Introduction
Hepatocellular carcinoma (HCC) is the most common
primary malignancy of the liver and the second leading cause of
cancer-related death worldwide. Globally, its incidence remains
high, with approximately 750,000 new cases reported per year.
Although significant improvement in diagnosis and treatment of HCC
has been achieved, the prognosis for HCC patients who were
surgically resectable remains poor. As estimated, the recurrence
rate is approximately 50% in 2 years and 75% in 5 years after
resection, respectively (1,2), and most patients present with
unresectable and advanced tumors. Therefore, it is vital to
understand the underlying mechanism of tumor growth and metastasis
and develop efficacious therapeutics for HCC.
Plac1, a known placenta-specific gene, is located on
the X-chromosome (Xq26.3) (3).
Plac1 gene encodes a 26-kDa protein, which is primarily expressed
in cells of the trophoblastic lineage, and plays an important role
in human placental development (4–6).
Although the expression is mostly restricted to placenta in normal
tissues, Plac1 is frequently activated and highly expressed in wide
variety of human tumor cell lines and tissues, including breast,
lung, colon, prostatic, cervical, gastric and hepatocellular cancer
(7–10). Emerging evidence indicates that
Plac1 is involved in the pathogenesis of cancer. Koslowski and
coworkers (10) recently disclosed
that knockdown of Plac1 suppressed breast cancer cell migration and
invasion, and resulted in an interception of breast cancer cell
proliferation by inducing a distinct G1 cell cycle arrest. Further,
silencing of Plac1 decreased cyclin D1 expression and
phosphorylation of AKT, which influenced cancer development. Since
Plac1 has effects on the development of placenta and cancer, some
scholars have given Plac1 designation cancer-placenta antigen 1
(CP1), which is regarded as a promising candidate for targeted
immunotherapy of cancer (7,8). Some surveys revealed that Plac1
elicited spontaneous antibody responses in HCC (8), and triggered anticancer immune
responses by inducing the generation of Plac1-reactive antibodies
and T cells in colorectal cancer patients (7). Nevertheless, the regulatory mechanism
of Plac1 in the development of HCC is still not well
elucidated.
In the present study, we discovered that Plac1
expression was significantly increased in HCC tissues, and
associated with metastasis in HCC patients. Moreover, Plac1 was
involved in the regulation of proliferation, cell cycle, apoptosis
and EMT, which were correlated with AKT signaling pathway in HCC.
Collectively, our findings indicate that Plac1 may be an effective
therapeutic target for HCC.
Materials and methods
Patients and tissues
Forty-six cases of paraffin-embedded HCC samples and
non-cancerous liver tissues were obtained from the Department of
Pathology of Affiliated Hospital of Guangdong Medical University.
All the specimens were validated by pathological diagnosis
complying with the World Health Organization standards. This study
was conducted with the approval of the Medical Ethics Committee of
Affiliated Hospital of Guangdong Medical University.
Cell lines
HepG2, Bel-7402, SMMC-7721, Huf7 and L02 cells were
obtained from the Cell Blank of Shanghai Biology Institute, Chinese
Academy of Sciences (Shanghai, China). HLE cell line was a gift
from Professor Yanhui Yin (Peking University Health Science Center,
Beijing, China). These cells were cultured in Dulbeccos modified
Eagles medium (DMEM; HyClone Laboratories, Thermo Fisher
Scientific, Waltham, MA, USA) and supplemented with 10% fetal
bovine serum (FBS; HyClone Laboratories, Thermo Fisher Scientific)
in a humidified incubator at 37°C with 5% CO2.
Cell transfection
The HepG2 and Bel-7402 cells were assigned to
Plac1-siRNA group, blank group, and negative control group (NC
siRNA). Cells in Plac1-siRNA (Sigma-Aldrich, St. Louis, MO, USA)
group were transfected with the following sequences: 5′ to
3′-GUGUUCAGCUUGUCACAGU, antisense, 5′ to 3′ ACUGUGACAAGCUGAACAC.
Cells in the negative control group were transfected with NC siRNA,
which was a non-specific scramble siRNA. Cells in the blank group
were exposed to normal medium. For transient transfection, HepG2
and Bel-7402 cells were transfected with siRNA using Lipofectamine
2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturers
instruction. Four hours after the transfection, cell were
transferred to fresh medium-containing 10% FBS and incubated for
24–36 h.
Immunohistochemistry
Tissue slides were dewaxed according to the standard
protocol. Paraffin sections of HCC were deparaffinized, hydrated
and washed in phosphate-buffered saline (PBS). After pretreatment
in a microwave oven for 8 min, the endogenous peroxidase was
inhibited with 3% H2O2 for 10 min, and the
sections were incubated with 10% normal goat serum for 30 min.
Subsequently, the sections were incubated with Plac1 primary
antibody (Abcam, 1:100) at 4°C overnight. After washing, the
sections were incubated for 30 min with secondary antibody (Dako,
Carpinteria, CA, USA) at room temperature. Following this
incubation, the sections were washed three times in PBS, and the
visualization signal was developed using diaminobenzidine
tetrahydrochloride (DAB), followed by counterstaining with
hematoxylin.
Western blot analysis
Cells were washed three times with cold PBS, and
lysed in RIPA-buffer (0.5% sodium deoxycholate, 0.1% SDS, 1% NP-40
and PBS) with PMSF and protease inhibitor cocktail (Sigma-Aldrich).
Equal amount of protein (30 µg) was separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto polyvinylidene fluoride (PVDF; Millipore Corp.,
Bedford, MA, USA) membrane after quantification. Following
incubation in 5% BSA, the membranes were incubated with primary
antibodies: CDK4 (1:1,000), snail1 (1:1,000), AKT (1:1,000), p-AKT
(1:2,000), E-cadherin (1:1,000), p53 (1;1,000) (Cell Signaling
Technology, Danvers, MA, USA), Twist (1:1,000), cyclin D1 (1:1,000)
(GeneTex, Inc., Irvine, CA, USA), Plac1 (1:1,000), vimentin
(1:1,000), Bax (1:1,000), Bcl-2 (1:500) (Abcam, Cambridge, UK), and
GAPDH (1:10,000) (KangChen Bio-tech Inc., Shanghai, China)
overnight at 4°C. Subsequently the membranes were incubated with a
horseradish-peroxidase-conjugated secondary antibody
(SouthernBiotech, Birmingham, AL, USA) at a dilution of 1:20,000 at
room temperature for 1 h and visualized by chemiluminescence. GAPDH
was used as an internal control for normalization.
Cell proliferation assay
Cell proliferation was performed using the Cell
Counting kit-8 (CCK-8) assay (Beyotime Institute of Biotechnology,
Shanghai, China) according to the manufacturers instructions. After
transfected with indicated siRNA, cells from Plac1-siRNA group,
blank group, and NC siRNA group were collected and seeded in
96-well plates (1×104 cells/well). Following further
incubated for 0, 24, 48 and 72 h, respectively, the cells were
incubated with CCK-8 reagents (10 ml/well) for 4 h. The optical
density values (OD) were measured at 450 nm. All experiments were
performed at least three times.
Cell cycle analysis
For the cell cycle analysis, 1×106 cells
from Plac1-siRNA group, blank group, and NC siRNA group were
collected after 48 h, washed twice in cold PBS, and fixed with cold
70% ethanol at 4°C overnight. Then, the cells were washed in cold
PBS, and resuspended in 500 µl PBS with propidium iodide (PI),
Triton X-100, and RNase A at 4°C for 30 min. The samples were
analyzed by a FACSCalibur flow cytometer (BD Biosciences, San Jose,
CA, USA).
Cell apoptosis assay
Cell apoptosis was measured by FACS analysis using
the Annexin V-FITC/PI apoptosis detection kit (Nanjing KeyGen
Biotech, Co., Ltd., Nanjing, China). Cells 5×105 from
Plac1-siRNA group, blank group, and NC siRNA group were collected
and washed twice in cold PBS. Subsequently, the cells were
resuspended in 500 µl binding buffer, and incubated with 1.25 µl
Annexin V-FITC and 10 µl propidium iodide in the dark at room
temperature for 15 min. The stained cells were analyzed by a
FACSCalibur flow cytometer (BD Biosciences).
Cell migration and invasion
assays
Cell migration and invasion were carried out by the
way of Transwell and Matrigel invasion (BD Biosciences),
respectively. For cell migration assay, 1×105 cells were
seeded on the upper of chambers in serum-free DMEM. The lower
chambers were filled with DMEM, supplemented with 10% FBS as a
chemoattractant. After incubation for 48 h, non-migrated cells on
the top of the well were removed with cotton swabs. The cells
migrated to the underside of the membrane were fixed with 4%
paraformaldehyde for 15 min, stained with 0.1% crystal violet, and
counted under a microscope. For the invasion assay, the procedures
were the same as those described above, except that the upper
chambers were coated with Matrigel. Each experiment was performed
in triplicate and the mean value was calculated from three
independent experiments.
Statistical analysis
SPSS 16.0 software was used for all the analysis.
The correlation between the Plac1 and the individual
clinicopathological factors was evaluated with the Chi-squared
test. For other data, the two or multiple group comparisons were
performed using the Students t-test or ANOVA. Data were reported as
mean ± SD. P<0.05 was considered statistically significant.
Results
Plac1 is upregulated in HCC tissues
and correlates with tumor metastasis
To clarify the underlying role of Plac1 in HCC
progression, we first determined the protein expression level of
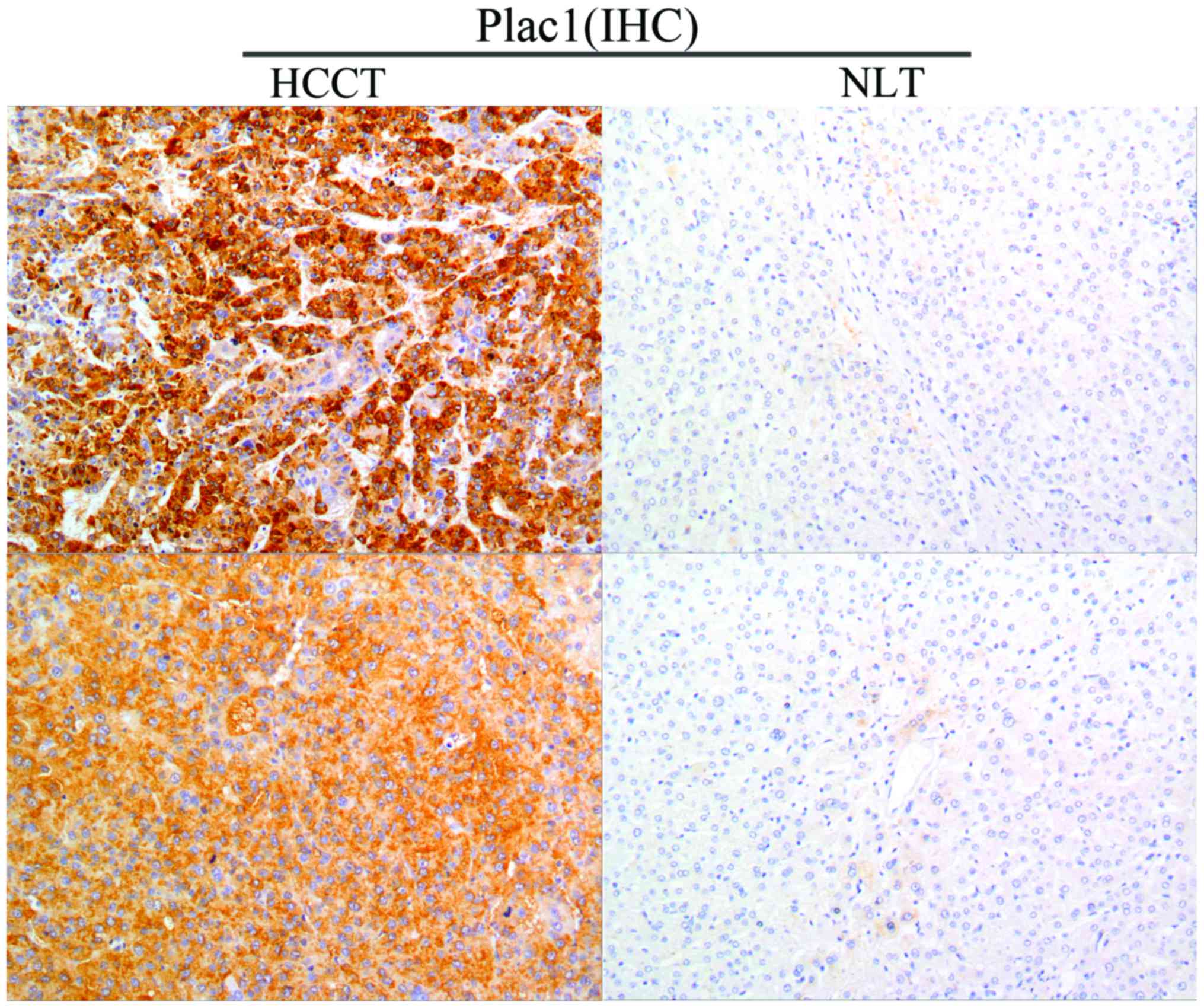
Plac1 in 46 tumor specimens from HCC patients. Immunohistochemistry
analysis revealed that Plac1 was highly expressed in 54.3% (25/46)
of HCC tissues, and the protein was located diffusely in the
cytoplasm and cell membrane. Noteworthy, non-cancerous liver
tissues exhibited negative or weak Plac1 staining (Fig. 1). The correlation between the Plac1
expression level and the clinicopathological features was further
evaluated, which is summarized in Table
I. The results showed that Plac1 expression was not associated
with the patient gender, age, HBsAg, serum AFP, tumor size or
cirrhosis, but significantly with tumor metastasis (P=0.015). These
data illustrate that Plac1 may be involved in the HCC pathogenesis
and aggressiveness.
 | Table I.Relationship between the Plac1
expression and the clinicopathological features. |
Table I.
Relationship between the Plac1
expression and the clinicopathological features.
| Characteristics | No. of patients | Plac1 (+) | Plac1 (−) | χ2 | P-value |
|---|
| Gender |
|
|
|
|
|
|
Male | 36 | 19 | 17 | 0.002 | 0.963 |
|
Female | 10 | 6 | 4 |
|
|
| Age (years) |
|
|
|
|
|
|
≤60 | 30 | 14 | 16 | 2.051 | 0.152 |
|
>60 | 16 | 11 | 5 |
|
|
| HBsAg |
|
|
|
|
|
|
Positive | 36 | 19 | 17 | 0.096 | 0.757 |
|
Negative | 10 | 6 | 4 |
|
|
| AFP (µg/l) |
|
|
|
|
|
|
≤400 | 24 | 12 | 12 | 0.382 | 0.536 |
|
>400 | 22 | 13 | 9 |
| Cirrhosis |
|
|
|
|
|
|
Absent | 33 | 17 | 16 | 0.378 | 0.539 |
|
Present | 13 | 8 | 5 |
|
|
| Tumor size
(cm) |
|
|
|
|
|
| ≤5 | 23 | 10 | 13 | 2.190 | 0.139 |
|
>5 | 23 | 15 | 8 |
|
|
|
Differentiation |
|
|
|
|
|
| Well
and moderate | 31 | 18 | 13 | 0.529 | 0.467 |
|
Poor | 15 | 7 | 8 |
|
|
| Metastasis |
|
|
|
|
|
|
Absent | 15 | 12 | 3 | 5.903 | 0.015 |
|
Present | 31 | 13 | 18 |
Plac1 is overexpressed in HCC cells
and knocked down by siRNA
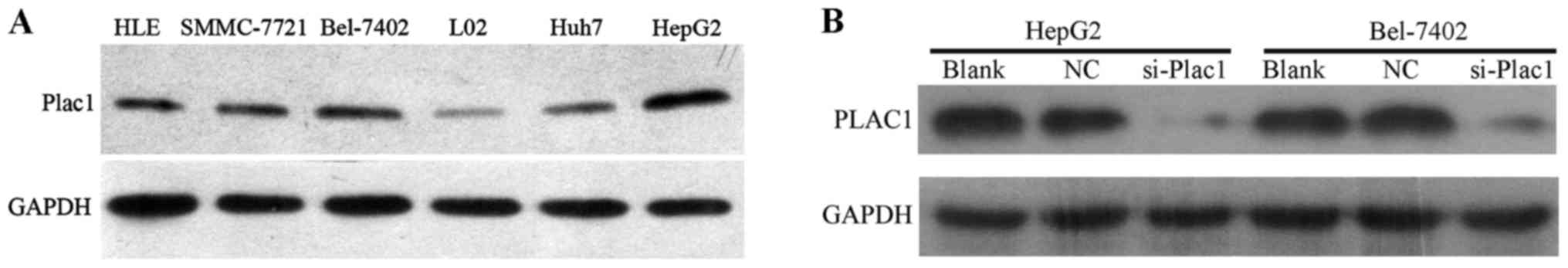
Next, we analyzed the Plac1 expression in five HCC
cell lines, HLE, Bel-7402, SMMC-7721, HepG2 and Huh7 by western
blot analysis. It was observed that Plac1 expression was
significantly upregulated in all HCC cell lines when compared to
normal liver cell line (Fig. 2A).
To further assess the impact of Plac1 on HCC, we employed Bel-7402
and HepG2 cells as model system, in which Plac1 expression is
higher than other HCC cell lines. In these cells, we downregulated
Plac1 expression by Plac1-specific siRNA, and a scrambled siRNA was
used as a negative control. As expected, Plac1 protein expression
was almost completely inhibited, as detected by western blotting
(Fig. 2B).
Knockdown of Plac1 inhibits cell
proliferation in HCC cells
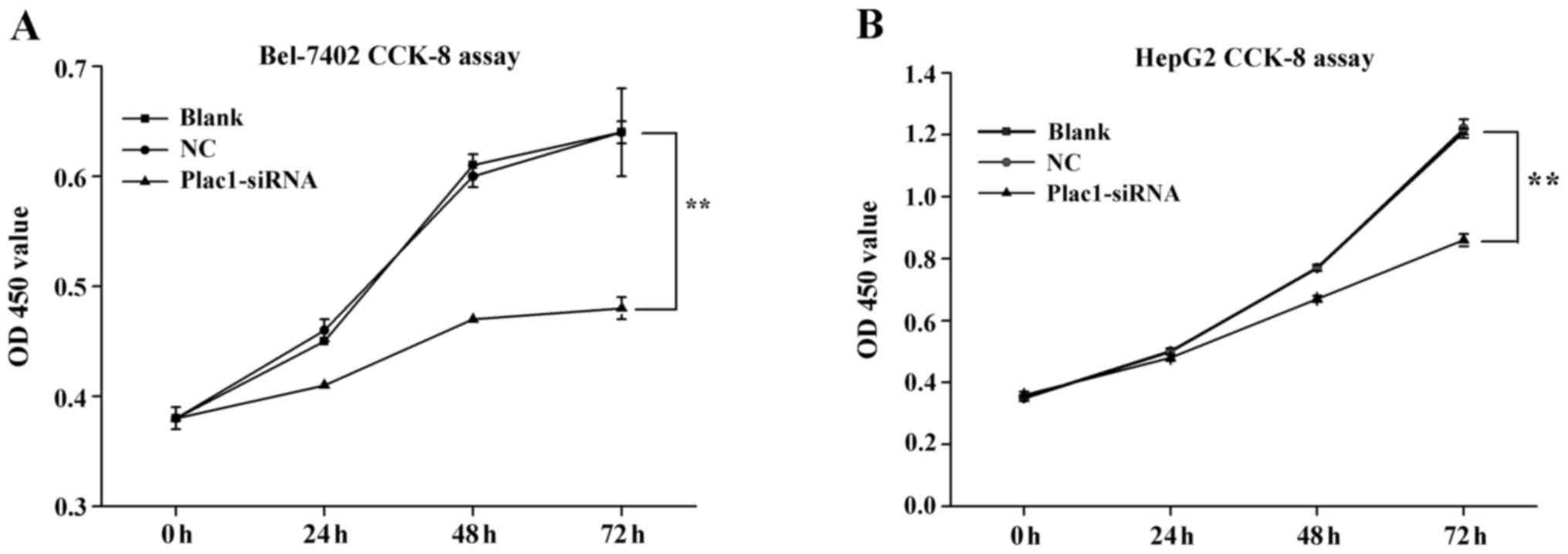
We carried out cell proliferation assay to explore
the effect of Plac1 on the growth of HCC cells. After Bel-7402 and
HepG2 cells were transiently transfected with Plac1 specific siRNA
(Plac1-siRNA) and a scrambled siRNA (siNC RNA, NC, negative
control) for 48 h, they were evaluated by using CCK-8 assays. The
cells treated with Plac1-siRNA exhibited a significant reduction in
cell proliferation compared to control cells (Fig. 3). These data supported that Plac1
promotes the growth of HCC cells in hepatocarcinogenesis.
Downregulation of Plac1 causes cell
cycle retardation and apoptosis of HCC cells
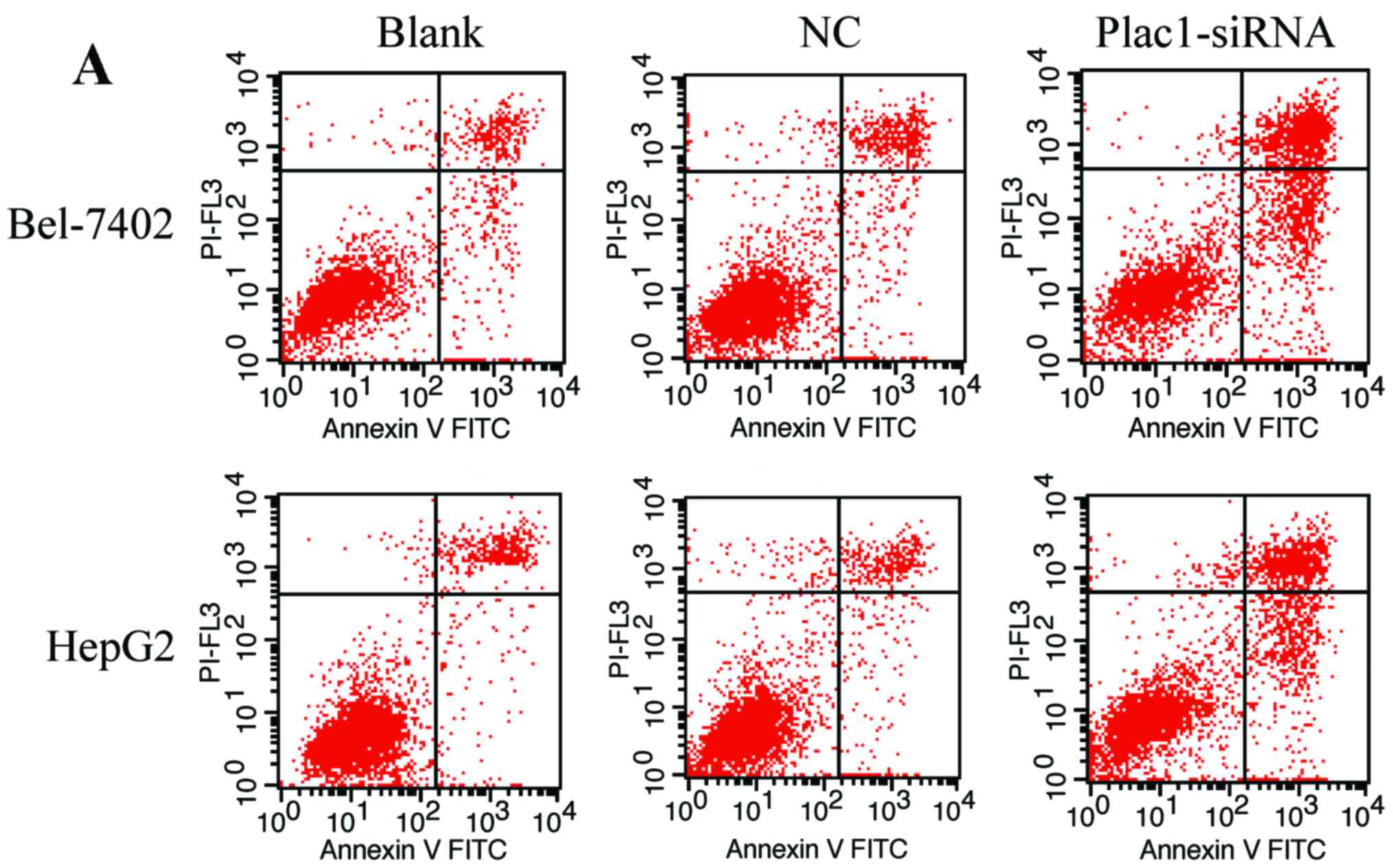
To verify if Plac1-siRNA-mediated suppression of
cell growth is associated with cell cycle arrest or an induction of
apoptosis, we performed cell cycle and apoptosis analysis using
flow cytometry. We observed that the amount of apoptotic cells was
more in cells transfected with Plac1-siRNA than those in control
groups. The average percentage of apoptotic Bel-7402 cells in
Plac1-siRNA, NC, and blank groups, was 45.96, 13.77 and 12.65%,
respectively, while apoptotic percentage was 30.84% in Plac1-siRNA
HepG2 cells, 10.36% in NC HepG2 cells and 11.51% in blank HepG2
cells (Fig. 4A and B). To further
investigate the molecular mechanism of these pro-apoptotic effects,
we measured the expression of apoptosis-related proteins, including
p53, Bcl-2 and Bax. In both Bel-7402 and HepG2 cells, silencing of
Plac1 caused p53 protein level increased, while the pro-apoptotic
protein Bax was downregulated. Notably, the anti-apoptotic protein
Bcl-2 was increased in HepG2-siPlac1 cells, but reduced in
Bel-7402-siPlac1 cells (Fig.
4G).
Next, we sought to determine cell cycle distribution
after cells were treated with Plac1-siRNA or siNC RNA by flow
cytometry. As shown in Fig. 4C-E,
knockdown of Plac1 significantly enhanced the percentage of
Bel-7402 and HepG2 cells in the G1 phase (52.64% in Bel-7402 cells
and 76.78% in HepG2 cells) in comparison to the NC (47.46% in
Bel-7402 cells and 69.13% in HepG2 cells) or blank group (46.72% in
Bel-7402 cells and 68.63% in HepG2 cells), at the same time,
accompanied by a decreased proportion of cells in S phase, which
was decreased at least 10% in Bel-7402 cells and 36% in HepG2
cells, but G2 phase had no change. These results indicated that
downregulation of Plac1 resulted in retardation of HCC cells in the
G1 phase. To elucidate the mechanism of G1 accumulation induced by
Plac1-siRNA, we used a western blot analysis to examine the
expression levels of cyclin D1 and CDK4. Compared to the control
groups, cells transfected with Plac1 siRNA exhibited inhibition of
cyclin D1 and CDK4 expression (Fig.
4F). Therefore, these data demonstrated that Plac1 enhanced
cell proliferation through promoting cell cycle progression in
HCC.
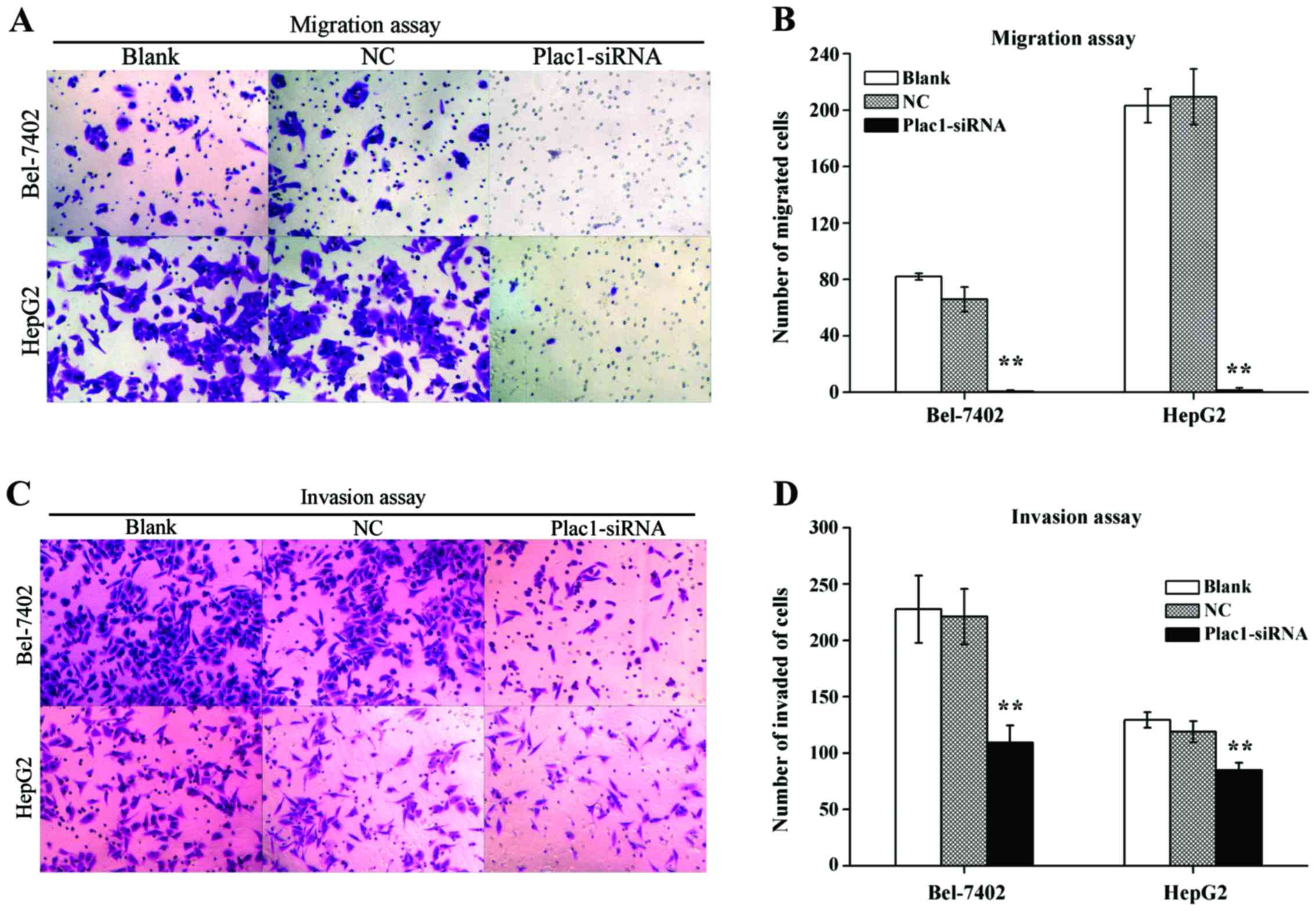
Decreased Plac1 expression represses
cell migration and invasion in HCC cells
Because Plac1 overexpression was significantly
associated with HCC metastasis, the effect of Plac1 in tumor cell
migration and invasion was investigated. Transwell migration assay
revealed a distinctly impaired migratory ability in Plac1-siRNA
transfected cells vs. control cells. The quantity of blank, NC and
Plac1-siRNA cells, which migrated to the lower face of the
Transwell membrane were 82±2.28, 65.83±8.73 and 0.67±0.82 in
Bel-7402 cells, respectively, and corresponding cell numbers were
203.17±11.94, 209.5±19.75 and 1.5±1.64 in HepG2 cells, respectively
(Fig. 5A and B). Concordantly,
knockdown of Plac1 also diminished cell invasion through Matrigel.
As shown in Fig. 5C and D,
Plac1-siRNA Bel-7402 cells demonstrated a decrease in the cell
quantity (109.17±15.3) vs. blank or NC cells (227.67±29.97 or
221±24.71, respectively), while invaded cells were numbered
(84.83±6.68) in Plac1-siRNA HepG2 cells, (129.33±6.92 and 119±9.36)
in blank and NC HepG2 cells, respectively. Thus, these findings
confirmed that Plac1 promotes the motility and invasiveness of HCC
cells.
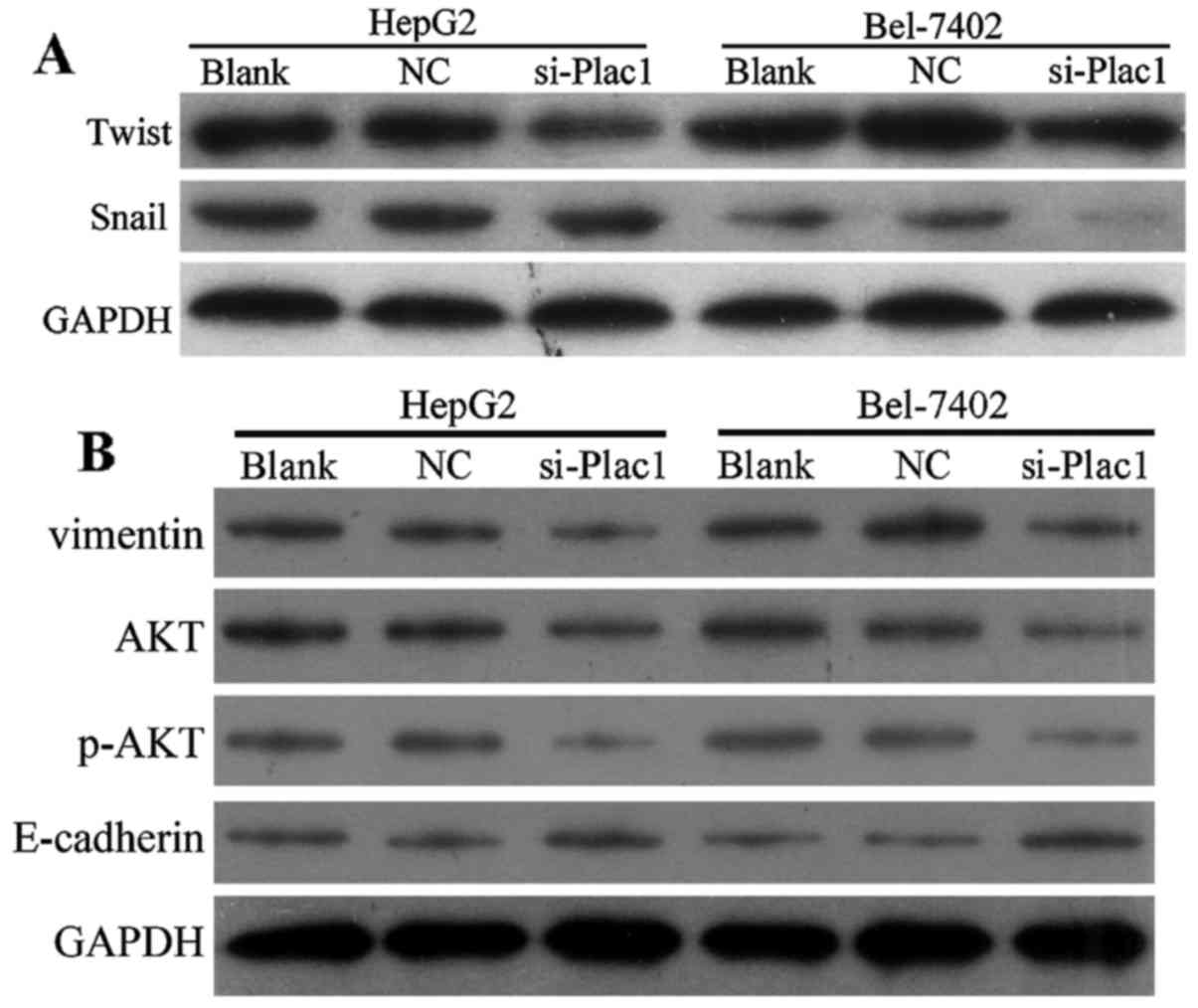
Depletion of Plac1 suppresses EMT and
leads to p-AKT reduction in HCC cells
As epithelial to mesenchymal transition (EMT) is one
of the key events in tumor invasion and metastasis, we next
conducted studies to investigate if EMT is responsible for
Plac1-mediated change in cell motility. Compared with control
cells, the epithelial marker E-cadherin expression had an increase
in the cells treated with Plac1-siRNA, accompanied by a reduction
of mesenchymal marker vimentin (Fig.
6B). Since Snail and Twist are transcription factors that have
been demonstrated as key EMT regulators by suppressing the
epithelial genes and inducing the mesenchymal genes, we further
examined whether increased E-cadherin expression is the result of
decreased Snail and Twist levels in the HCC cells treated with
Plac1 siRNA. It is suggested that downregulation of Plac1 repressed
Snail and Twist expression (Fig.
6A). Overall, our data are consistent with the hypothesis that
depletion of Plac1 elevated E-cadherin protein level via inhibiting
Snail and Twist expression. Considering the important regulator of
AKT activation in promoting cancer cell proliferation, motility and
invasiveness, we sought to evaluate the effect of Plac1 on AKT
signaling in HCC. In Bel-7402 and HepG2 cells, decreased Plac1
expression markedly reduced phosphorylation of AKT (Fig. 6B). Thus, we interpret these data to
support that Plac1 exerts carcinogenic effect possibly through the
activation of AKT pathway.
Discussion
The incidence of HCC has been increased in the
western and Asian countries. Although advances have been made in
treatment of HCC with surgery, chemotherapy and radiotherapy, the
mortality rate of HCC is still high. Good diagnostic markers, drug
targets and therapeutic strategies are in urgent need for a
successful treatment of HCC.
Plac1 is frequently expressed in the cells of the
trophoblast lineage and in different types of malignant tumors
(7–10). Additionally, overexpression of Plac1
is correlated with survival of gastric adenocarcinoma (11). Ghods et al (12) found that Plac1 presented
differential expression in prostate cancer, and positivity was
associated with the Gleason score, and negativity correlated with
prostate specific antigen (PSA) expression, thus highlighting the
potential usefulness of Plac1 for developing targeted prostate
cancer therapies, particularly for patients with advanced disease.
A recent study showed that Plac1 expression was absent in normal
tissues except for placenta, and expressed in partial HCC samples,
including mRNA and protein levels (8). Accordingly, we demonstrated that
increased expression of Plac1 was observed in 54.3% of HCC samples
and in several types of HCC cell lines, whereas non-cancerous liver
tissues and normal liver L02 cells barely expressed
Plac1 in this study. Furthermore, HCC patients who had Plac1
expression had higher incidence of metastasis than those without
Plac1 expression, which confirmed the effect of Plac1 on the
migration and invasion of tumor cells. Taken together, these
findings indicated that Plac1 may be an oncogene promoting HCC
progression.
To address the pathobiological role of Plac1 in the
HCC progess, we introduced Plac1 specific siRNA to silence Plac1 in
Bel-7402 and HepG2 cells, and assessed the impact of Plac1 on cell
proliferation, cell cycle, apoptosis and invasiveness. The present
study revealed that cell proliferation resulting from knockdown of
Plac1 was suppressed. Moreover, downregulation of Plac1 in HCC
cells resulted in a significant cell retardation in the G1 phase
and increased apoptosis, which implicated a role of Plac1 in the
resistance to apoptosis and enhancing HCC cell proliferation. These
results provide further evidence to confirm Plac1 as a candidate
oncogene in HCC. Notably, a study by Koslowski et al
(10) showed that Plac1 had no an
effect on apoptosis in breast cancer cell line MCF-7, the cell line
variability may explain the differences between their finding and
ours.
To elucidate how Plac1 affects the proliferation and
cell cycle of HCC cells, we explored the levels of cell
cycle-related protein. Cyclin D1 known as cyclin, and CDK4 called
the cyclin-dependent kinase, mediate cell proliferation and
differentiation through regulating the G1/S transition of the cell
cycle (13–15). We found that the expression of CDK4
and cyclin D1 were decreased when Bel-7402 and HepG2 cells were
treated with Plac1-siRNA. Considering the reported role of
Plac1-siRNA in inducing G1/S arrest and repressing CDK4 and cyclin
D1 expression of breast cancer cells (10), it is conceivable that Plac1
regulates cell cycle distribution by controlling the levels of cell
cycle-related proteins.
It is well known that cancer development and
progression are attributed to destruction of the balance between
cell proliferation and apoptosis (16). Since p53 possesses antitumorigenic
function by regulating cell cycle and apoptosis (17,18),
we were interested if this key cell apoptosis-signaling protein was
involved in the effect of Plac1 on apoptosis in HCC. Our findings
in HepG2 and Bel-7402 cells showed that downregulation of Plac1
enhanced p53 expression, raises the possibility that Plac1 exerts
biological behavior in HCC through suppressing the p53 level.
Previous studies have demonstrated that the p53 protein can
interact with the proteins of Bcl-2 family, which are classified as
pro-apoptotic (Bax, Bad, Bid and Bim) and anti-apoptotic (Bcl-2 and
Bcl-XL) (21) and induce apoptosis
(19,20). It has been shown that p53 induced
apoptosis through repressing the transcription of Bcl-2 (22). Bel-7402 cells treated with Plac1
siRNA showed decreased expression of Bcl-2, which was inversely
related to p53 expression, implying that downregulation of Bcl-2
expression in Bel-7402 cells might be suppressed by p53. This
hypothesis is supported by previous studies showing that Bcl-2 is
negatively regulated by the tumor suppressor p53 (23,24).
Surprisingly, instead of upregulation, Bax expression was inhibited
in Bel-7402 cells transfected with Plac1 siRNA. In fact, the
Bax/Bcl-2 ratio is more important in determining the sensitivity to
apoptotic stimuli in cancer cells than the expression of each
protein individually (25). In
contrast to Bel-7402 cells, we detected that silence of Plac1
strongly increased Bcl-2 expression in HepG2 cells, while Bax
expression had a reduction. These data disclose that the mechanism
of Plac1 on cell apoptosis in HepG2 and Bel-7402 cells are unlikely
identical. Bax and Bcl-2 does not account for the
Plac1-siRNA-mediated apoptosis in HepG2 cells, and another novel
mechanism mediating apoptosis in HepG2 cells must be presented.
The correlation between Plac1 expression and
metastasis of HCC in clinical specimens prompted our investigation
into the role of Plac1 in cell motility and invasion. Concordantly,
our data demonstrated that Plac1 promote migration and invasion of
HCC, supported by the fact that knockdown of Plac1 suppressed
Bel-7402 and HepG2 cell migration and invasion ability in
vitro by using Transwell assay. Our further focus in the
present study was to determine whether the changes in migration and
invasion were caused by EMT. EMT is a pivotal physiological process
contributing to cancer cell invasion and migration, including HCC
(26,27). It is by a dedifferentiation program
that epithelial cells lose their epithelial signatures and gain
mesenchymal characteristics (28).
Downregulation of E-cadherin, an epithelial cell adhesion molecule,
is regarded as the critical step of EMT (29,30).
Upregulation of mesenchymal marker vimentin and the most prominent
suppressors of E-cadherin such as Snail, Twist, ZEB-1 and ZEB-2,
which bind to E-boxes of E-cadherin promoter and suppress its
transcription, are intensively involved in the process of EMT
(29–32). In this study, downregulation of
Plac1 elevated E-cadherin expression, accompanied by a distinct
decreased of vimentin. This observation was reinforced by results
with downregulation of Snail and Twist expression, which repress
the expression of E-cadherin. Based on these observation, we
conclude that Plac1 likely exerts its carcinogenic effect in HCC
through development of EMT, thus, promoting cancer metastasis. We
hope to gain more detailed insights into the molecular mechanism,
in which Plac1 regulates EMT in our future research.
As PI3K/AKT pathway is an important regulatory
signal for cancer process (33,34),
we wondered whether PI3K/Akt participated in Plac1-mediated
biological behavior in HCC. We observed that phosphorylation of AKT
was attenuated in Plac1-siRNA HCC cells, suggesting Plac1 might be
involved in HCC carcinogenesis by activating PI3K/AKT signaling
pathway. The finding is consistent with the result that
phosphorylation of AKT was markedly decreased in breast cancer
MCF-7 cells treated with Plac1 siRNA, and less prominent in another
breast cancer cell line the BT-549 transfected with Plac1 siRNA,
which lack PTEN. There is evidence to show that PTEN, which can
bind to p53 and enhance p53-mediated functions, is a negative
regulator of AKT (34,35). Notably, the present study showed a
positive effect of Plac1 on the AKT signaling pathway and a
negative effect of Plac1 on p53. Little is known about Plac1 effect
on PTEN level, but we hypothesize that Plac1 caused an activation
of AKT pathway possibly through inhibition of PTEN and p53, thereby
promoting HCC cell proliferation and metastasis. However, the
detailed mechanism remains to be elucidated.
In conclusion, this study highlights that Plac1 is a
critical factor for HCC development by facilitating HCC
proliferation, cell cycle progression, cell migration through
enhancement of EMT and resisting tumor cell apoptosis. Thus, Plac1
may serve as a promising target molecule for the development of
novel therapeutic drugs in HCC.
Acknowledgements
The present study was supported by the Science and
Technology Planning Project of Guangdong Province, China (no.
2012B031800427), and the Science and Technology Planning Project of
Zhanjiang, Guangdong, China (no. 2013A01012).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maluccio M and Covey A: Recent progress in
understanding, diagnosing, and treating hepatocellular carcinoma.
CA Cancer J Clin. 62:394–399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cocchia M, Huber R, Pantano S, Chen EY, Ma
P, Forabosco A, Ko MS and Schlessinger D: PLAC1, an Xq26 gene with
placenta-specific expression. Genomics. 68:305–312. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fant M, Weisoly DL, Cocchia M, Huber R,
Khan S, Lunt T and Schlessinger D: PLAC1, a trophoblast-specific
gene, is expressed throughout pregnancy in the human placenta and
modulated by keratinocyte growth factor. Mol Reprod Dev.
63:430–436. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fant M, Barerra-Saldana H, Dubinsky W,
Poindexter B and Bick R: The PLAC1 protein localizes to membranous
compartments in the apical region of the syncytiotrophoblast. Mol
Reprod Dev. 74:922–929. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chang WL, Yang Q, Zhang H, Lin HY, Zhou Z,
Lu X, Zhu C, Xue LQ and Wang H: Role of placenta-specific protein 1
in trophoblast invasion and migration. Reproduction. 148:343–352.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu FF, Dong XY, Pang XW, Xing Q, Wang HC,
Zhang HG, Li Y, Yin YH, Fant M, Ye YJ, et al: The specific immune
response to tumor antigen CP1 and its correlation with improved
survival in colon cancer patients. Gastroenterology. 134:998–1006.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dong XY, Peng JR, Ye YJ, Chen HS, Zhang
LJ, Pang XW, Li Y, Zhang Y, Wang S, Fant ME, et al: Plac1 is a
tumor-specific antigen capable of eliciting spontaneous antibody
responses in human cancer patients. Int J Cancer. 122:2038–2043.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Silva WA Jr, Gnjatic S, Ritter E, Chua R,
Cohen T, Hsu M, Jungbluth AA, Altorki NK, Chen YT, Old LJ, et al:
PLAC1, a trophoblast-specific cell surface protein, is expressed in
a range of human tumors and elicits spontaneous antibody responses.
Cancer Immun. 7:18–25. 2007.PubMed/NCBI
|
|
10
|
Koslowski M, Sahin U, Mitnacht-Kraus R,
Seitz G, Huber C and Türeci O: A placenta-specific gene ectopically
activated in many human cancers is essentially involved in
malignant cell processes. Cancer Res. 67:9528–9534. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu F, Shen D, Kang X, Zhang C and Song Q:
New tumour antigen PLAC1/CP1, a potentially useful prognostic
marker and immunotherapy target for gastric adenocarcinoma. J Clin
Pathol. 68:913–916. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ghods R, Ghahremani MH, Madjd Z, Asgari M,
Abolhasani M, Tavasoli S, Mahmoudi AR, Darzi M, Pasalar P,
Jeddi-Tehrani M, et al: High placenta-specific 1/low
prostate-specific antigen expression pattern in high-grade prostate
adenocarcinoma. Cancer Immunol Immunother. 63:1319–1327. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Satyanarayana A and Kaldis P: Mammalian
cell-cycle regulation: Several Cdks, numerous cyclins and diverse
compensatory mechanisms. Oncogene. 28:2925–2939. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hochegger H, Takeda S and Hunt T:
Cyclin-dependent kinases and cell-cycle transitions: Does one fit
all? Nat Rev Mol Cell Biol. 9:910–916. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fung TK and Poon RY: A roller coaster ride
with the mitotic cyclins. Semin Cell Dev Biol. 16:335–342. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Avery-Kiejda KA, Bowden NA, Croft AJ,
Scurr LL, Kairupan CF, Ashton KA, Talseth-Palmer BA, Rizos H, Zhang
XD, Scott RJ, et al: P53 in human melanoma fails to regulate target
genes associated with apoptosis and the cell cycle and may
contribute to proliferation. BMC Cancer. 11:203–219. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang YQ, Xiao CX, Lin BY, Shi Y, Liu YP,
Liu JJ, Guleng B and Ren JL: Silencing of Pokemon enhances
caspase-dependent apoptosis via fas- and mitochondria-mediated
pathways in hepatocellular carcinoma cells. PLoS One. 8:e689812013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zand H, Rhimipour A, Bakhshayesh M,
Shafiee M, Mohammadi I Nour and Salimi S: Involvement of PPAR-gamma
and p53 in DHA-induced apoptosis in Reh cells. Mol Cell Biochem.
304:71–77. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sankari SL, Masthan KMK, Babu NA,
Bhattacharjee T and Elumalai M: Apoptosis in cancer - an update.
Asian Pac J Cancer Prev. 13:4873–4878. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Reed JC: Bcl-2 family proteins: Regulators
of apoptosis and chemoresistance in hematologic malignancies. Semin
Hematol 34 (Suppl 5). 9–19. 1997.
|
|
22
|
Yee KS and Vousden KH: Complicating the
complexity of p53. Carcinogenesis. 26:1317–1322. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakazawa K, Dashzeveg N and Yoshida K:
Tumor suppressor p53 induces miR-1915 processing to inhibit Bcl-2
in the apoptotic response to DNA damage. FEBS J. 281:2937–2944.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Borghetti G, Yamaguchi AA, Aikawa J,
Yamazaki RK, de Brito GA and Fernandes LC: Fish oil administration
mediates apoptosis of Walker 256 tumor cells by modulation of p53,
Bcl-2, caspase-7 and caspase-3 protein expression. Lipids Health
Dis. 14:94–98. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Raisova M, Hossini AM, Eberle J, Riebeling
C, Wieder T, Sturm I, Daniel PT, Orfanos CE and Geilen CC: The
Bax/Bcl-2 ratio determines the susceptibility of human melanoma
cells to CD95/Fas-mediated apoptosis. J Invest Dermatol.
117:333–340. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maheswaran T and Rushbrook SM:
Epithelial-mesenchymal transition and the liver: Role in
hepatocellular carcinoma and liver fibrosis. J Gastroenterol
Hepatol. 27:418–420. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li YM, Xu SC, Li J, Han KQ, Pi HF, Zheng
L, Zuo GH, Huang XB, Li HY, Zhao HZ, et al: Epithelial-mesenchymal
transition markers expressed in circulating tumor cells in
hepatocellular carcinoma patients with different stages of disease.
Cell Death Dis. 4:e8312013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Panebianco C, Saracino C and Pazienza V:
Epithelial-mesenchymal transition: Molecular pathways of hepatitis
viruses-induced hepatocellular carcinoma progression. Tumour Biol.
35:7307–7315. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zheng H and Kang Y: Multilayer control of
the EMT master regulators. Oncogene. 33:1755–1763. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhai X, Zhu H, Wang W, Zhang S, Zhang Y
and Mao G: Abnormal expression of EMT-related proteins, S100A4,
vimentin and E-cadherin, is correlated with clinicopathological
features and prognosis in HCC. Med Oncol. 31:9702014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: An alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Peinado H, Portillo F and Cano A:
Transcriptional regulation of cadherins during development and
carcinogenesis. Int J Dev Biol. 48:365–375. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Faes S and Dormond O: PI3K and AKT:
Unfaithful partners in cancer. Int J Mol Sci. 16:21138–21152. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Martini M, De Santis MC, Braccini L,
Gulluni F and Hirsch E: PI3K/AKT signaling pathway and cancer: An
updated review. Ann Med. 46:372–383. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Trotman LC, Wang X, Alimonti A, Chen Z,
Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo
C, Erdjument-Bromage H, et al: Ubiquitination regulates PTEN
nuclear import and tumor suppression. Cell. 128:141–156. 2007.
View Article : Google Scholar : PubMed/NCBI
|