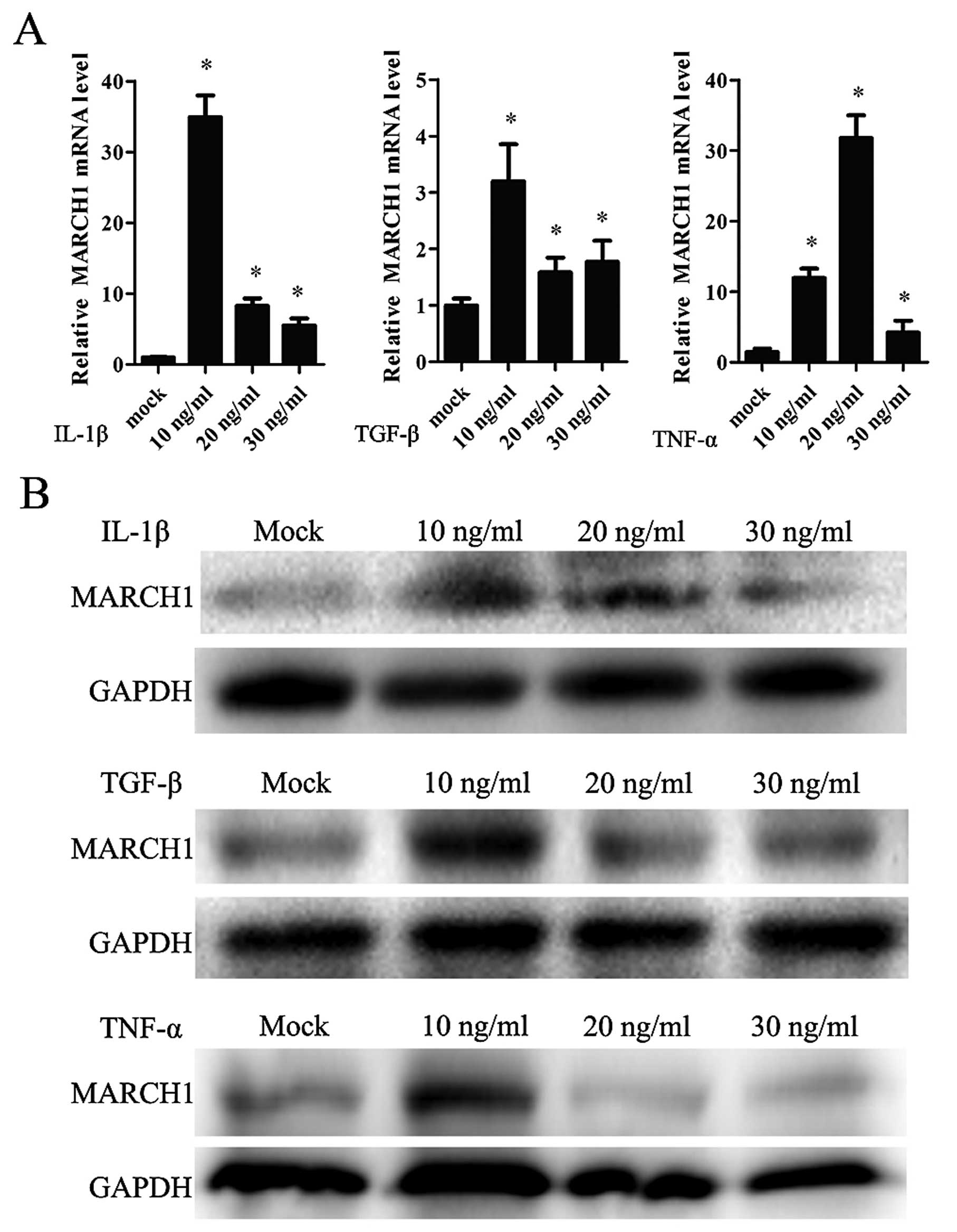
IL-1β, TNF-α and TGF-β positively
regulated MARCH1 expression
In order to determine whether IL-1β affects MARCH1
expression, cells were treated with IL-1β in a range of
concentrations: 0, 10, 20 and 30 ng/ml. MARCH1 expression,
validated by real-time qT-PCR and western blotting, was upregulated
by IL-1β, with the highest level noted correlating to a
concentration of 10 ng/ml (Fig. 4A and
B).
IL-1β affected tumor cells by activating the
canonical NF-κB pathway. TNF-α and TGF-β are also capable of
activating the NF-κB pathway. Thus, cells were treated with TNF-α
or TGF-β and MARCH1 was assayed. The results showed that TNF-α and
TGF-β upregulated MARCH1 expression at mRNA and protein levels
(Fig. 4A and B).
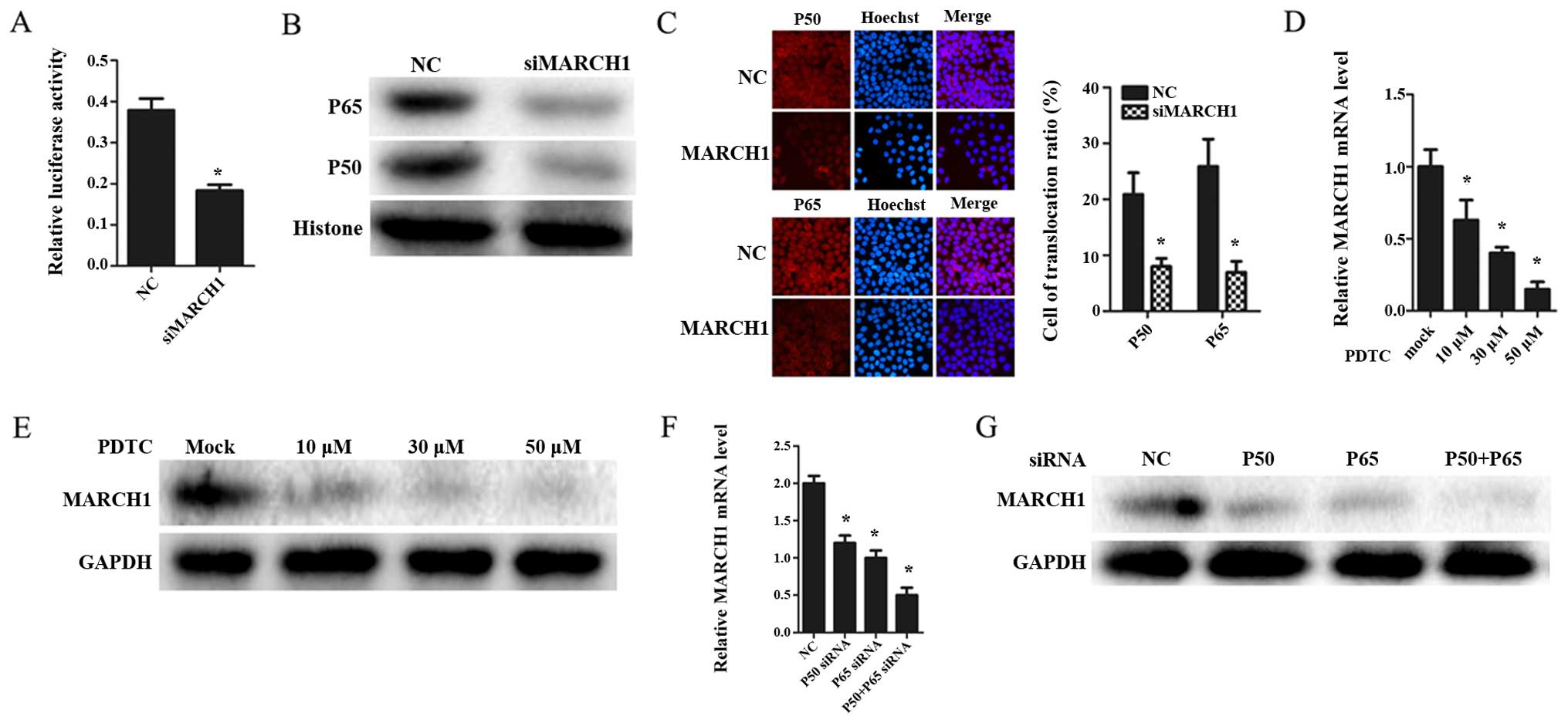
Knockdown of MARCH1 inhibits NF-κB
activity as well as the transportation of p65 and p50 to the
nucleus
In order to determine whether MARCH1 affects the
NF-κB pathway, NF-κB activity was measured using NF-κB luciferase
reporter. The results showed that NF-κB activity was inhibited in
MARCH1-silenced cells (Fig.
5A).
p50 and p65 are sub-units of the transcription
factor NF-κB (15). They can bind
DNA individually as a homodimer or as a p50-p65 heterodimer
(16), thus, activating
transcription of these genes. Therefore, we hypothesized that
MARCH1 may affect the NF-κB pathway by regulating p65 and p50. To
verify this hypothesis, p65 and p50 expression was validated by
western blotting. The MARCH1-silenced group was found to have a
lower total expression of p50 and p65, and a decrease in the
nucleus of p65 and p50 (Fig. 5B).
Moreover, the immunofluorescence assay indicated that silencing
MARCH1 attenuated nuclear translocation of p65 and p50 (Fig. 5C).
MARCH1 expression is mediated through an
NF-κB-dependent pathway
IL-1β can activate the NF-κB pathway (17). As IL-1β was proved to increase
MARCH1, we tested whether the IL-1-mediated induction of MARCH1
occurred through an NF-κB-dependent pathway. Pyrrolidine
dithiocarbamate (PDTC), an inhibitor of NF-κB (18), was used to block the NF-κB pathway.
MARCH1 expression decreased at both mRNA and protein levels
(Fig. 5D and E). Furthermore, when
NF-κB pathway was inhibited by silencing p65 and p50 with siRNAs,
MARCH1 was downregulated (Fig.
5F).
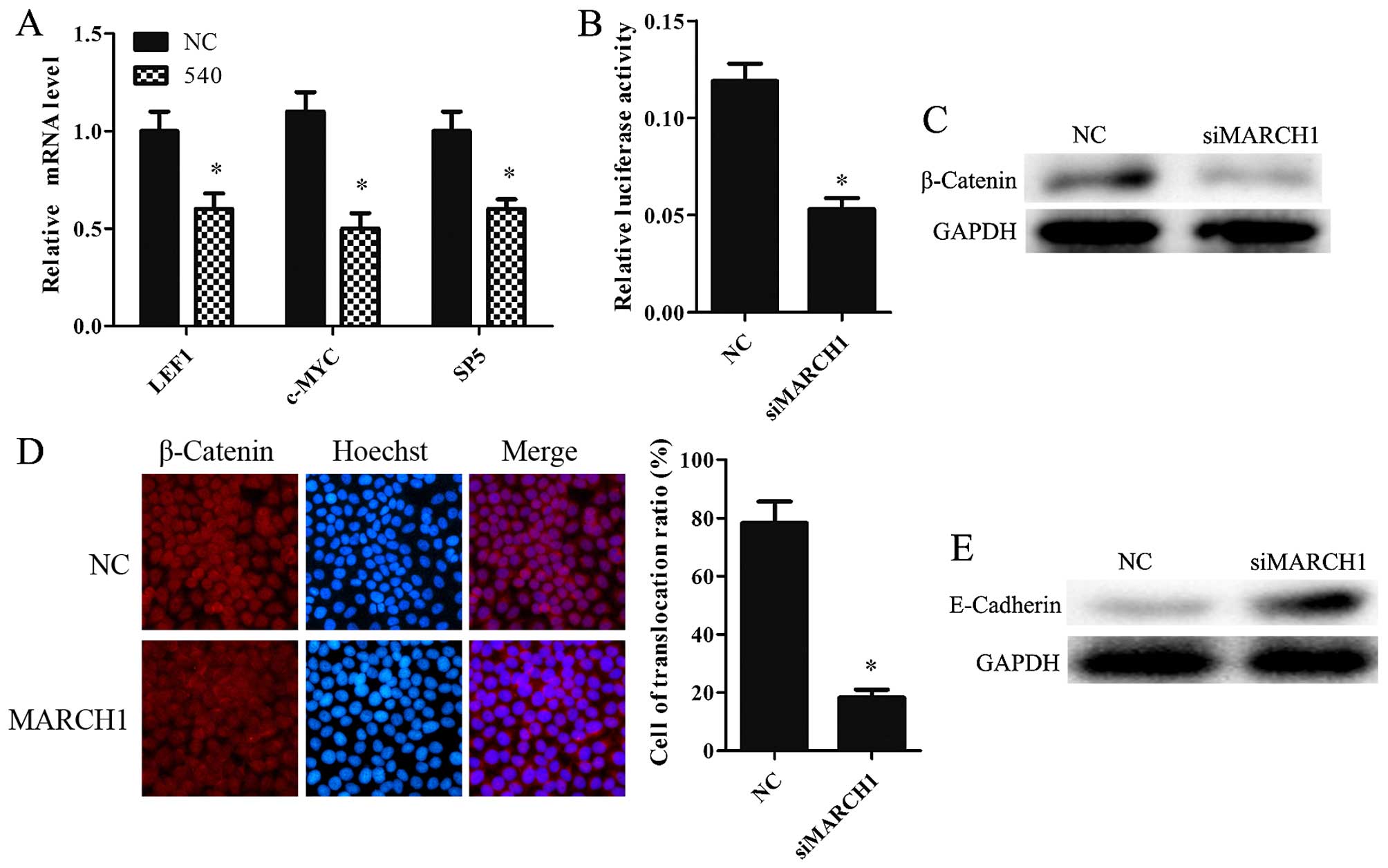
Knockdown of MARCH1 inhibits
Wnt/β-catenin pathway
Wnt targets LEF1, c-MYC and SP5 proteins (19–21).
Silencing MARCH1 was found to downregulate LEF1, c-MYC and SP5 at
the mRNA level (Fig. 6A). TOP-flash
luciferase reporter was used to determine whether MARCH1 mediated
the Wnt/β-catenin pathway. MARCH1-silenced cells showed lower
levels of activity compared to the NC group (Fig. 6B). We also examined β-catenin
expression in MARCH1-silenced cells. The data showed that silencing
MARCH1 significantly decreased β-catenin expression at the protein
level (Fig. 6C). Furthermore, the
immunofluorescence results indicated that silencing MARCH1 inhibits
the translocation of β-catenin to the nucleus (Fig. 6D). These results indicate that
MARCH1 can upregulate the Wnt/β-catenin pathway.
Knockdown of MARCH1 negatively regulated
E-cadherin
E-cadherin is a tumor suppressor (22,23).
It can bind and antagonize the nuclear signaling function of
β-catenin (24), a known
proto-oncogene. E-cadherin inhibits the Wnt/β-catenin pathway by
binding with β-catenin and inducing its degradation (25,26).
MARCH1 may affect E-cadherin expression. E-cadherin expression in
MARCH1 knockdown cells and NC cells was therefore observed. The
results showed that loss of MARCH1 protein resulted in higher
levels of E-cadherin protein expression (Fig. 6E).
Discussion
Previous studies of MARCH1 have mainly focused on
its function in the immune system. The role of MARCH1 in tumors has
thus remained unclear. The present study shows that MARCH1 is
overexpressed in ovarian cancer tissues, and that silencing MARCH1
inhibited proliferation, migration and invasion of ovarian cancer
SKOV3 cells. Additionally, silencing MARCH1 inhibits the NF-κB and
the Wnt/β-catenin pathways.
The immunohistochemistry results showed a high level
of MARCH1 expression in cancer tissues, while adjacent normal
ovarian tissues were weakly stained and normal ovarian tissues were
negatively stained. However, clinicopathological variables did not
correlate to MARCH1 expression. This may be due to the limited
sample size, and should be verified in a larger-scale trial.
Nevertheless, silencing MARCH1 was found to inhibit SKOV3 cell
proliferation, invasion and migration. These data suggest that
MARCH1 could play an important role in the formation, development
and metastasis of ovarian cancer.
Cytokines have an impact on the cancer progress via
modulating tumor microenvironment and cell signaling pathways
(27,28). For instance, L-1β, TNF-α and TGF-β
can activate the NF-κB pathway (29,30),
thereby regulating the biological behavior of cancer cells
(15,29,31).
The present study showed that L-1β, TNF-α and TGF-β all upregulate
MARCH1 expression. Silencing MARCH1 led to an inhibition of the
behavior of ovarian cancer cells. Thus, a correlation may exist
between MARCH1 and the NF-κB pathway, which was confirmed by the
findings in the cell signaling pathway reporter assays.
NF-κB can activate the transcription of several
genes involved in the regulation of numerous important processes
such as immune response, inflammation (30), apoptosis (32,33)
and cell proliferation (34,35).
Aberrant activation of NF-κB signaling is related to various human
cancers (15,36) including ovarian cancer (36,37).
Silencing MARCH1 can not only downregulate the expression of NF-κB,
p65 and p50, but also sequester p65-p50 in the cytoplasm. These
results imply that MARCH1 positively regulates the NF-κB pathway.
IL-1 induces NF-κB activation in a dose-dependent manner (17). IL-1β increases the protein level of
p65 in the nucleus at low concentration but decreases it at high
concentration (38). These findings
are consistent with the data generated by the present study. In
addition, we assumed that NF-κB regulated MARCH1, reversely. The
present study showed that inhibiting the NF-κB pathway, whether by
PDTC or by silencing p50 and p65, downregulated MARCH1 expression
at the protein level. In conclusion, IL-1β upregulates MARCH1,
which positively regulates the NF-κB pathway. In addition, the
NF-κB pathway can upregulate MARCH1.
The Wnt/β-catenin pathway is involved in the
aggressive behavior of cancer cells. E-cadherin inhibits the
Wnt/β-catenin pathway by binding with β-catenin and inducing its
degradation. Overexpression of β-catenin increases tumor migration
and invasion (39). MARCH1, as a
tumor promoter in ovarian cancer, was found to upregulate β-catenin
at a post-transcriptional level, thereby facilitating the
translocation of β-catenin into the nucleus. These results indicate
that MARCH1 downregulates E-cadherin, leading to an accumulation of
β-catenin in plasma, which contributes to upregulation of the
Wnt/β-catenin pathway.
In conclusion, our data provide clinical and
laboratory evidence that MARCH1 expression plays an important role
in the progression of ovarian cancer and, therefore, may be a novel
therapeutic target.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81172492), the
Specialized Research Fund fo rthe Doctoral Program of Higher
Education (grant no. 20125503110004) and the Key Project of Science
and Technology of Chongqing (grant no. CSTC2012JJB10030).
References
|
1
|
Kandukuri SR and Rao J: FIGO 2013 staging
system for ovarian cancer: What is new in comparison to the 1988
staging system? Curr Opin Obstet Gynecol. 27:48–52. 2015.
View Article : Google Scholar
|
|
2
|
Tew WP and Fleming GF: Treatment of
ovarian cancer in the older woman. Gynecol Oncol. 136:136–142.
2015. View Article : Google Scholar
|
|
3
|
Musto A, Grassetto G, Marzola MC, Rampin
L, Chondrogiannis S, Maffione AM, Colletti PM, Perkins AC, Fagioli
G and Rubello D: Management of epithelial ovarian cancer from
diagnosis to restaging: An overview of the role of imaging
techniques with particular regard to the contribution of 18F-FDG
PET/CT. Nucl Med Commun. 35:588–597. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nathan JA and Lehner PJ: The trafficking
and regulation of membrane receptors by the RING-CH ubiquitin E3
ligases. Exp Cell Res. 315:1593–1600. 2009. View Article : Google Scholar
|
|
5
|
Ohmura-Hoshino M, Goto E, Matsuki Y, Aoki
M, Mito M, Uematsu M, Hotta H and Ishido S: A novel family of
membrane-bound E3 ubiquitin ligases. J Biochem. 140:147–154. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jabbour M, Campbell EM, Fares H and
Lybarger L: Discrete domains of MARCH1 mediate its localization,
functional interactions, and posttranscriptional control of
expression. J Immunol. 183:6500–6512. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bourgeois-Daigneault MC and Thibodeau J:
Identification of a novel motif that affects the conformation and
activity of the MARCH1 E3 ubiquitin ligase. J Cell Sci.
126:989–998. 2013. View Article : Google Scholar
|
|
8
|
Corcoran K, Jabbour M, Bhagwandin C,
Deymier MJ, Theisen DL and Lybarger L: Ubiquitin-mediated
regulation of CD86 protein expression by the ubiquitin ligase
membrane-associated RING-CH-1 (MARCH1). J Biol Chem.
286:37168–37180. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tze LE, Horikawa K, Domaschenz H, Howard
DR, Roots CM, Rigby RJ, Way DA, Ohmura-Hoshino M, Ishido S,
Andoniou CE, et al: CD83 increases MHC II and CD86 on dendritic
cells by opposing IL-10-driven MARCH1-mediated ubiquitination and
degradation. J Exp Med. 208:149–165. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chattopadhyay G and Shevach EM:
Antigen-specific induced T regulatory cells impair dendritic cell
function via an IL-10/MARCH1-dependent mechanism. J Immunol.
191:5875–5884. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bourgeois-Daigneault MC and Thibodeau J:
Autoregulation of MARCH1 expression by dimerization and
autoubiquitination. J Immunol. 188:4959–4970. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Matsuki Y, Ohmura-Hoshino M, Goto E, Aoki
M, Mito-Yoshida M, Uematsu M, Hasegawa T, Koseki H, Ohara O,
Nakayama M, et al: Novel regulation of MHC class II function in B
cells. EMBO J. 26:846–854. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen R, Li M, Zhang Y, Zhou Q and Shu HB:
The E3 ubiquitin ligase MARCH8 negatively regulates IL-1β-induced
NF-κB activation by targeting the IL1RAP coreceptor for
ubiquitination and degradation. Proc Natl Acad Sci USA.
109:14128–14133. 2012. View Article : Google Scholar
|
|
14
|
van de Kooij B, Verbrugge I, de Vries E,
Gijsen M, Montserrat V, Maas C, Neefjes J and Borst J:
Ubiquitination by the membrane-associated RING-CH-8 (MARCH-8)
ligase controls steady-state cell surface expression of tumor
necrosis factor-related apoptosis inducing ligand (TRAIL) receptor
1. J Biol Chem. 288:6617–6628. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hayden MS and Ghosh S: Signaling to
NF-kappaB. Genes Dev. 18:2195–2224. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fujita T, Nolan GP, Ghosh S and Baltimore
D: Independent modes of transcriptional activation by the p50 and
p65 subunits of NF-kappa B. Genes Dev. 6:775–787. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Woronicz JD, Gao X, Cao Z, Rothe M and
Goeddel DV: IkappaB kinase-beta: NF-kappaB activation and complex
formation with IkappaB kinase-alpha and NIK. Science. 278:866–869.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schreck R, Albermann K and Baeuerle PA:
Nuclear factor kappa B: An oxidative stress-responsive
transcription factor of eukaryotic cells (a review). Free Radic Res
Commun. 17:221–237. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Logan CY and Nusse R: The Wnt signaling
pathway in development and disease. Annu Rev Cell Dev Biol.
20:781–810. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Neth P, Ries C, Karow M, Egea V, Ilmer M
and Jochum M: The Wnt signal transduction pathway in stem cells and
cancer cells: Influence on cellular invasion. Stem Cell Rev.
3:18–29. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Katoh M and Katoh M: WNT signaling pathway
and stem cell signaling network. Clin Cancer Res. 13:4042–4045.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cheng CW, Wu PE, Yu JC, Huang CS, Yue CT,
Wu CW and Shen CY: Mechanisms of inactivation of E-cadherin in
breast carcinoma: Modification of the two-hit hypothesis of tumor
suppressor gene. Oncogene. 20:3814–3823. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Berx G, Cleton-Jansen AM, Nollet F, de
Leeuw WJ, van de Vijver M, Cornelisse C and van Roy F: E-cadherin
is a tumour/invasion suppressor gene mutated in human lobular
breast cancers. EMBO J. 14:6107–6115. 1995.PubMed/NCBI
|
|
24
|
Gottardi CJ, Wong E and Gumbiner BM:
E-cadherin suppresses cellular transformation by inhibiting
beta-catenin signaling in an adhesion-independent manner. J Cell
Biol. 153:1049–1060. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nelson WJ and Nusse R: Convergence of Wnt,
beta-catenin, and cadherin pathways. Science. 303:1483–1487. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mantovani A, Allavena P, Sica A and
Balkwill F: Cancer-related inflammation. Nature. 454:436–444. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liotta LA and Kohn EC: The
microenvironment of the tumour-host interface. Nature. 411:375–379.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tak PP and Firestein GS: NF-kappaB: A key
role in inflammatory diseases. J Clin Invest. 107:7–11. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ghosh S, May MJ and Kopp EB: NF-kappa B
and Rel proteins: Evolutionarily conserved mediators of immune
responses. Annu Rev Immunol. 16:225–260. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Van Antwerp DJ, Martin SJ, Kafri T, Green
DR and Verma IM: Suppression of TNF-alpha-induced apoptosis by
NF-kappaB. Science. 274:787–789. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Micheau O, Lens S, Gaide O, Alevizopoulos
K and Tschopp J: NF-kappaB signals induce the expression of c-FLIP.
Mol Cell Biol. 21:5299–5305. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Thomas RP, Farrow BJ, Kim S, May MJ,
Hellmich MR and Evers BM: Selective targeting of the nuclear
factor-kappaB pathway enhances tumor necrosis factor-related
apoptosis-inducing ligand-mediated pancreatic cancer cell death.
Surgery. 132:127–134. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Guttridge DC, Albanese C, Reuther JY,
Pestell RG and Baldwin AS Jr: NF-kappaB controls cell growth and
differentiation through transcriptional regulation of cyclin D1.
Mol Cell Biol. 19:5785–5799. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen F, Castranova V and Shi X: New
insights into the role of nuclear factor-kappaB in cell growth
regulation. Am J Pathol. 159:387–397. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dolcet X, Llobet D, Pallares J and
Matias-Guiu X: NF-kB in development and progression of human
cancer. Virchows Arch. 446:475–482. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sirotkin AV, Alexa R, Kišová G, Harrath
AH, Alwasel S, Ovcharenko D and Mlynček M: MicroRNAs control
transcription factor NF-kB (p65) expression in human ovarian cells.
Funct Integr Genomics. 15:271–275. 2015. View Article : Google Scholar
|
|
38
|
Cafferata EG, Guerrico AM, Pivetta OH and
Santa-Coloma TA: NF-kappaB activation is involved in regulation of
cystic fibrosis transmembrane conductance regulator (CFTR) by
interleukin-1beta. J Biol Chem. 276:15441–15444. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lu Z, Ghosh S, Wang Z and Hunter T:
Downregulation of caveolin-1 function by EGF leads to the loss of
E-cadherin, increased transcriptional activity of beta-catenin, and
enhanced tumor cell invasion. Cancer Cell. 4:499–515. 2003.
View Article : Google Scholar
|