Introduction
Prostate cancer is the most prevalent cancer among
men in the United States, with an estimated annual incidence of
180,890 new cases and 26,120 deaths as reported in 2016 (1). Likewise, the incidence and mortality
rate of prostate cancer have sharply increased in China over the
past few decades (2). Androgen
deprivation is the mainstay of therapy for advanced prostate
cancer. Unfortunately, most androgen-dependent prostate cancer
patients progressed to a castration-resistant state after a median
time of 18–24 months (3).
Therefore, effective new therapies are needed to improve clinical
care of prostate cancer patients.
It is known that exogenous small activating RNAs
(saRNAs) are able to stimulate gene expression at the
transcriptional level by targeting specific promoter regions,
termed as RNA activation (RNAa) (4). Besides, the endogenous microRNAs
(miRNAs) have also been implicated in induction of gene expression
via the same mechanism (5). As a
class of small non-coding RNA molecules, miRNAs play important
roles in prostate cancer proliferation and apoptosis (6,7). In
addition, downregulation of specific miRNAs in tumor tissues can
lead to aberrant expression of their targeted genes, which are
often involved in prostate cancer initiation, development and
progression (8). Hereby,
restoration of the specific miRNA in prostate tumors could be
considered as a promising therapeutic strategy for prostate
cancer.
Increased proliferative activity, often caused by
impaired regulation of cell cycle, is the main characteristic of
cancer cells. The CDKN1A gene is a broad-acting
cyclin-dependent kinase inhibitor and encodes the p21 protein
(9). Upregulated expression of
CDKN1A by adenoviral vectors has been shown to inhibit
prostate cancer growth both in vitro and in vivo
(10). Thus, the CDKN1A gene
is an ideal target for gain-of-function manipulation to suppress
prostate cancer cells growth.
In the present study, a new miRNA (miR-3619-5p) was
selected and transfected into prostate cancer cell lines DU145 and
PC3. Our findings showed that miR-3619-5p had potent ability to
induce CDKN1A expression by directly targeting its promoter
region, and inhibiting prostate cancer cell proliferation.
Materials and methods
Tissue samples and cell culture
Prostate cancer tumor specimens and tumor normal
adjacent tissues were obtained from Tongji Hospital, Tongji Medical
College, Huazhong University of Science and Technology (Wuhan,
China) after informed consent and ethics committee approval. After
surgical removal, fresh samples were fixed in 4% paraformaldehyde
immediately and then paraffin-embedded for
immunohistochemistry.
Human prostate cancer cell lines DU145, PC3 and
LNcaP (ATCC) were maintained in RPMI-1640 medium (Hyclone, Logan,
UT, USA) and normal prostate epithelial cells RWPE-1 (ATCC) were
maintained in Keratinocyte-SFM (Gibco, Grand Island, NY, USA),
respectively. The medium was supplemented with 10% fetal bovine
serum (FBS) (Gibco) and cells were cultured in a humidified
atmosphere of 5% CO2 at 37°C.
Prediction and transfection of Target
miRNAs
A candidate miRNA complementary to CDKN1A
promoter was predicted by the miRanda program on the Linux
operating system (11). In
addition, the selected miRNA possessed no putative target sites in
3′ or 5′ untranslated regions (UTR) of the p21 transcript.
RNAs and 5′- or 3′-biotin covalently linked RNAs
were manufactured by Shanghai GenePharma Co., Ltd. (Shanghai,
China). A dsControl lacking significant homology to all known human
sequences was used as a non-specific control and dsP21-322 was used
as the positive control (Table I).
Cells were plated in growth medium at a density of 50–60%. The
transfection was carried out by using Lipofectamine RNAiMax
(Invitrogen, Carlsbad, CA, USA) 24 h later according to the
manufacturer's protocol. The final concentration of RNAs was 50 nM
for each well.
 | Table I.Sequences for RNAs used in present
study. |
Table I.
Sequences for RNAs used in present
study.
| Name | Sequences
(5′-3′) |
|---|
| dsControl
Sense |
ACUACUGAGUGACAGUAGA[dT][dT] |
| dsControl
Antisense |
UCUACUGUCACUCAGUAGU[dT][dT] |
| dsP21-322
Sense |
CCAACUCAUUCUCCAAGUA[dT][dT] |
| dsP21-322
Antisense |
UACUUGGAGAAUGAGUUGG[dT][dT] |
| siP21 Sense |
CUUCGACUUUGUCACCGAG[dT][dT] |
| siP21
Antisense |
CUCGGUGACAAAGUCGAAG[dT][dT] |
RNA extraction, real-time PCR and
miRNA analysis
Total RNA from cells was extracted by using TRIzol
reagent (Invitrogen). Total RNA (500 ng) was reverse transcribed
and real-time quantitative PCR was performed on the Mx3000P system
(Stratagene, Santa Clara, CA, USA) according to the manufacturer's
instructions. Mature miRNA were reverse transcribed and then
quantitated by using All-in-one™ miRNA qPCR kit (GeneCopoeia,
Guangdong, China). GAPDH and U6 snRNA were used as internal
controls. The primers were provided by Invitrogen or GeneCopoeia.
Some primer sequences used are available in Table II.
| Table II.Primers used in this study. |
Table II.
Primers used in this study.
| Name | Sequences
(5′-3′) | Assay used for |
|---|
| p21 | F:
GCCCAGTGGACAGCGAGCAG | PCR |
|
| R:
GCCGGCGTTTGGAGTGGTAGA | PCR |
| GAPDH | F:
TCCCATCACCATCTTCCA | PCR |
|
| R:
CATCACGCCACAGTTTCC | PCR |
| p21-1309/−1150 | F:
GAGCAGCCTGAGATGTCAGTAATT | ChIP |
|
| R:
TCCCCTGGACTTCACCTTTG | ChIP |
| p21-272/−27 | F:
GGAAATGTGTCCAGCGCACC | ChIP |
|
| R:
AGCGCGGCCCTGATATACAACC | ChIP |
| Cyclin D1 | F:
GCTGCGAAGTGGAAACCATC | PCR |
|
| R:
CCTCCTTCTGCACACATTTGAA | PCR |
| CDK4 | F:
ATGGCTACCTCTCGATATGAGC | PCR |
|
| R:
CATTGGGGACTCTCACACTCT | PCR |
| CDK6 | F:
TCTTCATTCACACCGAGTAGTGC | PCR |
|
| R:
TGAGGTTAGAGCCATCTGGAAA | PCR |
Immunohistochemistry (IHC)
Formalin-fixed, paraffin-embedded tissue sections (5
µm) were deparaffinized in xylene and rehydrated with gradient
concentrations of ethanol. The tissue sections were stained with
specific antibodies against p21 (Cell Signaling Technology,
Danvers, MA, USA) (1:400). The sections incubated with secondary
antibodies in the absence of primary antibodies were used as
negative control. Hematoxylin was used for counterstaining. Slides
were viewed and photographed under a light microscope.
Fluorescence in situ hybridization
(FISH)
FISH was determined as described previously
(12). Briefly, a 5′ and
3′-digoxin-conjugated miR-3619-5p probe (Exiqon, Vedbaek, Denmark)
was used for miRNA in situ hybridization of miR-3619-5p.
Sections were first blocked with prehybridization buffer for 20 min
at 22–25°C below the predicted probe melting temperature in a
humidified chamber, and then the miR-3619-5p probe (10 ng/ml) in
the hybridization buffer was hybridized with the tissue sections
overnight at the same temperature. After washing the slides with
washing buffer, the sections were stained with anti-digoxin
rhodamine conjugate (1:100, Exon Biotech Inc., China) at 37°C for 1
h away from light. Then, the sections were stained with DAPI for
nuclear staining. All fluorescence images were captured on a
fluorescence microscope (Leica, Mannheim, Germany).
Western blot analysis
Cells were harvested at 72 h following transfection.
Protein were separated by 10% SDS/PAGE and transferred on to PVDF
membranes (Millipore, Billerica, MA, USA). After blocking the
membranes were incubated overnight at 4°C with appropriate
dilutions of specific primary antibodies as follows: p21 (1/2000)
(Cell Signaling Technology), cyclin D1 (1/2000) (Affinity, USA),
CDK4 (1/1000) (Affinity), CDK6 (1/2000) (Affinity), GAPDH (1/500)
(Boster, Wuhan, China) and α-tubulin (1/500) (Boster). Then
membranes were incubated with corresponding second antibody and
detected by enhanced chemiluminescence (ECL) assay kit
(Millipore).
Chromatin immunoprecipitation (ChIP)
assay
Cells transfected with biotin-labelled RNAs were
processed for ChIP assay (Millipore) at 72 h according to the
manufacturer's instructions with further modifications. Approximate
3×106 cells were used for one immunoprecipitation.
Degradation of RNAs was avoided by RNase inhibitor (Thermo Fisher
Scientific, Waltham, MA, USA) with a final concentration of 50
U/ml. An antibody against biotin (Santa Cruz Biotechnology, Santa
Cruz, CA USA) and negative control normal rabbit IgG (Millipore)
were used for immunoprecipitation. Primers are available in
Table II.
Cell cycle analysis
At 72 h after transfection, cells were fixed by 70%
cold ethanol, incubated with RNase A (Sigma, St. Louis, MO, USA)
and stained by propidium iodide (PI) (Nanjing KeyGen Biotech Co.,
Ltd., Nanjing, China) staining solution. After staining, the cells
were analyzed on a FACSort flow cytometer (BD Biosciences, San
Diego, CA, USA). The data were processed by CELL Quest software (BD
Biosciences).
Clonogenic survival assay
After transfection for 24 h, cells were trypsinized,
counted and reseeded in 6-well plates at the density of 1000
cells/well for 10 days. The colonies were fixed and stained with
0.5% crystal violet (Sigma). The colony formation rate = number of
colonies/number of seeded cells × 100%.
Cell proliferation assay
Cell proliferation was assessed by using the
CellTiter 96 AQueous One Solution Cell Proliferation
Assay kit (Promega, Madison, WI, USA) as previously described
(13). Briefly, RNA transfected
cells were grown in 96-well plates at a density of 1000 cells/well.
Cell growth was measured daily for 4 days. At each time point, 20
µl of CellTiter 96 AQueous One Solution was added and
incubated. Absorbance was detected by a microplate reader (Bio-Rad,
Berkeley, CA, USA) at 490 nm.
Statistical analysis
The data are expressed as the mean ± standard
deviation (SD) for three independent experiments. Differences
between groups were analyzed by t-tests using SPSS version 13.0
software (SPSS Inc., Chicago, IL, USA). P-value <0.05 was
considered to be statistically significant.
Results
Expression patterns of miR-3619-5p and
CDKN1A in prostate cancer cell lines and tissues
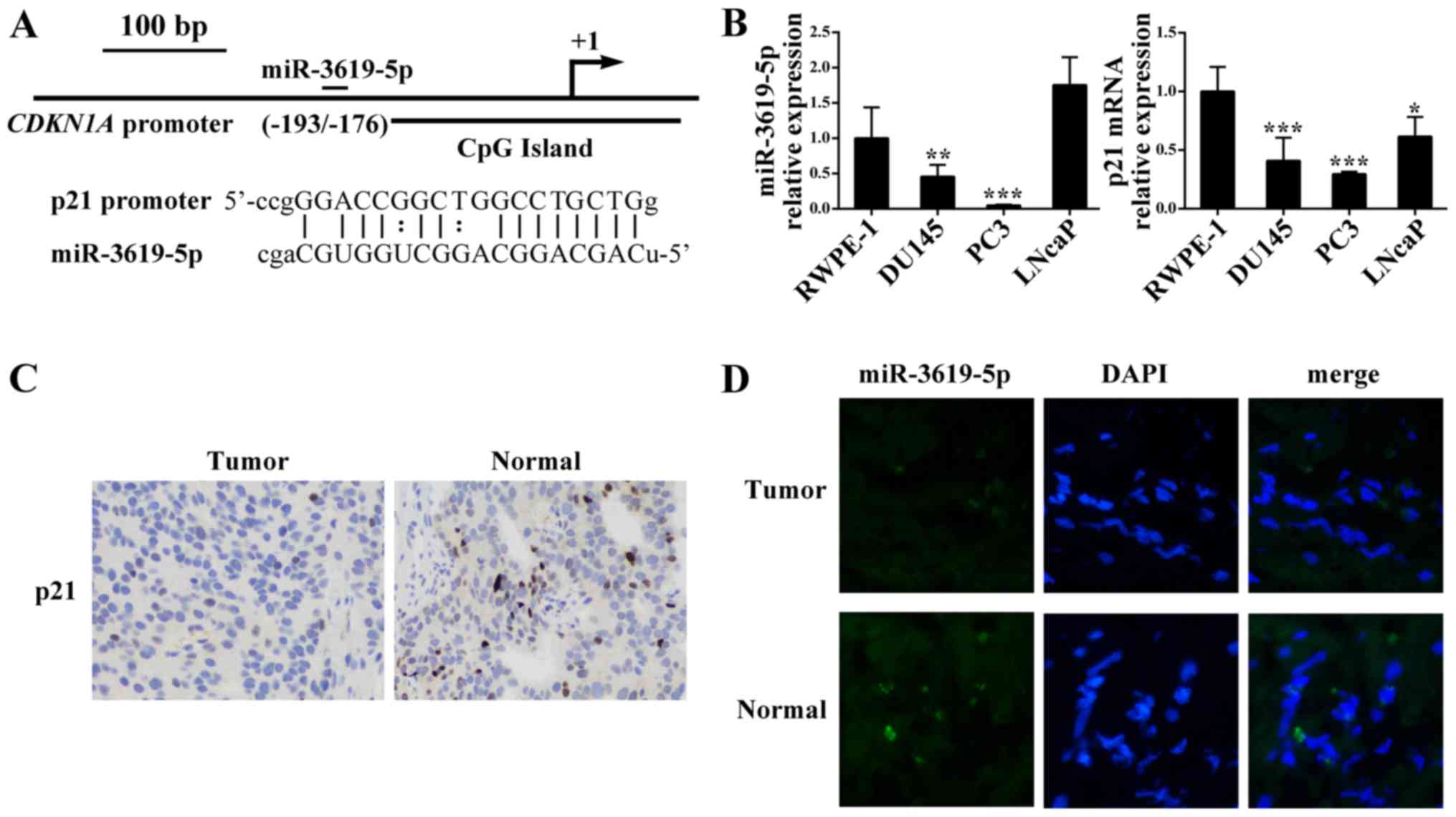
Firstly, we scanned 1 kb of CDKN1A promoter
for sites complementary to known human miRNAs from miRBase
database. A candidate miRNA (miR-3619-5p) was identified with a
higher sequence complementarity score (171) and a lower minimum
free energy (MFE) (−33.24 kcal/mol) through the miRanda algorithm.
In addition, the putative target of CDKN1A promoter position
−193/−176 relative to the transcription start site (Fig. 1A).
Then we performed quantitative real-time PCR to
analyze the miR-3619-5p and CDKN1A expression patterns in
prostate cancer cell lines. As shown in Fig. 1B, compared with normal prostate
epithelial cells RWPE-1, prostate cancer cell lines DU145 and PC3
had significant lower levels of miR-3619-5p expression). However,
the prostate cancer LNcaP cells exhibited higher expression of
miR-3619-5p than RWPE-1. Furthermore, CDKN1A mRNA expression
was downregulated in the 3 prostate cancer cell lines compared to
RWPE-1.
Moreover, we evaluated the p21 protein levels in
prostate tissues and we found that p21 was lower expressed in
prostate cancer than the corresponding adjacent control (Fig. 1C). Additionally, the FISH assay was
conducted to determine miR-3619-5p expression in tissues and
indicated that miR-3619-5p expression level in tumor tissues was
lower compared to adjacent non-tumor tissues (Fig. 1D). Based on these results,
miR-3619-5p may act as a tumor suppressor in prostate cancer
cells.
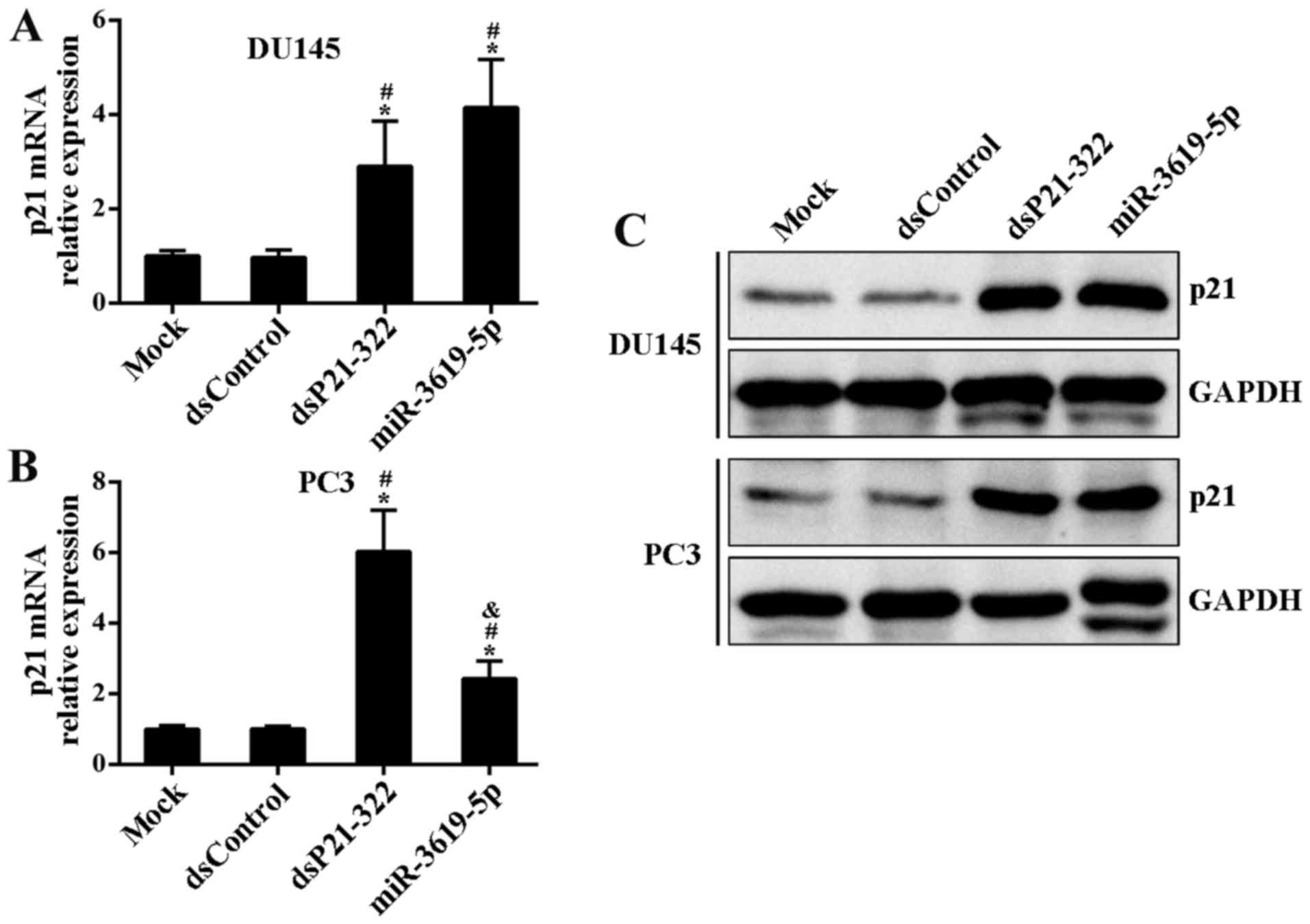
miR-3619-5p activates CDKN1A
expression by directly interacting the promoter
To identify candidate miR-3619-5p that may be
involved in positively regulating CDKN1A expression,
synthetic miR-3619-5p mimics or controls (dsP21-322 and dsControl)
were transfected into DU145 and PC3 cells. Compared to mock and
control treatment, dsP21-322 and miR-3619-5p led to a significant
induction of p21 mRNA levels in both cell lines at 72 h after
transfection (Fig. 2A and B).
Moreover, this induction was further confirmed by immunoblot
analysis (Fig. 2C).
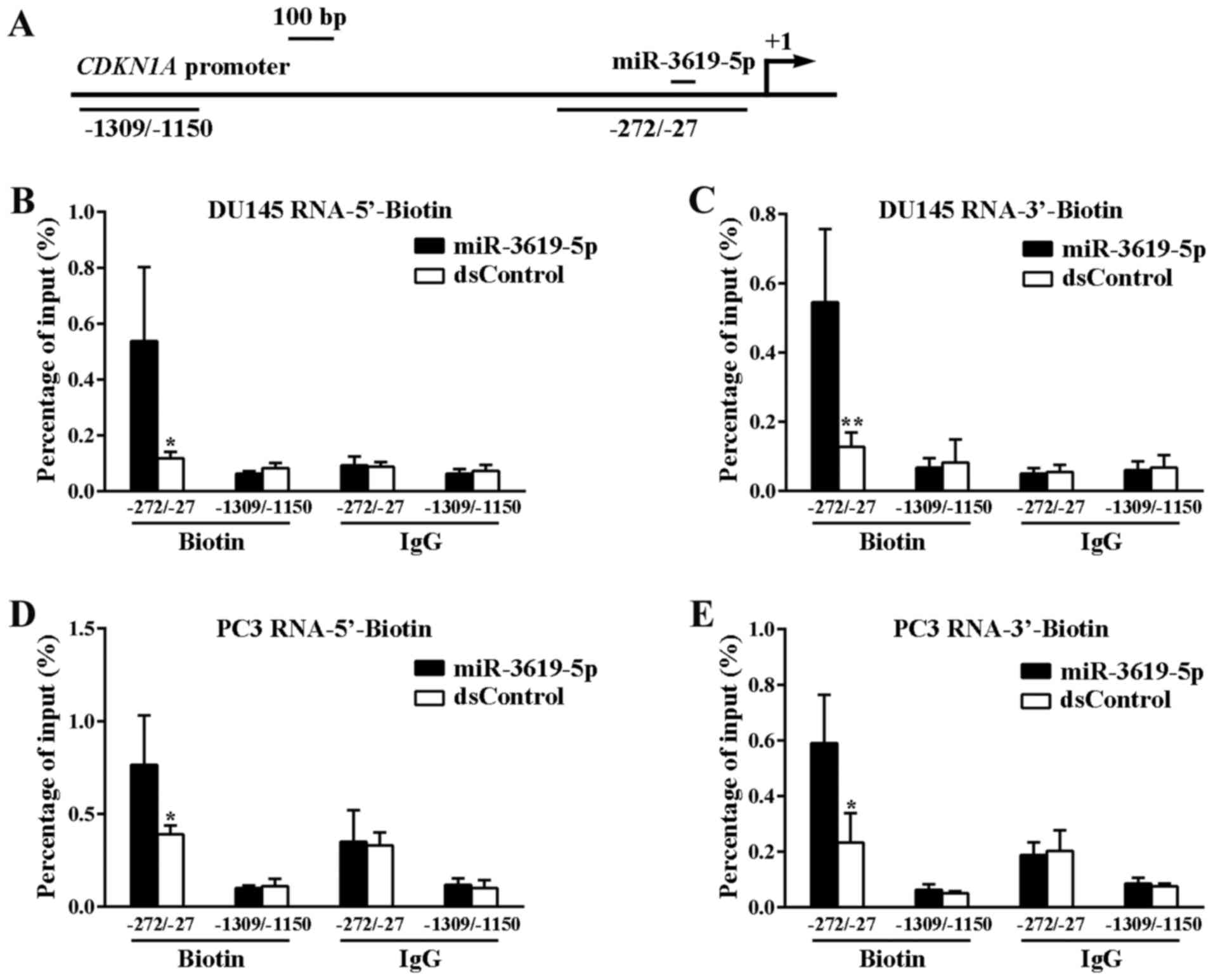
We and other researchers have reported that the
binding of miRNAs to the promoter is one of the possible mechanisms
to promoter-targeted specific gene activation (11,14).
To explore whether miR-3619-5p interact directly with CDKN1A
promoter, we performed ChIP assay via biotinylated RNAs. Biotin was
covalently linked to either 5′-end or 3′-end of miR-3619-5p and
dsControl antisense strand. After 72 h with individual
transfection, the target promoter DNA was pulled down by using a
well-characterized biotin antibody and then amplified by real-time
PCR. Two sets of primers were used to amplify promoter DNA
sequences at different locations, in which one primer set
amplifying sequences from −1309 bp to −1150 bp relative to the TSS
served as a negative control (Fig.
3A).
Finally, miR-3619-5p pulled down promoter DNA more
effectively than the dsControl RNA both in DU145 and PC3 cells
(Fig. 3B-E). In contrast, there was
no difference in the binding of miR-3619-5p and dsControl to DNA
upstream of the CDKN1A promoter (−1309 to −1150). These
results indicate that the miR-3619-5p interacts directly with
CDKN1A promoter and enhances its expression.
miR-3619-5p inhibits prostate cancer
cell growth mainly by activating p21 expression
Next, we investigated whether p21 was mainly
responsible for tumor suppression after miR-3619-5p transfection.
To address this, a small interfering RNA (siP21) was used to
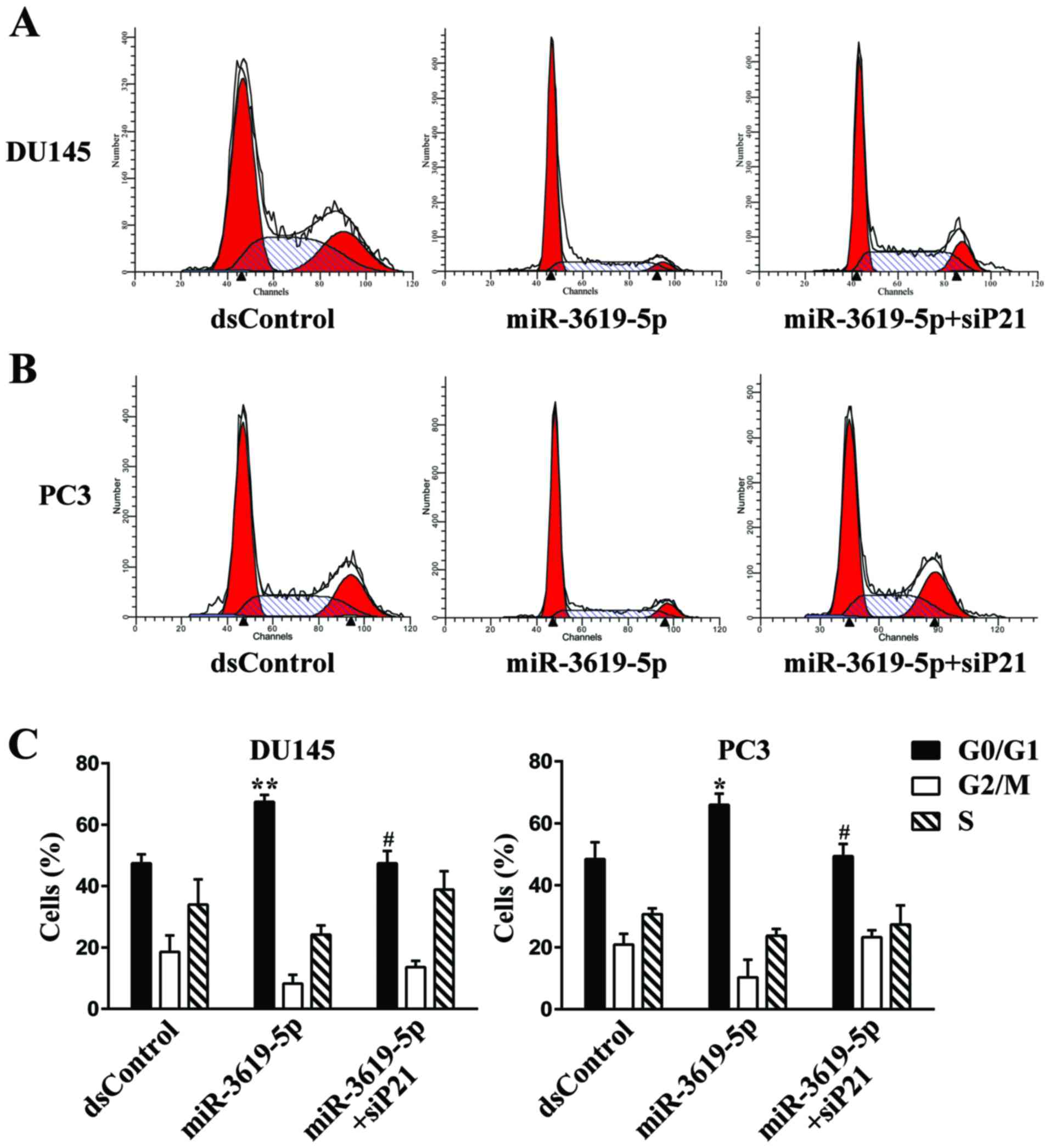
silence the p21 expression in DU145 and PC3 cells (Fig. 6A and B). Then we performed flow
cytometry to measure cell cycle distribution. In both tested cell
types, transfection with miR-3619-5p led to a significant increase
in the G0/G1 population compared with dsControl group (Fig. 4A-C). Moreover, knockout of p21
expression markedly attenuated G0/G1 cycle arrest mediated by
miR-3619-5p in DU145 (Fig. 4A and
C) and PC3 cells (Fig. 4B and
C).
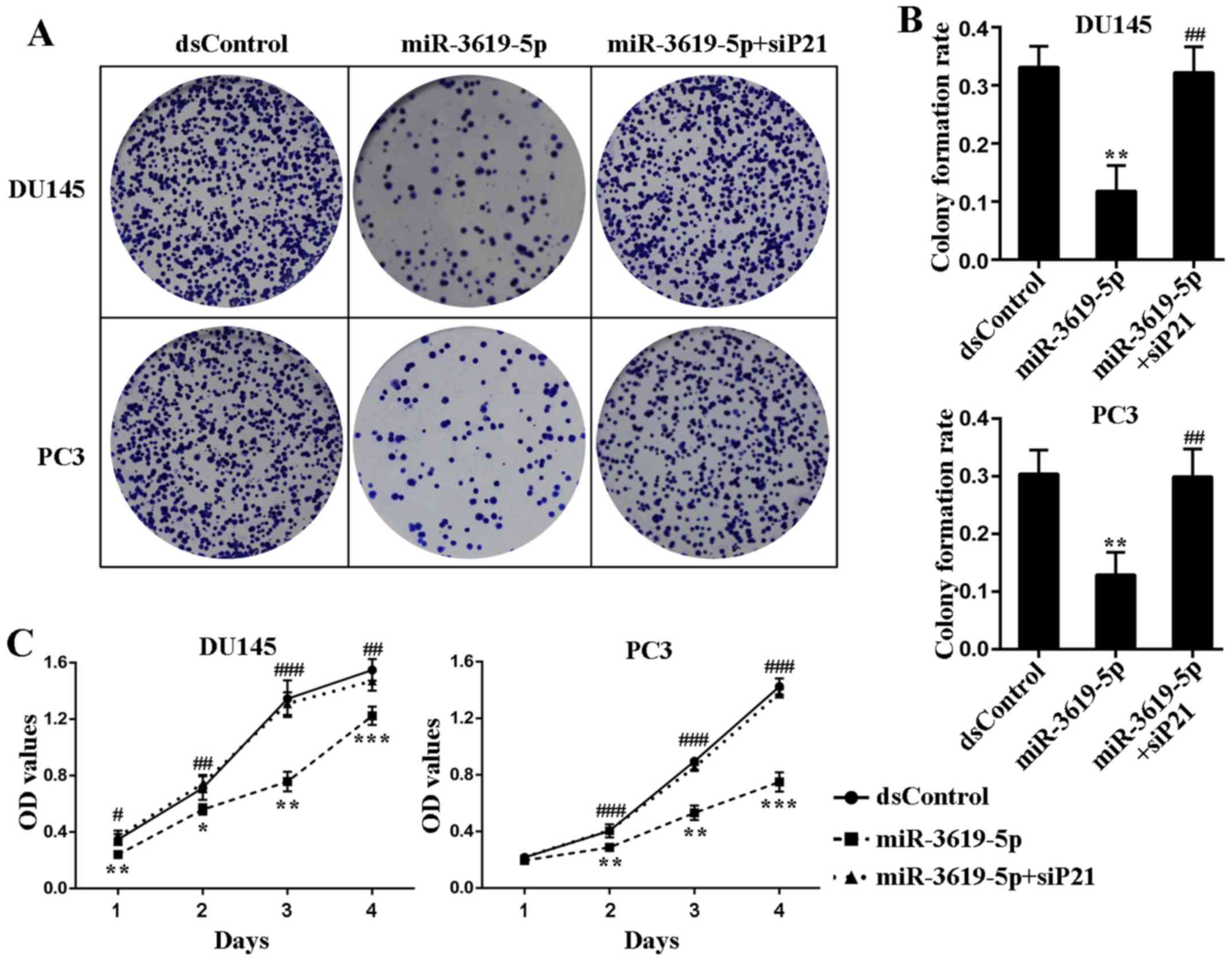
In order to assess the cell growth mode after p21
upregulation activated by miR-3619-5p, the colony formation assay
was applied and showed miR-3619-5p transfected cells formed
significantly fewer colonies in number and smaller in size
(Fig. 5A). Additionally, the colony
formation rates of miR-3619-5p transfected cells were remarkably
lower than dsControl treatment in both cell lines (Fig. 5B). Moreover, the colony formation
ability of the prostate cancer cells was restored after
co-treatment of siP21 (Fig. 5A and
B).
CellTiter 96 AQueous One Solution Cell
Proliferation Assay indicated that compared to dsControl group,
both DU145 and PC3 cells transfected with miR-3619-5p exhibited
progressive retarded growth from the next day following
transfection (Fig. 5C). Then we
verified whether depletion of p21 could affect the inhibitory
function of miR-3619-5p in prostate cancer cells. As illustrated in
Fig. 5C, silencing of p21 evidently
attenuated the anti-proliferative effect mediated by miR-3619-5p in
both cell types. Taken together, these results suggest that
miR-3619-5p possesses the capacity to inhibit prostate cancer cell
growth through activating CDKN1A expression.
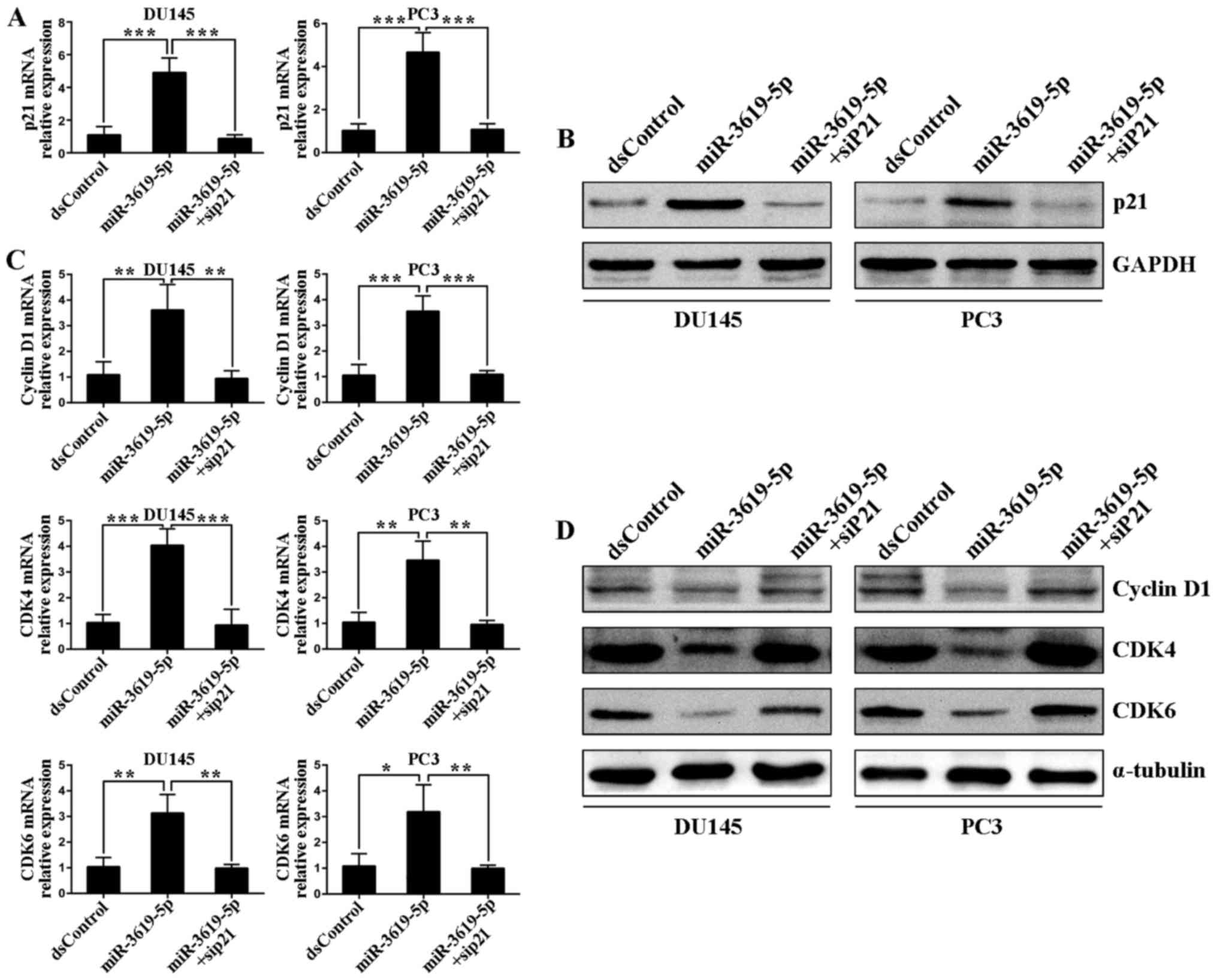
miR-3619-5p regulates prostate cancer
cell cycle-associated genes mainly by enhancing CDKN1A
Subsequently, we determined the effects of
miR-3619-5p transfection on the expression of downstream genes
associated with cell cycle in prostate cancer cells. As shown in
Fig. 6C, transfection of
miR-3619-5p caused a significant decrease of mRNA levels of cyclin
D1, CDK4 and CDK6 in both DU145 and PC3 cells. The suppression
effects on protein levels of these genes were further verified by
western blot analysis (Fig. 6D). We
also blocked p21 expression by cotransfecting the siP21 in both
cell lines. Real-time PCR revealed that miR-3619-5p failed to
downregulate cyclin D1, CDK4 and CDK6 mRNA levels after siP21
cotreatment (Fig. 6C). The protein
analysis of immunoblotting further proved this (Fig. 6D). These findings manifested that
miR-3619-5p manipulated prostate cancer cell cycle-associated genes
largely by upregulating CDKN1A expression.
Discussion
In the present study, we identified an endogenous
miR-3619-5p with lower expression in prostate cancer tissues and
cells than corresponding normal controls. Moreover, overexpression
of miR-3619-5p readily induces CDKN1A gene expression by
targeting the putative site in the promoter. Mechanistically, this
gene activation depends on miR-3619-5p directly interacting with
the CDKN1A promoter sequences. Besides, miR-3619-5p holds
considerable ability to inhibit prostate cancer DU145 and PC3 cell
growth, and downregulate several CDKN1A downstream genes,
such as cyclin D1, CDK4 and CDK6. Notably, this antitumor function
of miR-3619-5p is primarily achieved by activating CDKN1A
gene expression.
It is recognized that sustained proliferative
activity is a distinct feature of cancer cells, therefore blockade
of cell cycle is regarded as an promising strategy for treatment of
prostate cancer (15). Cell cycle
is mainly controlled by cyclin/cyclin-dependent kinase (CDK)
complexes (16). Accumulation of
cyclin D1 promotes its binding to CDK4/6 and activation of the
complexes, and resulting in the progression of cell cycle from
G0/G1 to S phase (17). The
well-known CDK inhibitor p21 can negatively regulate cell cycle
progression by inhibiting the activity of the cyclin D1 and CDK4/6
(18). Place et al have
reported that induction of p21 through RNAa inhibited prostate
cancer cells growth both in vitro and in vivo
(19). In this study, we
demonstrated that overexpression of endogenous miR-3619-5p induced
CDKN1A expression via RNAa and downregulated cyclin
D1-CDK4/6 levels, and consequently inhibited prostate cancer cell
proliferation.
Different from RNA interference, RNAa is a positive
gene regulation phenomenon. It is disappointing that the exact
molecular mechanism involved remains poorly understood. However,
based on current knowledge, the saRNA-mediated RNAa process
requires both transcriptional and epigenetic changes (20,21).
For miRNA-mediated RNAa, a simple model is that miRNA directly
binds to complementary DNA within gene promoter and triggers gene
expression. In this regard, miRNA may act as a transcription factor
and targets complementary motifs in the gene promoter.
Alternatively, cells may produce RNA copies of the target promoter
region. Complementary miRNA would directly interact with the
transcripts and finally lead to downstream gene expression
(5,11). However, further research is needed
to explore the molecular components required in miRNA mediated gene
activation.
High complementarity of sequences between candidate
miRNA and the target promoter DNA is necessary for RNAa, and some
natural mismatches are quite common and tolerable (14,22).
Herein, we selected miR-3619-5p as the research candidate, which
had a sequence complementarity score of 171 and an MFE value of
−33.24 kcal/mol. The MFE of hybridization between miRNA and its
predicted target site have been successfully used to predict miRNA
target sites within 3′ UTR (23).
Therefore, the MFE values may also be beneficial for predicting
miRNA targets within gene promoter (24). Another important criterion applied
in miRNA target prediction is sequence complementarity, which was
used to predict miRNA target sites within 3′ UTRs successfully
(25). These algorithms were
included in the miRanda program and facilitated to assess the
degree of sequence complementarity between the miR-3619-5p and
predicted target sites within CDKN1A promoter (25).
Prostate cancer development contains numerous
genetic alternations, such as inactivation of some critical tumor
suppressor genes (26,27). Therefore, reactivation of specific
tumor suppressor CDKN1A will provide a promising alternative
for prostate cancer therapeutics. Additionally, RNAa is a safe
method for manipulating certain gene expression as it does not
alter the expression profile of other non-specific genes (22).
Taken together, our results provide evidence that an
endogenous miR-3619-5p stimulated CDKN1A expression by
directly targeting promoter sequences. Moreover, miR-3619-5p also
has the capacity to induce cell cycle arrest and inhibits
proliferation of prostate cancer cells. Our study identified a new
miRNA which both mediate endogenous CDKN1A overexpression
and suppress prostate cancer.
Acknowledgements
This work was supported by the National Natural
Science Foundation of China (grant number 81302218, China).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Damber JE: Endocrine therapy for prostate
cancer. Acta Oncol. 44:605–609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li LC, Okino ST, Zhao H, Pookot D, Place
RF, Urakami S, Enokida H and Dahiya R: Small dsRNAs induce
transcriptional activation in human cells. Proc Natl Acad Sci USA.
103:17337–17342. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Place RF, Li LC, Pookot D, Noonan EJ and
Dahiya R: MicroRNA-373 induces expression of genes with
complementary promoter sequences. Proc Natl Acad Sci USA.
105:1608–1613. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Catto JW, Alcaraz A, Bjartell AS, De Vere
White R, Evans CP, Fussel S, Hamdy FC, Kallioniemi O, Mengual L,
Schlomm T, et al: MicroRNA in prostate, bladder, and kidney cancer:
A systematic review. Eur Urol. 59:671–681. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fendler A, Stephan C, Yousef GM and Jung
K: MicroRNAs as regulators of signal transduction in urological
tumors. Clin Chem. 57:954–968. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fabris L, Ceder Y, Chinnaiyan AM, Jenster
GW, Sorensen KD, Tomlins S, Visakorpi T and Calin GA: The potential
of microRNAs as prostate cancer biomarkers. Eur Urol. 70:312–322.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Warfel NA and El-Deiry WS: p21WAF1 and
tumourigenesis: 20 years after. Curr Opin Oncol. 25:52–58. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eastham JA, Hall SJ, Sehgal I, Wang J,
Timme TL, Yang G, Connell-Crowley L, Elledge SJ, Zhang WW, Harper
JW, et al: In vivo gene therapy with p53 or p21 adenovirus for
prostate cancer. Cancer Res. 55:5151–5155. 1995.PubMed/NCBI
|
|
11
|
Huang V, Place RF, Portnoy V, Wang J, Qi
Z, Jia Z, Yu A, Shuman M, Yu J and Li LC: Upregulation of Cyclin B1
by miRNA and its implications in cancer. Nucleic Acids Res.
40:1695–1707. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xiao H, Zeng J, Li H, Chen K, Yu G, Hu J,
Tang K, Zhou H, Huang Q, Li A, et al: MiR-1 downregulation
correlates with poor survival in clear cell renal cell carcinoma
where it interferes with cell cycle regulation and metastasis.
Oncotarget. 6:13201–13215. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang C, Ge Q, Chen Z, Hu J, Li F, Song X,
Xu H and Ye Z: A new double stranded RNA suppresses bladder cancer
development by upregulating p21 (Waf1/CIP1) expression. Biomed Res
Int. 2015:3047532015.PubMed/NCBI
|
|
14
|
Wang C, Chen Z, Ge Q, Hu J, Li F, Hu J, Xu
H, Ye Z and Li LC: Up-regulation of p21(WAF1/CIP1) by miRNAs and
its implications in bladder cancer cells. FEBS Lett. 588:4654–4664.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sperka T, Wang J and Rudolph KL: DNA
damage checkpoints in stem cells, ageing and cancer. Nat Rev Mol
Cell Biol. 13:579–590. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bloom J and Cross FR: Multiple levels of
cyclin specificity in cell-cycle control. Nat Rev Mol Cell Biol.
8:149–160. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hallstrom TC and Nevins JR: Balancing the
decision of cell proliferation and cell fate. Cell Cycle.
8:532–535. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Denicourt C and Dowdy SF: Cip/Kip
proteins: More than just CDKs inhibitors. Genes Dev. 18:851–855.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Place RF, Wang J, Noonan EJ, Meyers R,
Manoharan M, Charisse K, Duncan R, Huang V, Wang X and Li LC:
Formulation of small activating RNA into lipidoid nanoparticles
inhibits xenograft prostate tumor growth by inducing p21
expression. Mol Ther Nucleic Acids. 1:e152012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zheng L, Wang L, Gan J and Zhang H: RNA
activation: Promise as a new weapon against cancer. Cancer Lett.
355:18–24. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Portnoy V, Lin SH, Li KH, Burlingame A, Hu
ZH, Li H and Li LC: saRNA-guided Ago2 targets the RITA complex to
promoters to stimulate transcription. Cell Res. 26:320–335. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Portnoy V, Huang V, Place RF and Li LC:
Small RNA and transcriptional upregulation. Wiley Interdiscip Rev
RNA. 2:748–760. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stark A, Brennecke J, Russell RB and Cohen
SM: Identification of Drosophila MicroRNA targets. PLoS Biol.
1:E602003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Younger ST, Pertsemlidis A and Corey DR:
Predicting potential miRNA target sites within gene promoters.
Bioorg Med Chem Lett. 19:3791–3794. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Needleman SB and Wunsch CD: A general
method applicable to the search for similarities in the amino acid
sequence of two proteins. J Mol Biol. 48:443–453. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mitchell T and Neal DE: The genomic
evolution of human prostate cancer. Br J Cancer. 113:193–198. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Barbieri CE and Tomlins SA: The prostate
cancer genome: Perspectives and potential. Urol Oncol.
32:53.e15-222014. View Article : Google Scholar : PubMed/NCBI
|