Introduction
Epigenetics is the study of inherited changes in
phenotype (appearance) or gene expression that are caused by
mechanisms other than changes in the underlying DNA sequence
(1,2). These changes may persist through
multiple cell divisions, even for the remainder of the cell's life,
and may also last for multiple generations. However, to reiterate,
there is no change in the underlying DNA sequence of the organism.
The most significant epigenetic mechanisms include DNA methylation,
histone modifications, and the processes mediated by the most
recently discovered class, the non-coding RNAs (3). DNA methylation is defined as the
selective methylation (addition of a methyl group) of cytosine
within a CpG dinucleotide, thereby forming 5-methylcytosine
(4,5). There are two types of DNA
methyltransferases (DNMTs). The first type, DNMT1, mainly plays a
role in the maintenance of methylation, can methylate the
hemi-methylated cytosine in double-stranded DNA molecules, and may
be involved in the methylation of the newly synthesized strand
during replication of duplex DNA (6). However, DNMT3a and DNMT3b play a major
role in de novo methylation, in which methylation can be
performed on double-stranded DNA that is not methylated. DNA
methylation is generally associated with gene silencing (7), and DNA demethylation is usually
connected with gene activation (8–10).
Histone modification is the process of modification
of histone proteins by enzymes, including post-translational
modifications, such as methylation, acetylation, phosphorylation,
and ubiquitination. These modifications constitute a rich ‘histone
code’ (11). Histone modifications
play a vital role in gene expression by modulating the degree of
tightness, or compaction, of chromatin (12). Methylation, which frequently occurs
on histones H3 (13) and H4 on
specific lysine (K) and arginine (A) residues, is one important
method for the modification of histone proteins. Histone lysine
methylation can lead to activation and can also lead to inhibition,
usually depending on the situation in which it is located. For
instance, H3K9, H3K27, and H4K20 are well known by scholars as
important ‘inactivation’ markers, i.e., repressive marks, because
of the relationship between these methylations and heterochromatin
formation. However, the methylation of H3K4 and H3K36 are
considered to be ‘activation’ marks (14,15).
Acetylation, which in most cases occurs in the N-terminal conserved
lysine residues, is also an important way to modify the histone
proteins, for example, acetylations of lysine residues 9 and 14 of
histone H3 and of lysines 5, 8, 12, and 16 of histone H4. Both
acetylations are associated with the activation or opening of the
chromatin. On the contrary, de-acetylation of the lysine residues
leads to chromatin compression and inactivation of gene
transcription. Different histone modifications can affect each
other and can have interactions with DNA methylations (16,17).
Non-encoding RNAs (non-coding RNAs) that are not
translated into proteins can be divided into housekeeping
non-coding RNAs and regulatory non-coding RNAs. RNA that has a
regulatory role is mainly divided into two categories based on size
(18,19): short chain non-coding RNAs
(including siRNAs, miRNAs, and piRNAs) and long non-coding RNA
(lncRNAs) (Table I). In recent
years, a large number of studies have shown that non-encoding RNAs
play a significant role in epigenetic modification and can regulate
expression at the level of the gene and the level of chromosome to
control cell differentiation (20–23)
(Fig. 1). Therefore, in this review
we will focus on the above four kinds of non-coding RNAs and their
regulatory roles in epigenetics.
 | Table I.Main non-coding RNAs in regulation of
epigenetics. |
Table I.
Main non-coding RNAs in regulation of
epigenetics.
| Name | Size | Source | Main functions |
|---|
| siRNA | 19–24 bp | Double stranded
RNA | Silent
transcription gene |
| miRNA | 19–24 bp | pri-miRNA | Silent
transcription gene |
| piRNA | 26–31 bp | Long single chain
precursor | Transposon
repression DNA methylation |
| lncRNA | >200 bp | Multiple ways | Genomic imprinting
X-chromosome inactivation |
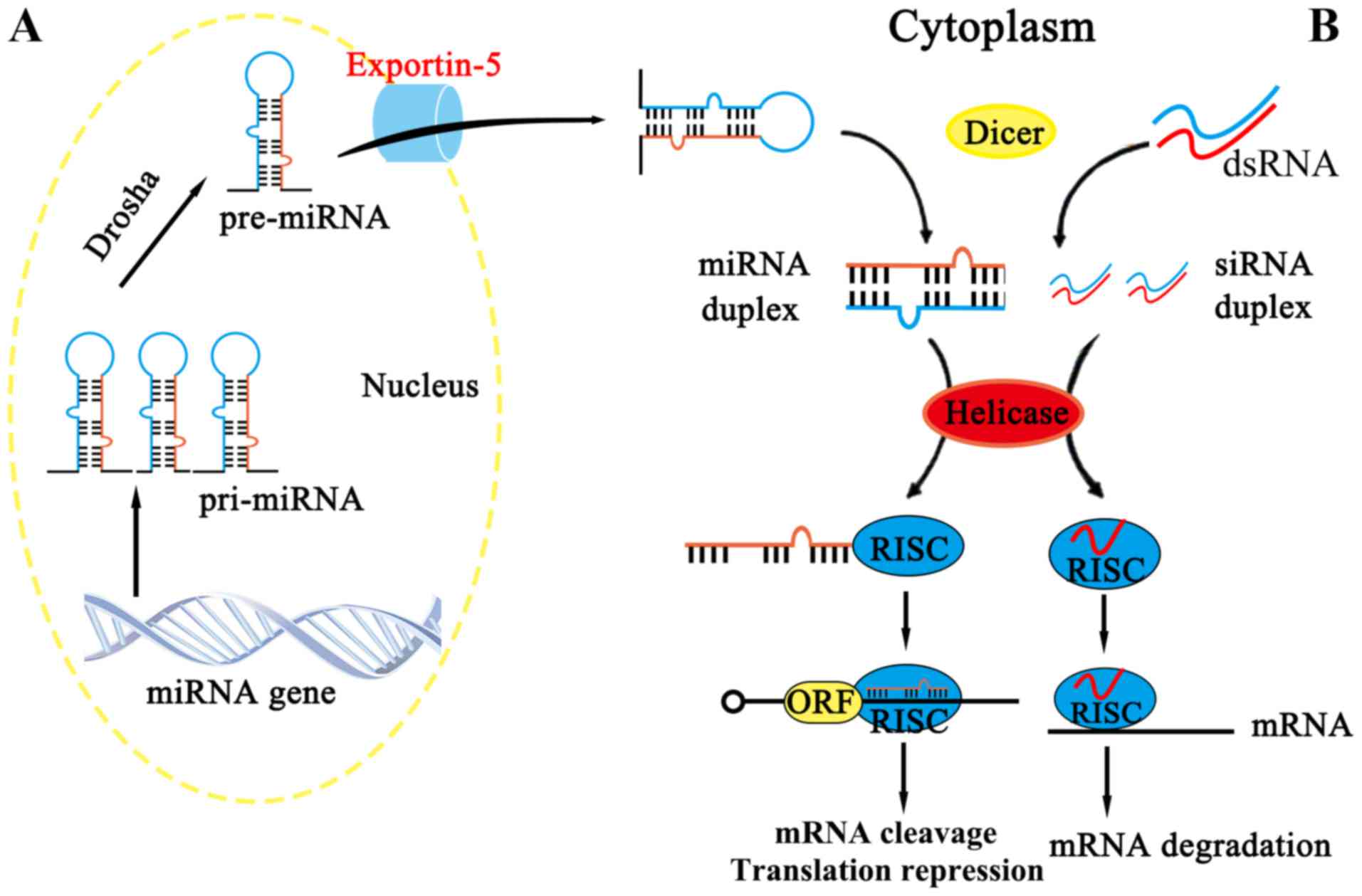
siRNA
siRNA is derived from long double-stranded RNA
molecules (including RNAs arising from virus replication,
transposon activity or gene transcription), which can be cut by the
Dicer enzyme into RNA fragments of 19–24 nt (nt: nucleotides), with
the resulting RNA fragments exercising their functions when loaded
onto Argonaute (AGO) proteins (24,25),
as summarized in Fig. 2B. (also
indicate Fig. 2A
appropriately)
Recent studies showed that siRNA can lead to
transcriptional gene silencing (TGS) in cells by means of DNA
methylation and histone modification in cells (26–28).
Zhou et al (29)
demonstrated that siRNA could silence EZH2 and then reverse
cisplatin-resistance in human non-small cell lung and gastric
cancer cells. EZH2, as a histone methyltransferase, can cause H3K27
methylation, and then the methylated H3K27 can serve as an anchor
point for CpG methylation, leading to the formation of silent
chromatin, and ultimately, to TGS (30). Chromatin immunoprecipitation
experiments showed that the binding of DNMTs to a given gene
inhibited by EZH2 depended on the presence of EZH2. Bisulfite
sequencing results also proved that EZH2 was required for the
methylation of an EZH2-repressed target promoter, suggesting that
EZH2 participated in DNA methylation by way of recruiting a DNA
methyltransferase.
Several teams have demonstrated that depletion of
Dicer, Argonaute, and RNA-dependent RNA polymerase homolog Rdp1
(the key components of the RNA interference silencing complex,
RISC) can cause the aberrant accumulation of long non-coding RNAs,
resulting in the loss of H3K9me, thus impairing centromere function
(31,32). siRNA-mediated epigenetic regulation
is also found in Arabidopsis (33),
in which case AGO4, a specific argonaute family protein, is
required for siRNA accumulation and DNA and histone methylation
(34). Consistent with these
results, H3K9me-marked pericentric heterochromatin is maintained by
an RNA component (35). Moreover,
with the development of sequencing technology, our efforts bring to
light the forceful correlation between H3K9 methylation and
repetitive elements (36), which
occupy two-thirds of the human genome. These outstanding findings
bring us an intriguing possibility that the RNA interference
pathways may have impressive roles in maintenance and regulation of
the epigenome. Deep mechanistic research of the siRNAs involved in
this process may contribute to helping us accurately understand the
inheritance of epigenetic regulation.
miRNA
miRNAs are single-stranded RNAs of approximately
19–24 nt, of which 50% are located in chromosomal regions that are
prone to structural changes (37).
Originally, it was thought that there were two main points of
difference between siRNA and miRNA as classes of regulatory RNAs.
One is that miRNA is endogenous and is the expression product of
the biological gene but that siRNA is exogenous, originating from
viral infection, the point of gene transfer, or the gene target.
The other point of difference is that miRNA consists of incomplete
hairpin-shaped double stranded RNA, which is processed by Drosha
and Dicer, whereas siRNA is the product of a fully complementary,
long double-stranded RNA, which is processed by Dicer (38). In spite of these differences, it is
speculated that miRNA and siRNA have a similar mechanism of action
in mediating transcriptional gene silencing because of the close
relationship between miRNA and siRNA, e.g., the size of the two
fragments are similar (Fig. 2).
Recently, in the human genome, almost 1,800 putative miRNAs have
been identified, and the number of miRNAs is still increasing
rapidly due to the development of high-throughput sequencing
technologies (39–41).
The current model is that the regulatory mechanism
of miRNA reflects the degree of complementarity between the
specific loading protein AGO, a given miRNA, and the target mRNA
(42). Usually only a very small
number of miRNAs are almost completely complementary with their
mRNA targets; in this case, a targeted mRNA can be directly cleaved
and degraded. However, the overwhelming number of miRNAs and their
target mRNAs are only partially complementary, generally for 6–7
nucleotides, which are located in the 5′ end of the 2 to 7
nucleotide stretch called the ‘seed region’. The ‘seed region’ is
the most basic and decisive factor in the selection of the target
because of the seed region's important role in the binding of the
target. At the same time, many studies suggest that a given miRNA
can regulate even hundreds of different genes (42–44).
Through the study of more than 13,000 human genes, Lewis et
al (44) made a further
speculation: that histone methyltransferases, methyl CpG binding
proteins, chromatin domain proteins, and histone deacetylases are
potential targets for miRNAs.
Although miRNAs that directly participate in
epigenetic regulation have not been reported in mammalian cells,
several scholars have found that aberrant expression of miRNAs can
change the whole DNA or chromatin state by restricting chromatin
remodeling enzyme activity (45,46).
Existing studies have shown that miRNAs can induce chromatin
remodeling through the regulation of histone modification. Some
scholars have reported that histone deacetylase 4 (HDAC4) is a
specific target for miR-140 in mouse embryonic cartilage
tissue. These two examples suggest that miRNA may be involved in
TGS via the modification of histone proteins (47). Kim et al (48) found that miR-320, one of the
most conserved miRNAs, can recruit AGO1 to the POLR3D promoter.
Moreover, EZH2 and H3K27 trimethylation are involved in TGS. This
further confirms that miRNAs can cause TGS.
Considering the large number of miRNAs that base
pair with transcribed RNAs, it is not surprising that the miRNA
class of noncoding RNAs may directly take part in epigenetic
control of gene expression. Two current studies demonstrated the
existence of miRNA-mediated DNA methylation changes in plants
(49,50). Other recent studies have shown that
miRNA can affect DNA methylation through the regulation of DNA
methylases. Fabbri et al (51) found that the expression of the
miR-29 family (miR-29a, miR-29b and miR-29c
included) is downregulated but that DNMT3a and DNMT3b are highly
expressed in non-small cell carcinoma. The complementarity between
the miR-29 family and the 3′UTRs of DNMT3a and 3b suggests
that mRNAs of DNMTs are the target of the miR-29 family.
Benetti et al (52) and Sinkkonen et al (53) both demonstrated that the
downregulation of DNMT3a and 3b activity depended on the
miR-29 family in mouse embryonic stem cells lacking Dicer.
In this case, the main mechanism for the absence of DNA methylation
was that retinoblastoma-like protein 2 (Rbl2) inhibited the
activity of DNMT3a and DNMT3b. Rbl2 can be inhibited by the
miRNA-29 family, but the miRNA-29 family was downregulated
in the absence of Dicer activity. These observations also raise the
possibility that miR-29 can inhibit tumors by enhancing the
expression of tumor suppressor genes. Work by Gonzalez et al
(54) shows that tumor miRNAs
(miR17-5p and miR-20a) have the ability to induce the
formation of heterochromatin, providing another example of the
existence of new mechanisms of chromatin remodeling and gene
transcription mediated by miRNA regulation. The above-mentioned
kinds of research will help us to clarify the mechanisms of
tumorigenesis and broaden our perspective of what may constitute
promising new targets for cancer therapy.
piRNA
piRNAs are a class of RNA molecules that are
approximately 26–31 nt in length. The name, piRNA (Piwi-interacting
RNA), reflects the fact that piRNAs bind to Piwi proteins under
physiological conditions (55,56).
Reflecting its role as an epigenetic regulatory factor, the Piwi
protein silences the homeobox gene by binding to genomic PcG
response elements together with PcGs (polycomb group proteins).
Thus, it has been speculated that the piRNAs that are associated
with the Piwi protein should also have important roles in
epigenetic regulation (57).
Relevant research shows that piRNAs can be divided
into two sub-clusters (58,59). One is the pachytene piRNA cluster,
which mainly occurs during meiosis and continues to be expressed up
through the haploid spermatid stage. The other is the pre-pachytene
piRNA cluster, which appears mainly in premeiotic germ cells.
Although pre-pachytene piRNAs have the molecular characteristics of
the pachytene piRNA cluster, pre-pachytene piRNAs come from a
completely different cluster and contain repetitive sequence
elements.
The biosynthesis and mechanism(s) of action of the
piRNA class of regulators are still unclear at present. Each piRNA
comes from a single chain precursor, rather than from a
double-stranded RNA, implying that the functions of piRNAs do not
depend on Dicer enzyme activity in cells. Another difference is the
feature of strand asymmetry in the pachytene piRNA cluster
(60). Consequently, there are two
emerging hypotheses: one is that each piRNA is generated from a
long single-stranded molecule, and the other hypothesis is that the
piRNA may serve as a primary transcript. Aravin et al
(58) proposed a model of piRNA
self-amplification in cells based on experimental evidence in rats.
It is different from the amplification model in Drosophila,
in which the gene cluster is not posited to be the main source of
the primary piRNAs. Instead, the primary piRNAs are thought instead
to be produced by transposon mRNAs in amplification cycles.
In contrast to gene-silencing mediated by small
RNAs, piRNAs were originally found to have the ability to promote
euchromatic histone modifications in Drosophila melanogaster
(61). Moreover, a current
genome-wide survey indicates that piRNAs and Piwi are strongly
associated with chromatin regulation and that piRNAs efficiently
recruit the HP1a protein to specific genomic loci in order to
repress RNA polymerase II transcription (62). Studies have shown that the DNA
methyltransferase family (DNMT3a, DNMT3b, and DNMT3L) plays a major
role in transposon methylation. Thus, the catalytic activities of
DNMT3a and DNMT3b are very important in germ cells and somatic
cells, while DNMT3L is a key promoter of methylation in germ cells
(63). Experiments show that two
Piwi proteins, MILI and MIWI2, are necessary for silencing
LINE-1 and IAP transposons in the testis and the
deletion of MILI or MIWI2 reduces the level of transposon
methylation; MIWI2 is always located in the nucleus during the
critical period of methylation; small RNA sequence analysis showed
that MILI and MIWI2 both have a role upstream of DNMT3L and then
act on DNMT3a and DNMT3b. The above experimental results confirmed
that the complex of Piwi protein/piRNA can mediate the methylation
of transposons and that piRNA is a specific determinant of DNA
methylation in germ cells (64). A
similar regulatory mode may also exist in other kinds of somatic
tissues, but this notion still remains unclear.
lncRNA
LncRNAs represent another class of non-coding
regulatory RNAs. LncRNAs are generally >200 nt in length, are
located in the nucleus or cytoplasm, and rarely encode proteins
(65,66). LncRNAs usually can be divided into
five categories: Sense, Antisense, Bidirectional, Intronic, and
Intergenic lncRNA (19). However,
these five categories mainly involve only four archetypal lncRNA
mechanisms for regulating gene expression: Signals, Decoys, Guides,
and Scaffolds (67).
There are many different sources of lncRNAs
(19): (I) Arising by the
disruption of the translational reading frame of a protein-encoding
gene (II); Resulting from chromosomal reorganization, for example,
by the joining of two non-transcribed DNA regions in such a fashion
as to promote transcription of the merged, non-coding sequences;
(III) Produced by replication of a non-coding gene by
retrotransposition; (IV) Generation of a non-encoding RNA
containing adjacent repeats via a partial tandem duplication
mechanism; and (V) Arising from the insertion of a transposable
element(s) into a gene in such a way as to produce a functional,
transcribed non-encoding RNA. Studies have indicated that lncRNAs
play a similar role in the regulation of gene expression in spite
of fact that there is no common, shared mechanism by which all the
numerous lncRNAs originated (65).
Studies of genomic imprinting and X chromosome
inactivation were the first to reveal a role for lncRNAs in
epigenetic regulation, identifying roles for two lncRNAs,
H19 RNA and Xist RNA, respectively (68). H19 is a genomic imprinting
lncRNA that can be transported to the cytoplasm, is spliced and
polyadenylated, and achieves a high cytosolic concentration. The
function of H19 is still not clear, even if it is the first gene to
be associated closely with genomic imprinting. Recently, H19
RNA was found to contain a precursor of miR-675 in human and
rat cells (69), indicating that
H19 RNA may also regulate gene expression through a
miRNA-based mechanism. Genomic imprinting is associated not only
with the H19 gene cluster, but also with Kcnq1ot1,
Air, and Nespas lncRNAs (70,71).
Experimental results of promoter deletion in Kcnq1ot1 and
Air suggest that lncRNA transcripts are necessary for
silencing Kcnq1ot1- and lgf2r/Air-imprinted genes and
that they are essential for the methylation of H3K27 and H3K9 and
for DNA methylation of some genes (72).
Xist RNA, which is a 17 kb long non-coding
RNA, is very important for X chromosome inactivation. Xist
does not travel to the cytoplasm; instead, this RNA interacts
physically with the X chromosome that is about to be inactivated by
Xist. Xist RNA silences genes in cis by
‘coating’ the surface of the inactivated X chromosome (73). Recent research by Zhao et al
(74) shows that RepA (a 1.6
kb fragment of Xist RNA) can induce H3K27 trimethylation by
recruiting the Polycomb repressive complex 2 (PRC2) to the
inactivated X. In addition, RepA has a crucial importance not only
for methylation of H3K27 but also for expression of Xist
RNA.
Recent studies show that some gene clusters and
lncRNAs may have dual roles, participating both in the process of X
chromosome inactivation and in the process of genomic imprinting.
In this context, gene silencing may involve both cis and
trans mechanisms. For instance, the HOTAIR RNA (Hox
transcript antisense RNA) originates from the HOXC cluster
and can silence gene transcription in a region of up to 40 kb in
HOXD; the main mechanism is through recruitment of Polycomb
repressive complex PRC2 to HOXD through HOTAIR, which
then triggers the formation of heterochromatin and TGS via
trimethylation of H3K9 (75–77).
Other studies (78)
indicate that there is another mechanism of X chromosome
inactivation. According to this mechanism, Xist and
Tsix can form an RNA dimer that is processed by Dicer to
yield siRNAs. These different approaches may be able to coordinate
the roles of lncRNAs and small RNAs in chromatin remodeling,
indicating that there is a more complex and interactive network of
regulation by non-coding RNAs.
Conclusion and prospective
With the attention on non-coding RNAs in the
etiology of diseases, the noncoding RNA has become a ‘hot’ issue in
modern genetics research, especially as a new mechanism of
epigenetic regulation. However, thus far, scientists have only a
very limited understanding of the mechanisms by which non-coding
RNAs regulate gene expression.
At first, piRNAs, miRNAs and siRNAs were thought to
function independently, and this supposition was reinforced by the
obvious differences between them. However, in recent years, the
roles of these pathways have begun to blur, and some of these
pathways appear to interact, thereby constituting a regulatory
network. So how have living organisms adapted to these multiple
regulatory mechanisms? Before we can solve this problem, we must
first understand the details of the non-coding RNAs themselves. At
present, a key problem is the respective roles and
interrelationships between this large, complex RNA regulatory
network and protein-based regulatory mechanisms. Analysis of this
regulatory network for non-coding RNAs is very difficult work, so
we need to systematically find and analyze non-coding RNAs. This
will require gradual improvements and new methods in gene and
genome scanning technologies to reveal all the information and
functions of non-coding RNAs. The ultimate goal is to clarify the
detailed mechanisms of regulation by the non-coding RNAs and their
interactions with normal cells and disease states.
Acknowledgements
This work was supported by the National High
Technology Research and Development Program 863 (2014AA021102),
China National Natural Scientific Fund (81372703, 81572932).
References
|
1
|
Egger G, Liang G, Aparicio A and Jones PA:
Epigenetics in human disease and prospects for epigenetic therapy.
Nature. 429:457–463. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bird A: Perceptions of epigenetics.
Nature. 447:396–398. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Peschansky VJ and Wahlestedt C: Non-coding
RNAs as direct and indirect modulators of epigenetic regulation.
Epigenetics. 9:3–12. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Goll MG and Bestor TH: Eukaryotic cytosine
methyltransferases. Annu Rev Biochem. 74:481–514. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bird A: DNA methylation patterns and
epigenetic memory. Genes Dev. 16:6–21. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weber M, Hellmann I, Stadler MB, Ramos L,
Pääbo S, Rebhan M and Schübeler D: Distribution, silencing
potential and evolutionary impact of promoter DNA methylation in
the human genome. Nat Genet. 39:457–466. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Okano M, Bell DW, Haber DA and Li E: DNA
methyltransferases Dnmt3a and Dnmt3b are essential for de novo
methylation and mammalian development. Cell. 99:247–257. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Berger SL: The complex language of
chromatin regulation during transcription. Nature. 447:407–412.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bernstein BE, Meissner A and Lander ES:
The mammalian epigenome. Cell. 128:669–681. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goldberg AD, Allis CD and Bernstein E:
Epigenetics: A landscape takes shape. Cell. 128:635–638. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Harr JC, Gonzalez-Sandoval A and Gasser
SM: Histones and histone modifications in perinuclear chromatin
anchoring: From yeast to man. EMBO Rep. 17:139–155. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jenuwein T and Allis CD: Translating the
histone code. Science. 293:1074–1080. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lu C, Jain SU, Hoelper D, Bechet D, Molden
RC, Ran L, Murphy D, Venneti S, Hameed M, Pawel BR, et al: Histone
H3K36 mutations promote sarcomagenesis through altered histone
methylation landscape. Science. 352:844–849. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mellor J, Dudek P and Clynes D: A glimpse
into the epigenetic landscape of gene regulation. Curr Opin Genet
Dev. 18:116–122. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Grewal SI and Elgin SC: Transcription and
RNA interference in the formation of heterochromatin. Nature.
447:399–406. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reik W: Stability and flexibility of
epigenetic gene regulation in mammalian development. Nature.
447:425–432. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tchurikov NA: Molecular mechanisms of
epigenetics. Biochemistry (Mosc). 70:406–423. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zaratiegui M, Irvine DV and Martienssen
RA: Noncoding RNAs and gene silencing. Cell. 128:763–776. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ponting CP, Oliver PL and Reik W:
Evolution and functions of long noncoding RNAs. Cell. 136:629–641.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Costa FF: Non-coding RNAs, epigenetics and
complexity. Gene. 410:9–17. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Amaral PP, Dinger ME, Mercer TR and
Mattick JS: The eukaryotic genome as an RNA machine. Science.
319:1787–1789. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ghildiyal M and Zamore PD: Small silencing
RNAs: An expanding universe. Nat Rev Genet. 10:94–108. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu H: Epigenetics: Advances of non-coding
RNAs regulation in mammalian cells. Yi Chuan. 31:1077–1086.
2009.(In Chinese). View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Moazed D: Small RNAs in transcriptional
gene silencing and genome defence. Nature. 457:413–420. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jinek M and Doudna JA: A three-dimensional
view of the molecular machinery of RNA interference. Nature.
457:405–412. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Morris KV, Chan SW, Jacobsen SE and Looney
DJ: Small interfering RNA-induced transcriptional gene silencing in
human cells. Science. 305:1289–1292. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kawasaki H and Taira K: Induction of DNA
methylation and gene silencing by short interfering RNAs in human
cells. Nature. 431:211–217. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bayne EH and Allshire RC: RNA-directed
transcriptional gene silencing in mammals. Trends Genet.
21:370–373. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou W, Wang J, Man WY, Zhang QW and Xu
WG: siRNA silencing EZH2 reverses cisplatin-resistance of human
non-small cell lung and gastric cancer cells. Asian Pac J Cancer
Prev. 16:2425–2430. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Viré E, Brenner C, Deplus R, Blanchon L,
Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden
JM, et al: The Polycomb group protein EZH2 directly controls DNA
methylation. Nature. 439:871–874. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hall IM, Shankaranarayana GD, Noma K,
Ayoub N, Cohen A and Grewal SI: Establishment and maintenance of a
heterochromatin domain. Science. 297:2232–2237. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Volpe TA, Kidner C, Hall IM, Teng G,
Grewal SI and Martienssen RA: Regulation of heterochromatic
silencing and histone H3 lysine-9 methylation by RNAi. Science.
297:1833–1837. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen S, He H and Deng X: Allele-specific
DNA methylation analyses associated with siRNAs in Arabidopsis
hybrids. Sci China Life Sci. 57:519–525. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zilberman D, Cao X and Jacobsen SE:
ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and
histone methylation. Science. 299:716–719. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Maison C, Bailly D, Peters AH, Quivy JP,
Roche D, Taddei A, Lachner M, Jenuwein T and Almouzni G:
Higher-order structure in pericentric heterochromatin involves a
distinct pattern of histone modification and an RNA component. Nat
Genet. 30:329–334. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lippman Z, Gendrel AV, Black M, Vaughn MW,
Dedhia N, McCombie WR, Lavine K, Mittal V, May B, Kasschau KD, et
al: Role of transposable elements in heterochromatin and epigenetic
control. Nature. 430:471–476. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ruvkun G: Molecular biology. Glimpses of a
tiny RNA world. Science. 294:797–799. 2001.
|
|
38
|
Carthew RW and Sontheimer EJ: Origins and
mechanisms of miRNAs and siRNAs. Cell. 136:642–655. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Luo M, Hao L, Hu F, Dong Y, Gou L, Zhang
W, Wang X, Zhao Y, Jia M, Hu S, et al: MicroRNA profiles and
potential regulatory pattern during the early stage of
spermatogenesis in mice. Sci China Life Sci. 58:442–450. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kozomara A and Griffiths-Jones S: miRBase:
Annotating high confidence microRNAs using deep sequencing data.
Nucleic Acids Res. 42(D1): D68–D73. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Griffiths-Jones S, Grocock RJ, van Dongen
S, Bateman A and Enright AJ: miRBase: microRNA sequences, targets
and gene nomenclature. Nucleic Acids Res. 34:D140–D144. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rajewsky N: microRNA target predictions in
animals. Nat Genet. 38:(Suppl). S8–S13. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Denis H, Ndlovu MN and Fuks F: Regulation
of mammalian DNA methyltransferases: A route to new mechanisms.
EMBO Rep. 12:647–656. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yuan JH, Yang F, Chen BF, Lu Z, Huo XS,
Zhou WP, Wang F and Sun SH: The histone deacetylase
4/SP1/microRNA-200a regulatory network contributes to aberrant
histone acetylation in hepatocellular carcinoma. Hepatology.
54:2025–2035. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tuddenham L, Wheeler G, Ntounia-Fousara S,
Waters J, Hajihosseini MK, Clark I and Dalmay T: The cartilage
specific microRNA-140 targets histone deacetylase 4 in mouse cells.
FEBS Lett. 580:4214–4217. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kim DH, Saetrom P, Snøve O Jr and Rossi
JJ: MicroRNA-directed transcriptional gene silencing in mammalian
cells. Proc Natl Acad Sci USA. 105:16230–16235. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Khraiwesh B, Arif MA, Seumel GI, Ossowski
S, Weigel D, Reski R and Frank W: Transcriptional control of gene
expression by microRNAs. Cell. 140:111–122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wu L, Zhou H, Zhang Q, Zhang J, Ni F, Liu
C and Qi Y: DNA methylation mediated by a microRNA pathway. Mol
Cell. 38:465–475. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fabbri M, Garzon R, Cimmino A, Liu Z,
Zanesi N, Callegari E, Liu S, Alder H, Costinean S,
Fernandez-Cymering C, et al: MicroRNA-29 family reverts aberrant
methylation in lung cancer by targeting DNA methyltransferases 3A
and 3B. Proc Natl Acad Sci USA. 104:15805–15810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Benetti R, Gonzalo S, Jaco I, Muñoz P,
Gonzalez S, Schoeftner S, Murchison E, Andl T, Chen T, Klatt P, et
al: A mammalian microRNA cluster controls DNA methylation and
telomere recombination via Rbl2-dependent regulation of DNA
methyltransferases. Nat Struct Mol Biol. 15:9982008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sinkkonen L, Hugenschmidt T, Berninger P,
Gaidatzis D, Mohn F, Artus-Revel CG, Zavolan M, Svoboda P and
Filipowicz W: MicroRNAs control de novo DNA methylation through
regulation of transcriptional repressors in mouse embryonic stem
cells. Nat Struct Mol Biol. 15:259–267. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gonzalez S, Pisano DG and Serrano M:
Mechanistic principles of chromatin remodeling guided by siRNAs and
miRNAs. Cell Cycle. 7:2601–2608. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lau NC, Seto AG, Kim J, Kuramochi-Miyagawa
S, Nakano T, Bartel DP and Kingston RE: Characterization of the
piRNA complex from rat testes. Science. 313:363–367. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Grivna ST, Beyret E, Wang Z and Lin H: A
novel class of small RNAs in mouse spermatogenic cells. Genes Dev.
20:1709–1714. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lin H: piRNAs in the germ line. Science.
316:3972007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Aravin AA, Sachidanandam R, Bourc'his D,
Schaefer C, Pezic D, Toth KF, Bestor T and Hannon GJ: A piRNA
pathway primed by individual transposons is linked to de novo DNA
methylation in mice. Mol Cell. 31:785–799. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Aravin AA, Hannon GJ and Brennecke J: The
Piwi-piRNA pathway provides an adaptive defense in the transposon
arms race. Science. 318:761–764. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Houwing S, Kamminga LM, Berezikov E,
Cronembold D, Girard A, van den Elst H, Filippov DV, Blaser H, Raz
E, Moens CB, et al: A role for Piwi and piRNAs in germ cell
maintenance and transposon silencing in Zebrafish. Cell. 129:69–82.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yin H and Lin H: An epigenetic activation
role of Piwi and a Piwi-associated piRNA in Drosophila
melanogaster. Nature. 450:304–308. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Huang XA, Yin H, Sweeney S, Raha D, Snyder
M and Lin H: A major epigenetic programming mechanism guided by
piRNAs. Dev Cell. 24:502–516. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Bourc'his D and Bestor TH: Meiotic
catastrophe and retrotransposon reactivation in male germ cells
lacking Dnmt3L. Nature. 431:96–99. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kuramochi-Miyagawa S, Watanabe T, Gotoh K,
Totoki Y, Toyoda A, Ikawa M, Asada N, Kojima K, Yamaguchi Y, Ijiri
TW, et al: DNA methylation of retrotransposon genes is regulated by
Piwi family members MILI and MIWI2 in murine fetal testes. Genes
Dev. 22:908–917. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cao X, Yeo G, Muotri AR, Kuwabara T and
Gage FH: Noncoding RNAs in the mammalian central nervous system.
Annu Rev Neurosci. 29:77–103. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: Insights into functions. Nat Rev Genet.
10:155–159. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wang KC and Chang HY: Molecular mechanisms
of long noncoding RNAs. Mol Cell. 43:904–914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Yang PK and Kuroda MI: Noncoding RNAs and
intranuclear positioning in monoallelic gene expression. Cell.
128:777–786. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Cai X and Cullen BR: The imprinted H19
noncoding RNA is a primary microRNA precursor. RNA. 13:313–316.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wu HA and Bernstein E: Partners in
imprinting: Noncoding RNA and polycomb group proteins. Dev Cell.
15:637–638. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Pandey RR, Mondal T, Mohammad F, Enroth S,
Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D and Kanduri C:
Kcnq1ot1 antisense noncoding RNA mediates lineage-specific
transcriptional silencing through chromatin-level regulation. Mol
Cell. 32:232–246. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Regha K, Sloane MA, Huang R, Pauler FM,
Warczok KE, Melikant B, Radolf M, Martens JH, Schotta G, Jenuwein
T, et al: Active and repressive chromatin are interspersed without
spreading in an imprinted gene cluster in the mammalian genome. Mol
Cell. 27:353–366. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Clemson CM, McNeil JA, Willard HF and
Lawrence JB: XIST RNA paints the inactive X chromosome at
interphase: Evidence for a novel RNA involved in nuclear/chromosome
structure. J Cell Biol. 132:259–275. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhao J, Sun BK, Erwin JA, Song JJ and Lee
JT: Polycomb proteins targeted by a short repeat RNA to the mouse X
chromosome. Science. 322:750–756. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhou X, Ren Y, Zhang J, Zhang C, Zhang K,
Han L, Kong L, Wei J, Chen L, Yang J, et al: HOTAIR is a
therapeutic target in glioblastoma. Oncotarget. 6:8353–8365. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhang K, Sun X, Zhou X, Han L, Chen L, Shi
Z, Zhang A, Ye M, Wang Q, Liu C, et al: Long non-coding RNA HOTAIR
promotes glioblastoma cell cycle progression in an EZH2 dependent
manner. Oncotarget. 6:537–546. 2015.PubMed/NCBI
|
|
77
|
Rinn JL, Kertesz M, Wang JK, Squazzo SL,
Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et
al: Functional demarcation of active and silent chromatin domains
in human HOX loci by noncoding RNAs. Cell. 129:1311–1323. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Ogawa Y, Sun BK and Lee JT: Intersection
of the RNA interference and X-inactivation pathways. Science.
320:1336–1341. 2008. View Article : Google Scholar : PubMed/NCBI
|