Introduction
Lung cancer is the most common cause of
cancer-related death in males and second most common cause in
females (1). Recently, the
morbidity and mortality of lung carcinoma has noticeably increased.
Lung cancer is classified as non-small cell lung cancer (NSCLC) and
small cell lung cancer (SCLC). NSCLC represents approximately 85%
(2) of cases and has a five-year
survival of only 15% (3).
Pain relief is a fundamental and formidable task in
the treatment of cancer patients because most cancer patients have
severe pain (4). The µ-opioid
receptor was proposed as the third step on the analgesic ladder and
is used to relieve pain for terminal cancer patients according to
WHO guidelines.
Fentanyl is widely used for relieving pain and
narcotizin. Specifically, it is considered a good analgesic that is
effective against cancer pain in terminal cancer patients (5). Recently, the effects of opioid
analgesics on cancer patients have gained increasing attention.
Basic research indicates that opioids induce tumour growth, inhibit
apoptosis, and promote angiogenesis. However, the effect of
fentanyl on autophagy has not been reported. We found that fentanyl
increases autophagy in lung cancer cells. Autophagy is an
evolutionarily conserved catabolic process that involves the
degradation of cellular components through the lysosomal machinery
(6). Autophagy may remove and
assist long-standing proteins in metabolic waste recycling to
maintain a stable cell environment. A recent study reported that
autophagy exerts a protective effect when cells experience
increased pressure and contributes to cellular survival by
providing nutrients and energy to help cells adapt to starvation or
stress (such as hypoxia, X-rays and anticancer drugs) (7).
LC3 and Beclin-1 are two key proteins involved in
autophagy. In this study, we demonstrate that fentanyl induces
higher levels of LC3-II. LC3-II formation is regarded as reliable
biochemical evidence of autophagy as the amount of LC3-II usually
correlates well with the number of autophagosomes. In our study,
Beclin-1 expression also increased in a dose-dependent manner.
Beclin-1 plays a role in the initiation step and is essential for
autophagosome formation (8). In a
previous study, the epidermal growth factor receptor (EGFR)
inhibitors gefitinib and erlotinib both induced autophagy in lung
cancer cells, and hypoxia-induced autophagy mediates cisplatin
resistance in lung cancer cells (9). Therefore, in the present study, we
investigated whether fentanyl, which is commonly used, alters
sensitivity to cisplatin in the human lung cancer cell line
A549.
Chemotherapy is the main method of treating lung
cancer in the early stages of disease, particularly as
postoperative adjuvant chemotherapy. Platinum drugs are the most
common chemotherapy agents used to treat lung cancer. Combining
cisplatin (DDP) and other chemotherapy drugs for middle-late lung
cancer patients has become a standardized treatment. Although the
rate of survival has improved, sensitivity has decreased, which is
a major obstacle in the clinical application of chemotherapy. The
direct effects of opioids on cancer cell sensitivity to
chemotherapy have not been adequately examined. Therefore, in the
present study, we investigated whether fentanyl, which is commonly
used during general anaesthesia, alters sensitivity to cancer
chemotherapy in a human lung cancer cell line (10).
Increasing evidence indicates that multiple stress
conditions, such as oxidative stress (11), endoplasmic reticulum (ER) stress and
pathogen infection (12), induce
autophagy through different molecular pathways. Among them,
reactive oxygen species (ROS) function as signalling molecules not
only in cell growth, differentiation, proliferation and apoptosis
(13) but also in autophagy
(14). ROS are active forms of
oxygen generated as by-products from cellular metabolism.
Furthermore, ROS affect various signalling pathways, such as the
MAPK (including ERK, JNK and p38) signal transduction cascades.
Recently, it was demonstrated that p38 MAPK mediates autophagy in
response to chemotherapeutic agents (15). JNK, also known as the
stress-activated protein kinase, has been implicated in apoptosis
and autophagy (15–17). In mammals, there are three JNK
genes: JNK1, JNK2 and JNK3 (18).
JNK1 and JNK2 are ubiquitously expressed, whereas JNK3 is primarily
expressed in the brain, cardiac smooth muscle and testes (18). In particular, ROS induces
JNK-dependent autophagy (19,20).
Our data demonstrated that fentanyl, via the
activation of ROS-JNK-mediated autophagy, inhibits the
chemotherapeutic sensitivity of DDP. The autophagy inhibitor
3-methyladenine (3-MA) may weaken this effect.
Materials and methods
Reagents and antibodies
Fentanyl hydrochloride and DDP was obtained from
Northeast Pharmaceutical Group Co., Ltd., Shenyang, China).
N-Acetyl-L-cysteine (NAC) were purchased from Beyotime Institute of
Biotechnology (Shanghai, China), JNK inhibitor (SP600125), and 3-MA
were purchased from Selleck Chemicals (Houston, TX, USA).
Cell culture
The human NSCLC cell lines A549 and SPC-A1 were
obtained from the Shanghai Institute of Biochemistry and Cell
Biology, Chinese Academy of Sciences (Shanghai, China) and
maintained in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% fetal bovine serum and 1%
penicillin/streptomycin (all from Gibco-BRL, Invitrogen Life
Technologies, Gaithersburg, MD, USA) in a humidified cell culture
incubator at 37°C and in 5% CO2.
Cell viability assay
The cells (3×103/well) were seeded in
96-well plates and incubated overnight. After treatment, 10 µl of
the Cell Counting Kit-8 (CCK-8) reagent (Dojindo Molecular
Technologies, Inc., Kumamoto, Japan) was added to 90 µl of media in
each well, followed by a 2-h incubation, after which the absorbance
was measured at 450 nm using a microplate reader (Thermo Fisher
Scientific, Waltham, MA, USA). Each assay was conducted in
quadruplicate and repeated at least three times.
Brightfield images
Cell monolayers were cultured for 24 h in 2-well
glass-covered chamber slides and then treated with various drugs.
After washing with PBS, micrographs were taken using an Olympus
inverted fluorescence microscope (Olympus, Tokyo, Japan). The
number of cells with increased acidic vesicular organelles was
determined by counting at least three representative fields per
treatment condition, and a minimum of three replicate experiments
was conducted.
Apoptosis assay
Apoptosis was assessed by Annexin V-binding analysis
of flow cytometry. For flow cytometric analysis, cells
(1×105) were seeded in 6-cm dishes overnight before
treatment. Both adherent and floating cells were harvested and
combined, washed twice with PBS, resuspended in 500 µl of binding
buffer, and stained using an Annexin V-FITC/PI kit (Nanjing KeyGen
Biotech Co., Ltd., Nanjing, China) according to the manufacturer's
instructions. After incubation in the dark for 30 min, the cells
were analyzed using FACSCalibur instrument (FACSCalibur; BD
Biosciences, San Jose, CA, USA). All experiments were performed in
duplicate with reproducibility.
DAPI staining
Cells were grown on glass coverslips. After washing
with PBS, cells were fixed in 4% paraformaldehyde-PBS for 30 min
and treatment followed with 0.1% Triton X-100 for 10 min at 4°C.
Then stained with DAPI (Beyotime Institute of Biotechnology),
washed with PBS. Images were captured with the inverted and
confocal microscope (Olympus IX71; Olympus) (Leica TCS SP5 II;
Leica, Mannheim, Germany).
Western blot analysis
Cells were washed with PBS (pH 7.4), and incubated
with 2X concentrated electrophoresis sample buffer (125 mM
Tris-HCl, pH 6.8, 5% glycerol, 2% SDS, 1% β-mercaptoethanol) for 30
min on ice. Protein concentration was determined with Coomassie
protein assay reagent using bovine serum albumin (BSA) as a
standard, and equal aliquots of protein (40 mg) were separated
using 8–15% SDS-PAGE. The proteins were transferred onto a
nitrocellulose membrane (Millipore Corp., Billerica, MA, USA),
which was blocked in TBST (TBS containing 0.1% Tween-20) with 5%
non-fat dry milk for 2 h at room temperature and was then incubated
overnight at 4°C with the respective antibodies diluted in
TBS/T/BSA (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.05% Tween-20,
0.1% BSA). The membranes were immunoblotted with the respective
antibodies and then incubated with horseradish
peroxidase-conjugated secondary antibodies. Enhanced
chemiluminescence (ECL) detection system (Bio-Rad Laboratories,
Richmond, CA, USA) was used to visualize immunoreactive bands.
Antibodies against Beclin-1, P62 were purchased from ProteinTech
Group, Inc. (Wuhan, China), LC3B was purchased from Sigma-Aldrich
(St. Louis, MO, USA), p-c-JUN and c-JUN were from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). LC3B and phospho-JNK,
JNK were from Cell Signaling Technology, Inc. (Beverly, MA, USA).
All of the primary antibodies were used at a 1:1,000 dilution. All
of the western blot analyses were replicated a minimum of three
times, and densitometry comparing the proteins of interest with
GAPDH as a loading control was conducted using ImageJ software.
LysoTracker Red
After washing twice with PBS, the monolayer A549
cells were labeled with 50 nM LysoTracker Red (Beyotime Institute
of Biotechnology) for 30 min in serum-free DMEM under 5%
CO2 at 37°C. Images were obtained using the fluorescence
microscope (Olympus).
Monodansylcadaverine (MDC)
staining
Cell monolayers were cultured for 24 h in 2-well
glass-covered chamber slides and then treated with various drugs.
The slides were washed with culture medium without serum. Then, the
cells were exposed to 50 mM MDC (Nanjing KeyGen Biotech Co., Ltd.),
an autofluorescent dye, for 10 min at 37°C and visualized using a
confocal laser scanning microscope (Olympus IX71 and Leica TCS SP5
II). At least 3 areas per well were analysed. Two wells were
analysed per treatment and per time. The experiment was repeated
three times.
Transmission electron microscopy
A549 cells were fixed with 2.5% glutaraldehyde and
post-fixed with 1% osmium tetroxide. After being dehydrated in
increasing concentrations of alcohol, the cell pellets were
embedded in epon. Representative areas were chosen for ultrathin
sectioning and examined on a transmission electron microscope
(JEM-2000EX; Jeol, Ltd., Tokyo, Japan).
DCFH-DA
Intracellular ROS generation was measured withan ROS
assay with dichlorofluorescein diacetate (DCFH-DA; Beyotime
Institute of Biotechnology). Briefly, the cells were plated at a
density of 2×105 cells/well in six-well plates.
Following treatment, intracellular ROS were detected via
fluorescence microscope using DCFH-DA staining. Then, the cells
were incubated with DCFH-DA (5 µM) for 30 min at 37°C in the dark
and were washed three times with serum-free medium. The levels of
ROS were determined by Olympus BX83 fluorescence microscope
(Olympus).
Statistical analysis
The data are expressed as the means ± standard
deviations (SDs) of at least 3 separate experiments. SPSS 19.0
software was used for statistical analysis. Multiple group
comparisons were analysed by one-way ANOVA; 2-group comparisons
were performed with Student's t-test. p<0.05 was considered
statistically significant.
Results
Fentanyl reduces the sensitivity of
cisplatin in lung cancer cells
The viabilities of A549 and SPC-A1 cells
concurrently treated with fentanyl and cisplatin for 24 h were
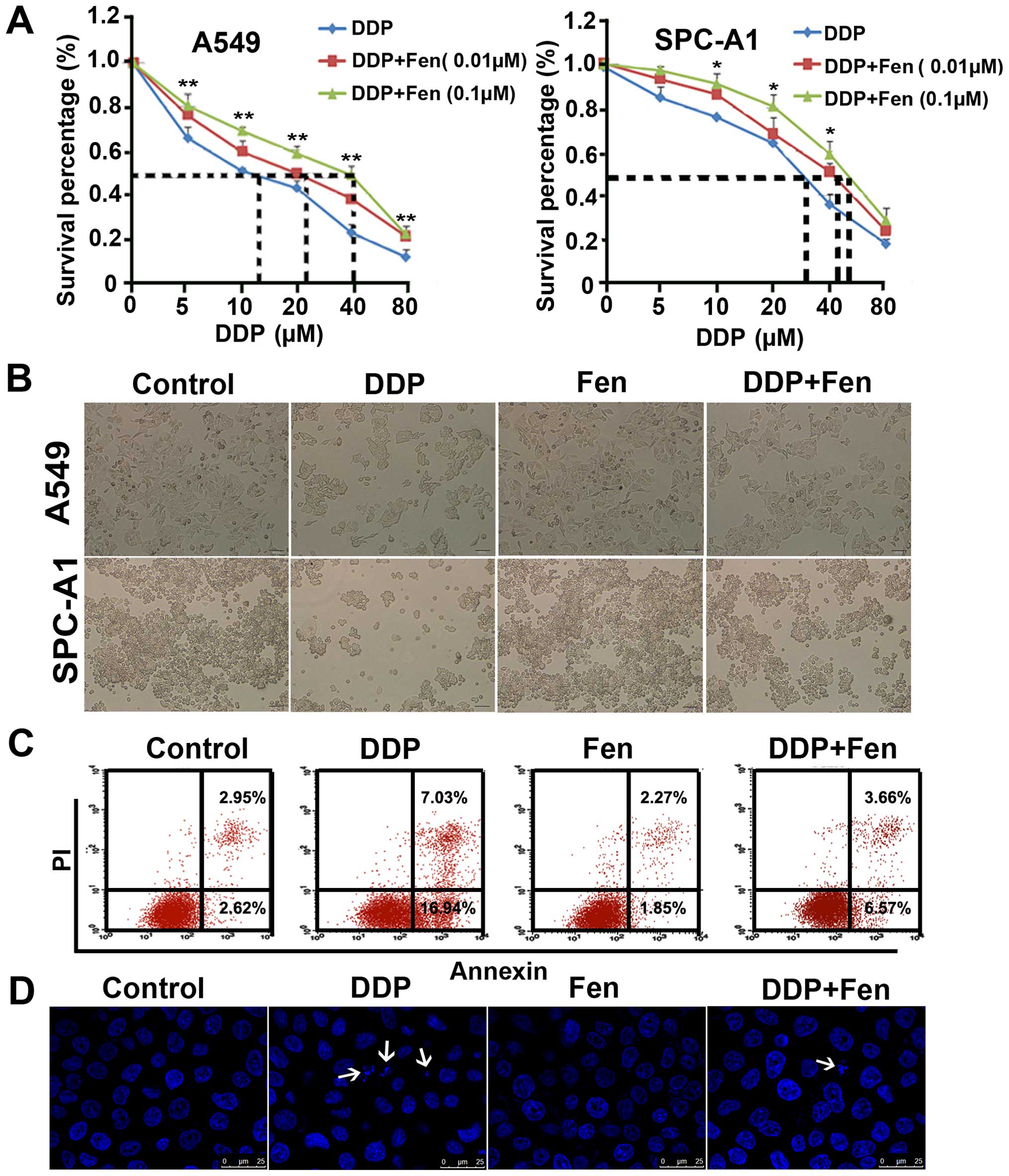
analysed using a CCK-8 assay. As shown in Fig. 1A, fentanyl reduced the
cisplatin-induced inhibition of cell viability in a
concentration-dependent manner in the two cell lines, and
brightfield images yielded the same result (Fig. 1B). Cisplatin-induced cell death in
lung cancer cells potentially involves multiple signalling
pathways. Among these pathways, apoptotic events play a critical
role. To confirm this possibility, we assayed cell apoptosis by
flow cytometry. The data, as indicated by the Annexin V-FITC assay,
revealed that the apoptosis rates were 5.57, 23.97, 4.12 and 10.23%
in the control, cisplatin, fentanyl and combined groups,
respectively (Fig. 1C), indicating
that fentanyl attenuates the apoptosis induced by cisplatin.
Nuclear condensation was investigated by DAPI staining in fentanyl
and cisplatin-treated lung cancer cells. As shown in Fig. 1D, cisplatin increased the frequency
of apoptotic nuclei in the A549 cell lines, and fentanyl reversed
this process.
Fentanyl promotes autophagy in A549
cells
We proposed that the impact of fentanyl on cisplatin
was exerted primarily through the promotion of autophagy (21). To determine whether fentanyl induces
autophagy, we treated A549 cells with fentanyl at two
concentrations. The conjugation of the soluble form of LC3 (LC3-I)
with phosphatidylethanolamine and the conversion of LC3-I to a
nonsoluble form (LC3-II) have been generally recognized as useful
signs of autophagy. Thus, we examined LC3B-II expression. After
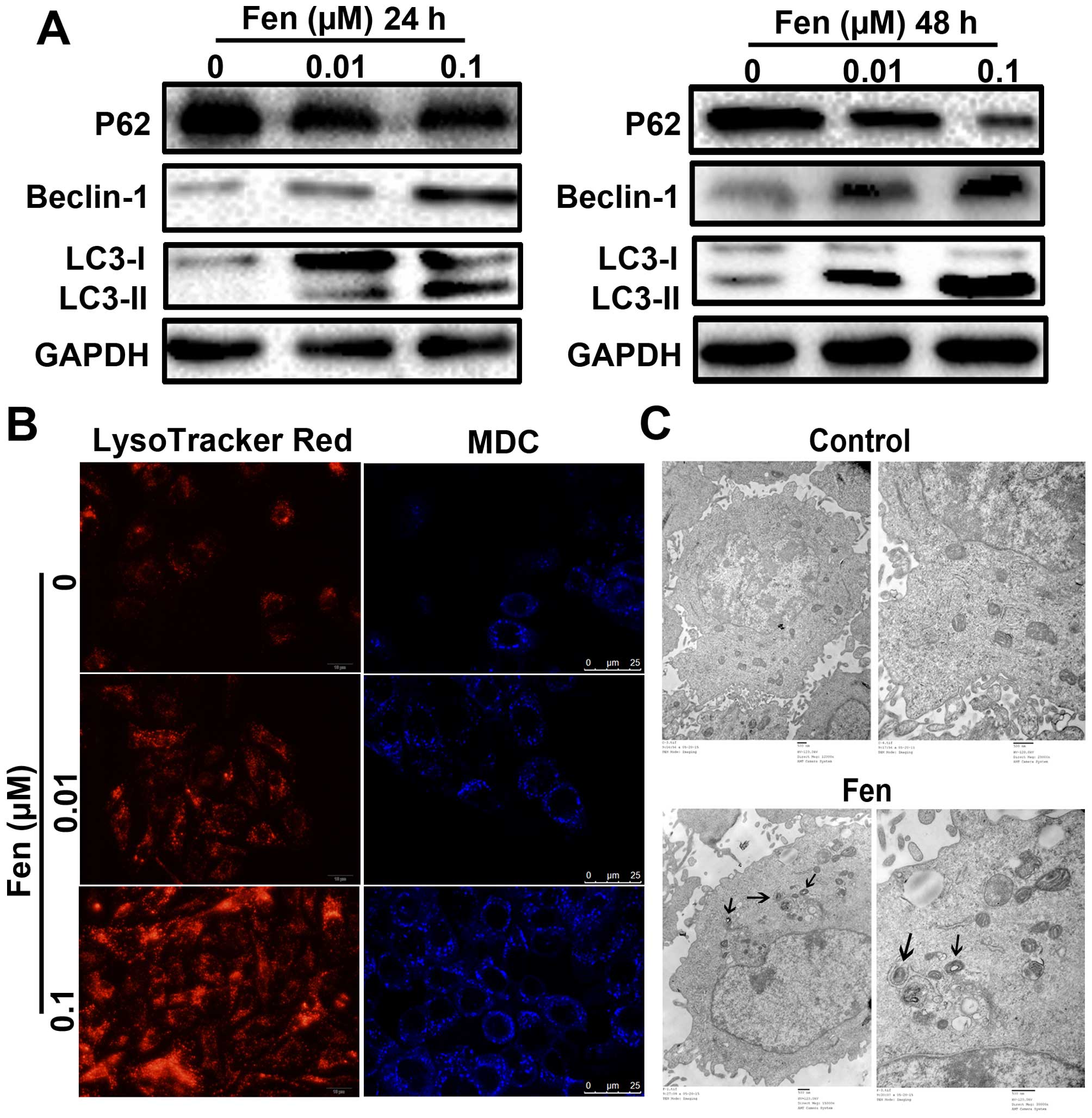
treatment with fentanyl (0.01 or 0.1 µM), LC3B-II levels increased
in a concentration-dependent manner (Fig. 2A). However, we could not ascertain
whether the increase in LC3B-II levels was due to the activation of
autophagy or the blockade of autophagosome-lysosome fusion. Thus,
we subsequently measured the protein level of p62, a selective
substrate of autophagy, because activation of the autophagic flux
leads to p62 degradation. It is now recognized that although
autophagosome formation is a necessary component of the autophagic
process, formation can occur without completion of autophagy and
degradation of the autophagosomal content (22). To evaluate autophagic flux, p62
protein levels were monitored by western blot analysis (21). As Fig.
2A shows, a marked decrease in p62 was detected (23). LysoTracker Red and MDC are
fluorescent substances often used to detect the occurrence of
autophagy. As shown in Fig. 2B,
compared with the control group, fentanyl induced far more red
spots and a stronger punctate fluorescence of MDC at the 24-h
recovery point in the cytoplasms of A549 cells. To further evaluate
the effects of fentanyl on A549 cells at the ultrastructural level,
TEM was performed on the fentanyl-treated cells (8). Compared with the control group, the
number of autophagic vacuoles of the fentanyl group were markedly
increased. Taken together, these results demonstrate that fentanyl
promotes autophagy in A549 cells (24).
ROS mediates fentanyl-induced
autophagy in A549 cells
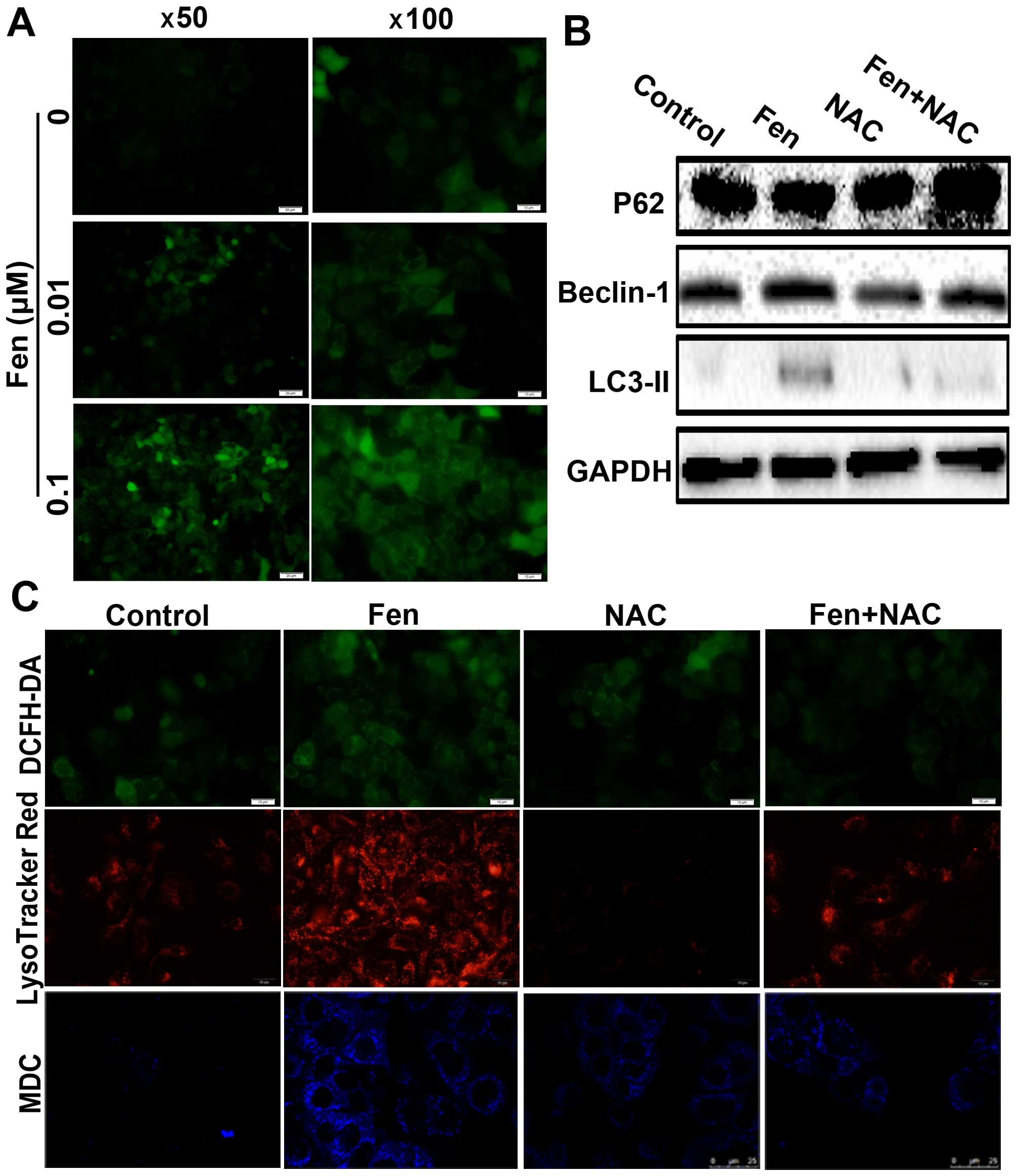
To investigate whether ROS play a role in
fentanyl-induced autophagy, A549 cells were labelled using DCFH-DA,
a fluorochrome that detects hydroperoxide generation. The
percentage of A549 cells exhibiting increased hydroperoxide was
greatly increased after one day of exposure to fentanyl compared
with the control cells (Fig. 3A).
Pre-incubation with NAC (5 mmol/l), a typical antioxidant, markedly
inhibited fentanyl-induced ROS generation (Fig. 3C). We then tested the effect of NAC
on autophagy by western blot analysis, LysoTracker Red and MDC
staining. As shown in Fig. 3B, NAC
inhibited fentanyl-induced autophagy, LC3BII and Beclin-1 protein
levels were downregulated and P62 levels were upregulated.
LysoTracker Red and MDC staining indicated similar results. Taken
together, fentanyl induces ROS overproduction, which contributes to
autophagy in A549 cells (23).
The ROS-activated JNK pathway
contributes to fentanyl-induced autophagy
ROS are inducers or mediators of the activation of
MAPK family members, including JNK, p38 and ERK1/2 (25). Additionally, studies have shown that
MAPKs play a pivotal role in autophagy (16,26,27).
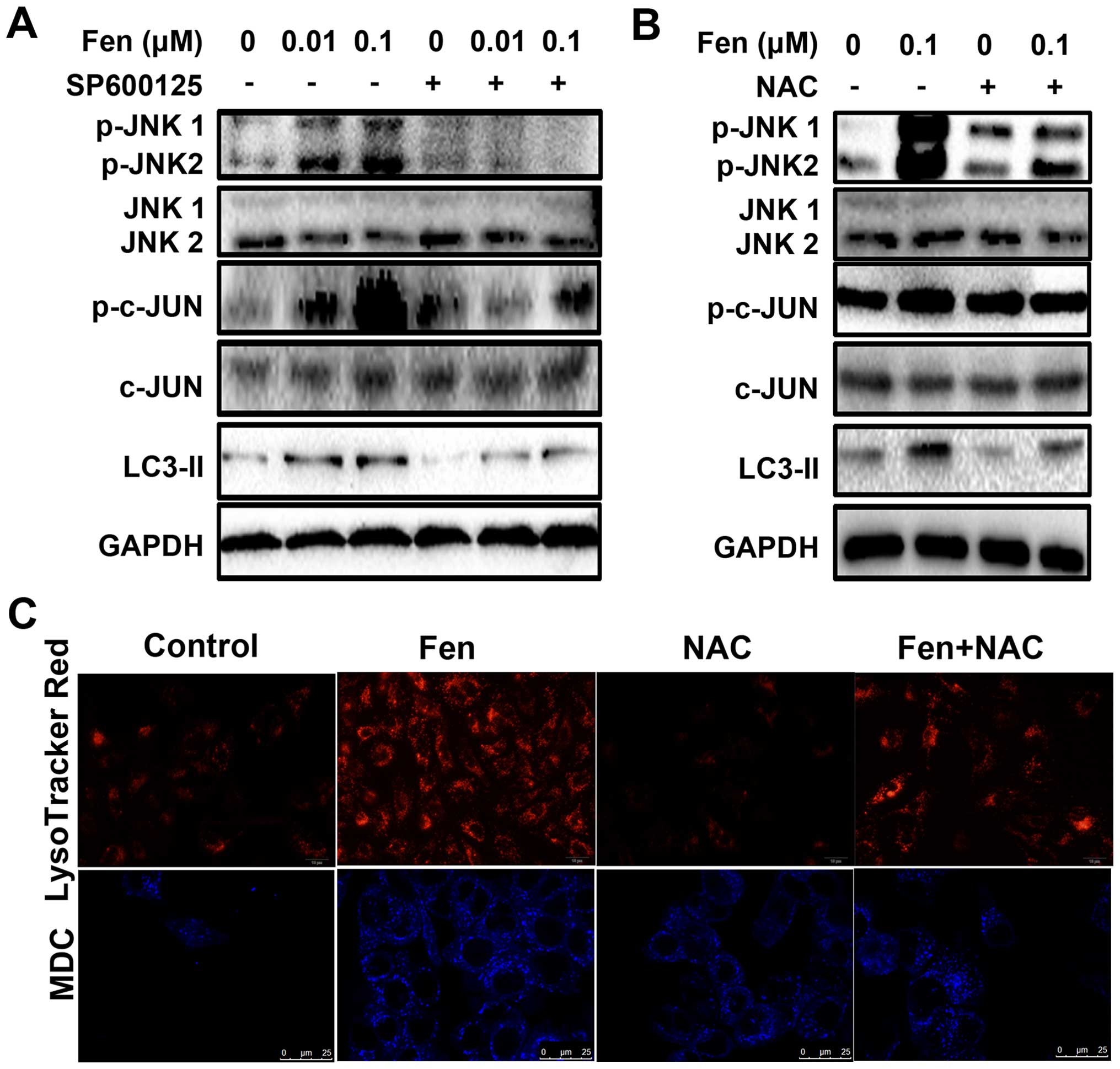
In this study, we observed that fentanyl induced JNK
phosphorylation in a concentration-dependent manner (Fig. 4A). Therefore, we next asked whether
fentanyl-induced autophagy involves JNK. To this end, a selective
inhibitor of JNK, SP600125, was employed. As shown in Fig. 4A, pretreatment with SP600125
potently inhibited the activation of the JNK pathway and the LC3-II
expression induced by fentanyl. MDC and LysoTracker Red staining
yielded similar results (Fig. 4C).
To investigate the role of ROS induction in fentanyl-induced
autophagy and the activation of the JNK pathway more extensively,
the ROS scavenger NAC was utilized. As shown in Fig. 4B, upregulation of fentanyl-induced
c-Jun phosphorylation, and LC3-II expression was attenuated
significantly by pretreatment with NAC. In conclusion, the ROS/JNK
axis is involved in fentanyl-associated autophagy.
Inhibition of autophagy restores lung
cancer cell sensitivity to cisplatin under fentanyl exposure
The data above demonstrated that fentanyl protected
A549 and SPC-A1 cells from cisplatin. It has recently been reported
that under adverse conditions, such as hypoxia, cancer cells make
themselves adaptive by activating autophagy (9); thus, we hypothesized that autophagy
may contribute to the fentanyl-induced cisplatin sensitivity
decline in lung cancer cells. To test this hypothesis, we inhibited
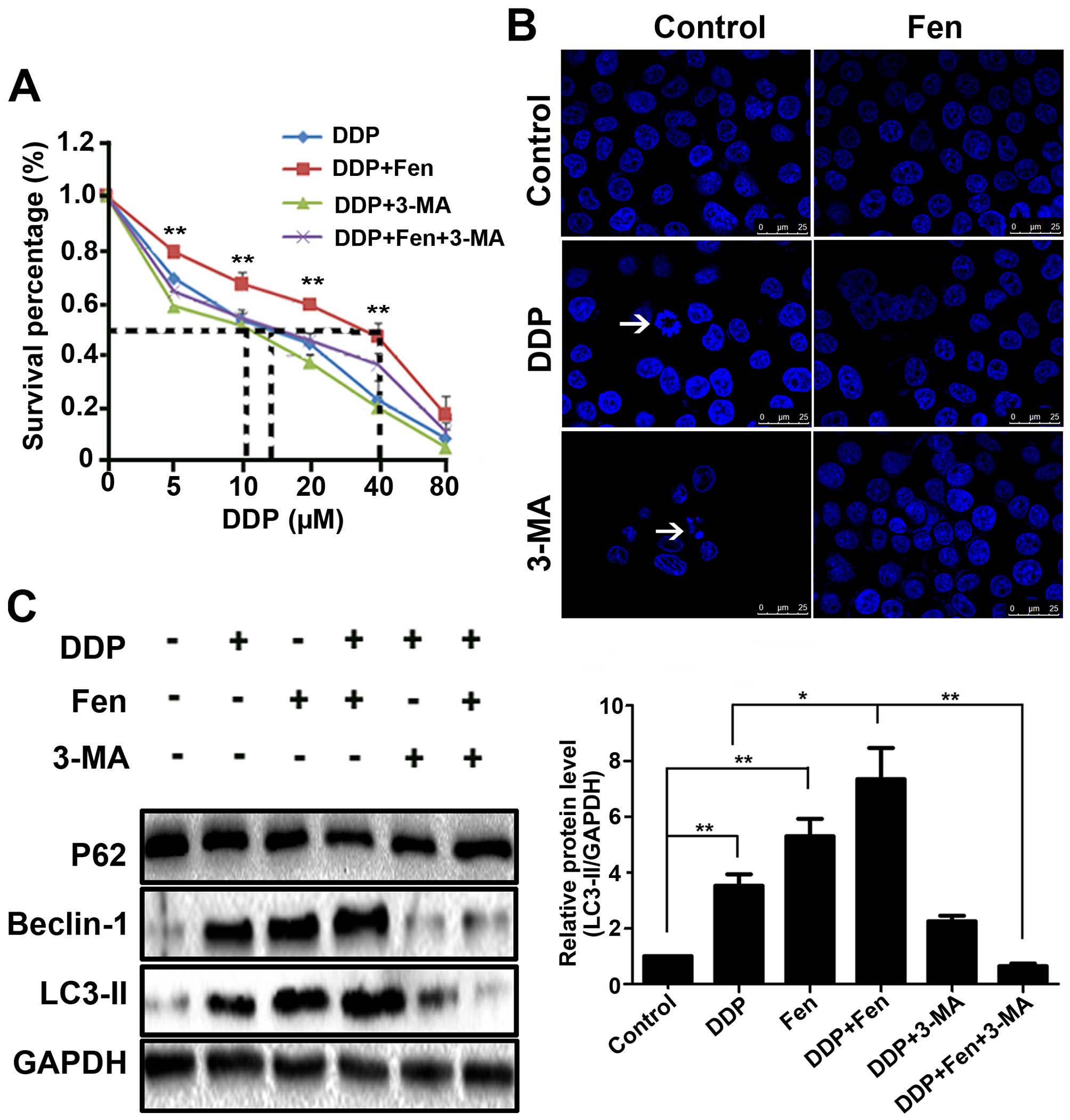
autophagy using 3-MA. Cell viability was assessed by CCK-8, DAPI
staining was performed to evaluate nuclear condensation, and
autophagy-related proteins were detected by western blot analysis.
First, we found that pretreatment with 3-MA for 1 h followed by DDP
treatment for 24 h greatly increased DDP-induced A549 cell death
(Fig. 5A). DAPI had similar result
(Fig. 5B). These data indicate that
autophagy may act as a suppressor of A549 cells in conjunction with
DDP treatment (28). Next, we found
that cisplatin significantly induced the expression of LC3-II and
Beclin-1 protein levels in A549 cells. Interestingly,
cisplatin-induced expression levels of the above proteins were
significantly increased when combined with fentanyl (Fig. 5C). These data imply that autophagy
mediates the sensitivity decline of cisplatin resistance in lung
cancer cells and that the inhibition of autophagy restores lung
cancer cell sensitivity to cisplatin therapy.
Discussion
Cancer patients often need to use both analgesic and
antitumour drugs simultaneously. As a result, research focused on
how an analgesic influences the effect of an antitumour drug is
necessary. However, there have been very few studies describing the
role of fentanyl in the chemoresistance of cancer cells (29). Therefore, this study aimed to
investigate whether fentanyl reduces the sensitivity of cisplatin
in NSCLC cells.
It is believed that autophagy plays a critical role
in the pathogenesis of diverse diseases, such as inflammatory bowel
disease, neuronal degeneration, ageing and cancer (28,30).
In recent years, autophagy has become increasingly relevant in
cancer research (31). One study
found that the activation of autophagy induces cell death, but
autophagy protects the effect of antitumour therapy by inhibiting
tumour cell apoptosis (22).
Recent studies have struggled to reveal the complex
paradoxical role of autophagy in cancer development and cancer
therapy (32). Wu et al
demonstrated that the augmented induction of autophagy by hypoxia
decreased lung cancer cell susceptibility to cisplatin-induced
apoptosis (9). Activated autophagy
protects tumour cells from targeted therapies, including
trastuzumab in breast cancer, imatinib mesylate in Philadelphia
chromosome-positive cells, and proteasome inhibitors in prostate
cancer (33). Li et al
reported that autophagy was activated as a protective mechanism
against the cellular effects of 5-fluorouracil (5-FU)-treatment and
that the inhibition of autophagy by 3-MA augmented 5-FU-induced
apoptosis in colon cancer cells (34). Tang et al found that
glycerrhetinic acid (GA) induces cytoprotective autophagy in NSCLC
cells by activating the IRE1-JNK/c-jun pathway. Autophagy induced
by gefitinib in lung cancer cells is cytoprotective (35,36).
Furthermore, crizotinib activates autophagy in lung cancer cells
(37).
However, there is no research focused on the
analgesic effect of autophagy in cancer cells. Our study is the
first to demonstrate that fentanyl improves the level of autophagy
in A549 cells and then reduces the sensitivity to cisplatin. After
treatment with cisplatin for 24 h, the IC50 values of
the A549 and SPC-A1 cells were 10 and 5 µM; when combined with 0.01
or 0.1 µM fentanyl, the IC50 values were higher.
Brightfield images demonstrated similar results. Flow cytometry and
DAPI staining all indicate that fentanyl weakens cisplatin-induced
apoptosis. In this study, we investigated the functional role of
autophagy in A549 cells exposed to fentanyl, as evidenced by the
accumulation of LysoTracker Red and MDC-positively-stained acidic
vesicles, the formation of autophagosomes observed via TEM and the
enhanced conversion of LC3-II and Beclin-1.
Autophagy occurs when cisplatin induces apoptosis in
A549 lung cancer cells, which ultimately results in cancer cell
survival. Similarly, Fig. 5 shows
that cisplatin itself induced autophagy and that when the cells
were pretreated with 3-MA for 1 h followed by DDP treatment for 24
h, DDP-induced A549 cell death was greatly increased (Fig. 5A). These data indicated that
autophagy may act in a protective manner to suppress A549 cells in
conjunction with cisplatin treatment. Cisplatin-induced autophagy
was significantly increased when combined with fentanyl, resulting
in reduced sensitivity to cisplatin.
Recent investigations demonstrate that autophagic
cell death is associated with alterations in ROS levels and/or the
JNK signalling pathway (38). ROS
are highly reactive molecules formed by incomplete one-electron
reduction of oxygen, including oxygen ions and peroxides. ROS form
as a natural by-product of the normal metabolism of oxygen and
participate in cell signalling and homeostasis at low levels.
However, under environmental stress, high levels of ROS cause
irreversible oxidative damage to cellular structures (39). In this study, we found that fentanyl
treatment resulted in an increase in ROS in A549 cells. The ROS
inhibitor, NAC, attenuated fentanyl-induced autophagy.
Several studies report that the activation of the
JNK signalling pathway by ROS represents a novel mechanism of
autophagic induction (40,41). In the present study, fentanyl
induced significant increases in ROS generation and JNK
phosphorylation. The ROS inhibitor, NAC, completely reversed the
fentanyl-induced inhibition of cell proliferation, apoptosis and
autophagy. In addition, the JNK inhibitor also significantly
attenuated these effects. In the present study, for the first time,
we demonstrate that ROS-JNK-autophagy was activated rather than
suppressed in fentanyl-treated cells compared with controls, which
was blocked by the inhibitors NAC and SP600125 (42).
Blocking cancer cell autophagy is emerging as a
novel approach to enhancing the efficiency of chemotherapy in
cancer treatment. Currently, inhibitors of autophagy that sensitize
chemoresistant cells to anticancer therapy are being investigated
in clinical trials (43,44). As activation of hVps34/PtdInsKC3 is
important for the development of the autophagosome, we evaluated
whether the class III PtdIns3K inhibitor, 3-MA, inhibits autophagy
and improves the cytotoxic effects of cisplatin (45). When lung cancer cells were treated
with 3-MA, the cytotoxic effects of cisplatin were significantly
enhanced, and the rate of apoptosis increased, which opposed the
effects of fentanyl (9).
In conclusion, our study is the first to demonstrate
that fentanyl reduces the sensitivity of cisplatin through the
ROS-JNK-autophagy pathway. This compelling evidence expands our
understanding of the benefits and clinical applications of fentanyl
therapy and suggests that adding an autophagy inhibitor maybe an
effective way of solving this problem.
Acknowledgements
The study was supported by the National Natural
Science Foundation of China Research Grant (81273923).
References
|
1
|
Zinner R, Visseren-Grul C, Spigel DR and
Obasaju C: Pemetrexed clinical studies in performance status 2
patients with non-small cell lung cancer (Review). Int J Oncol.
48:13–27. 2016.PubMed/NCBI
|
|
2
|
Calbó J, Meuwissen R, van Montfort E, van
Tellingen O and Berns A: Genotype-phenotype relationships in a
mouse model for human small-cell lung cancer. Cold Spring Harb Symp
Quant Biol. 70:225–232. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Verdecchia A, Francisci S, Brenner H,
Gatta G, Micheli A, Mangone L and Kunkler I: EUROCARE-4 Working
Group: Recent cancer survival in Europe: A 2000-02 period analysis
of EUROCARE-4 data. Lancet Oncol. 8:784–796. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ryan M, Moynihan TJ and Loprinzi CL:
As-needed morphine: Yes, but at what dose and at what interval? J
Clin Oncol. 23:3849–3852. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang XL, Chen ML and Zhou SL: Fentanyl
increases colorectal carcinoma cell apoptosis by inhibition of
NF-κB in a Sirt1-dependent manner. Asian Pac J Cancer Prev.
15:10015–10020. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wirawan E, Berghe T Vanden, Lippens S,
Agostinis P and Vandenabeele P: Autophagy: For better or for worse.
Cell Res. 22:43–61. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ma J, Wan J, Meng J, Banerjee S,
Ramakrishnan S and Roy S: Methamphetamine induces autophagy as a
pro-survival response against apoptotic endothelial cell death
through the Kappa opioid receptor. Cell Death Dis. 5:e10992014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu HM, Jiang ZF, Ding PS, Shao LJ and Liu
RY: Hypoxia-induced autophagy mediates cisplatin resistance in lung
cancer cells. Sci Rep. 5:122912015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nomura Y, Kawaraguchi Y, Sugimoto H,
Furuya H and Kawaguchi M: Effects of morphine and fentanyl on
5-fluorouracil sensitivity in human colon cancer HCT116 cells. J
Anesth. 28:298–301. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee J, Giordano S and Zhang J: Autophagy,
mitochondria and oxidative stress: Cross-talk and redox signalling.
Biochem J. 441:523–540. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jia K, Thomas C, Akbar M, Sun Q,
Adams-Huet B, Gilpin C and Levine B: Autophagy genes protect
against Salmonella typhimurium infection and mediate insulin
signaling-regulated pathogen resistance. Proc Natl Acad Sci USA.
106:14564–14569. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maryanovich M and Gross A: A ROS rheostat
for cell fate regulation. Trends Cell Biol. 23:129–134. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Scherz-Shouval R and Elazar Z: Regulation
of autophagy by ROS: Physiology and pathology. Trends Biochem Sci.
36:30–38. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang
Q, He C and Pan H: p38 and JNK MAPK pathways control the balance of
apoptosis and autophagy in response to chemotherapeutic agents.
Cancer Lett. 344:174–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
He W, Wang Q, Srinivasan B, Xu J, Padilla
MT, Li Z, Wang X, Liu Y, Gou X, Shen HM, et al: A JNK-mediated
autophagy pathway that triggers c-IAP degradation and necroptosis
for anticancer chemotherapy. Oncogene. 33:3004–3013. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lorin S, Pierron G, Ryan KM, Codogno P and
Djavaheri-Mergny M: Evidence for the interplay between JNK and
p53-DRAM signalling pathways in the regulation of autophagy.
Autophagy. 6:153–154. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wong CH, Iskandar KB, Yadav SK, Hirpara
JL, Loh T and Pervaiz S: Simultaneous induction of non-canonical
autophagy and apoptosis in cancer cells by ROS-dependent ERK and
JNK activation. PLoS One. 5:e99962010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ni Z, Wang B, Dai X, Ding W, Yang T, Li X,
Lewin S, Xu L, Lian J and He F: HCC cells with high levels of Bcl-2
are resistant to ABT-737 via activation of the ROS-JNK-autophagy
pathway. Free Radic Biol Med. 70:194–203. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bristol ML, Di X, Beckman MJ, Wilson EN,
Henderson SC, Maiti A, Fan Z and Gewirtz DA: Dual functions of
autophagy in the response of breast tumor cells to radiation:
Cytoprotective autophagy with radiation alone and cytotoxic
autophagy in radiosensitization by vitamin D3.
Autophagy. 8:739–753. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bo Q, Ma S, Han Q, Wang FE, Li X and Zhang
Y: Role of autophagy in photoreceptor cell survival and death. Crit
Rev Eukaryot Gene Expr. 25:23–32. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu J, Chang F, Li F, Fu H, Wang J, Zhang
S, Zhao J and Yin D: Palmitate promotes autophagy and apoptosis
through ROS-dependent JNK and p38 MAPK. Biochem Biophys Res Commun.
463:262–267. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li J, Wang F, Xia Y, Dai W, Chen K, Li S,
Liu T, Zheng Y, Wang J, Lu W, et al: Astaxanthin pretreatment
attenuates hepatic ischemia reperfusion-induced apoptosis and
autophagy via the ROS/MAPK pathway in mice. Mar Drugs.
13:3368–3387. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kyriakis JM and Avruch J: Mammalian MAPK
signal transduction pathways activated by stress and inflammation:
A 10-year update. Physiol Rev. 92:689–737. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cagnol S and Chambard JC: ERK and cell
death: Mechanisms of ERK-induced cell death - apoptosis, autophagy
and senescence. FEBS J. 277:2–21. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cui Q, Tashiro S, Onodera S, Minami M and
Ikejima T: Oridonin induced autophagy in human cervical carcinoma
HeLa cells through Ras, JNK, and P38 regulation. J Pharmacol Sci.
105:317–325. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Donadelli M, Dando I, Zaniboni T, Costanzo
C, Dalla Pozza E, Scupoli MT, Scarpa A, Zappavigna S, Marra M,
Abbruzzese A, et al: Gemcitabine/cannabinoid combination triggers
autophagy in pancreatic cancer cells through a ROS-mediated
mechanism. Cell Death Dis. 2:e1522011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang XL, Chen ML and Zhou SL: Fentanyl
inhibits proliferation and invasion of colorectal cancer via
β-catenin. Int J Clin Exp Pathol. 8:227–235. 2015.PubMed/NCBI
|
|
30
|
Echiburú-Chau C, Alfaro-Lira S, Brown N,
Salas CO, Cuellar M, Santander J, Ogalde JP and Rothhammer F: The
selective cytotoxicity elicited by phytochemical extract from
Senecio graveolens (Asteraceae) on breast cancer cells is enhanced
by hypoxia. Int J Oncol. 44:1357–1364. 2014.PubMed/NCBI
|
|
31
|
Thorburn A, Thamm DH and Gustafson DL:
Autophagy and cancer therapy. Mol Pharmacol. 85:830–838. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tan Q, Wang H, Hu Y, Hu M, Li X,
Aodengqimuge Ma Y, Wei C and Song L: Src/STAT3-dependent heme
oxygenase-1 induction mediates chemoresistance of breast cancer
cells to doxorubicin by promoting autophagy. Cancer Sci.
106:1023–1032. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vazquez-Martin A, Oliveras-Ferraros C and
Menendez JA: Autophagy facilitates the development of breast cancer
resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS
One. 4:e62512009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li J, Hou N, Faried A, Tsutsumi S and
Kuwano H: Inhibition of autophagy augments 5-fluorouracil
chemotherapy in human colon cancer in vitro and in vivo model. Eur
J Cancer. 46:1900–1909. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tang ZH, Zhang LL, Li T, Lu JH, Ma DL,
Leung CH, Chen XP, Jiang HL, Wang YT and Lu JJ: Glycyrrhetinic acid
induces cytoprotective autophagy via the inositol-requiring enzyme
1α-c-Jun N-terminal kinase cascade in non-small cell lung cancer
cells. Oncotarget. 6:43911–43926. 2015.PubMed/NCBI
|
|
36
|
Sui X, Kong N, Zhu M, Wang X, Lou F, Han W
and Pan H: Cotargeting EGFR and autophagy signaling: A novel
therapeutic strategy for non-small-cell lung cancer. Mol Clin
Oncol. 2:8–12. 2014.PubMed/NCBI
|
|
37
|
You L, Shou J, Deng D, Jiang L, Jing Z,
Yao J, Li H, Xie J, Wang Z, Pan Q, et al: Crizotinib induces
autophagy through inhibition of the STAT3 pathway in multiple lung
cancer cell lines. Oncotarget. 6:40268–40282. 2015.PubMed/NCBI
|
|
38
|
Kim AD, Kang KA, Kim HS, Kim DH, Choi YH,
Lee SJ, Kim HS and Hyun JW: A ginseng metabolite, compound K,
induces autophagy and apoptosis via generation of reactive oxygen
species and activation of JNK in human colon cancer cells. Cell
Death Dis. 4:e7502013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Martindale JL and Holbrook NJ: Cellular
response to oxidative stress: Signaling for suicide and survival. J
Cell Physiol. 192:1–15. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kawabe T: G2 checkpoint abrogators as
anticancer drugs. Mol Cancer Ther. 3:513–519. 2004.PubMed/NCBI
|
|
41
|
Nurse P: Universal control mechanism
regulating onset of M-phase. Nature. 344:503–508. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou H, Shen T, Shang C, Luo Y, Liu L, Yan
J, Li Y and Huang S: Ciclopirox induces autophagy through reactive
oxygen species-mediated activation of JNK signaling pathway.
Oncotarget. 5:10140–10150. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Livesey KM, Tang D, Zeh HJ and Lotze MT:
Autophagy inhibition in combination cancer treatment. Curr Opin
Investig Drugs. 10:1269–1279. 2009.PubMed/NCBI
|
|
44
|
Amaravadi RK, Lippincott-Schwartz J, Yin
XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT and White E:
Principles and current strategies for targeting autophagy for
cancer treatment. Clin Cancer Res. 17:654–666. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
O'Donovan TR, O'Sullivan GC and McKenna
SL: Induction of autophagy by drug-resistant esophageal cancer
cells promotes their survival and recovery following treatment with
chemotherapeutics. Autophagy. 7:509–524. 2011. View Article : Google Scholar : PubMed/NCBI
|