Introduction
Among the four main histopathological types of
gliomas, glioblastoma multiforme (GBM, grade IV astrocytoma) is the
most aggressive subtype, leading to death within 12–15 months
(1,2). The most common oncogenic events
encountered in GBM include, amplification of the epidermal growth
factor receptor (EGFR) (3,4);
overexpression (OE) of components of the platelet derived growth
factor (PDGF) pathway (PDGF-A, PDGF-B, PDGFR-α and PDGFR-β)
(4,5); mutations of genes residing in
chromosome 10, including the tumor suppressor phosphatase and
tensin homolog (PTEN) (4,6,7).
Recent evidence suggests however that stem and/or progenitors cells
in the adult brain may give rise to ‘glioma stem cells’ (GSCs)
representing transformed normal neural stem cells (NSCs) (8–10).
These cells are descendents of the bipotent radial glial cells
(RGCs), acting as the main progenitor cell type in the developing
cortex that are specified and controlled by the expression of the
transcription factor (TF) Pax6 (11–16,29).
In vertebrate CNS, Pax6 exists in three isoforms; the canonical
Pax6, a longer Pax6-5a isoform and the paired-less Pax6 form (∆PD).
Previous functional in vivo analyses suggested that, while
canonical Pax6 regulates neurogenesis, cell proliferation, and
patterning in developing telencephalon, the spliced Pax6-5a isoform
affects specifically cell proliferation (17). However, in murine embryonic stem
cells in vitro, Pax6-5a strongly stimulates neuronal
differentiation, while Pax6 failed to show such a robust influence
(18).
In humans with GBM, low PAX6 level is considered a
negative prognostic marker for the patient (19,20).
In highly malignant tumors, the low level of PAX6 correlates with
the elevated expression of vascular endothelial growth factor
(VEGF), while PAX6 overexpression (OE) in subcutaneous
xenographs suppresses the formation of new blood vessels (19). Along the same track, in glioma cell
lines, a stable OE of PAX6 suppresses the tumorigenic potential and
improves significantly the survival of nude mice, intra-cranially
implanted with these cells (20).
Together with the reported strong dose-dependent effect for
controlling the balance between NSC self-renewal and neurogenesis
(21), TF Pax6 is a good candidate
that may also act as a mediator of neurogenic versus certain
aspects of tumorigenic capacity of NSC in the adult brain.
MicroRNAs (miRNAs) have been identified as ‘tools’
for the in vivo regulation of the endogenous expression
level of molecular determinants that are acting as tumor suppressor
genes or oncogenes (22,23). Hierarchical clustering analysis
based on the expression profiles of 71 miRNAs in human gliomas
defined similar signatures in brain tumors and normal neural stem
cells (NSCs), suggesting that specific miRNAs may enforce
tumorigenesis at the expense of normal brain neurogenesis (24). The clusters/families that displayed
NSC-like miRNA signatures include miR-17, miR183-96-182,
miR367-302, miR371-373 (24).
miR-183-96-182 is a polycistronic, paralogous cluster of miRNAs,
which have initially been identified as sensory organ specific
(25,26), but also shown to be upregulated in
brain cancer (27–32). In glioma tumor biopsies, miR-182
showed specifically elevated expression (33) with the highest level attained in GBM
(grade IV) (33). miR-183 has been
associated with the targeting of isocitrate dehydrogenase 2 (IDH2)
in glioma (34), while knockdown
(KD) of the whole miRNA cluster induced oxidative apoptosis thus
inhibiting the survival of glioma cells in vitro (35).
Herein, we show that all members of the
miR-183-96-182 cluster are highly upregulated in 29 human glioma
tumor biopsies and 3 glioma cell lines, which correlates with a
simultaneous dramatic reduction of both PAX6 and PAX6-5a isoforms,
implying that they could be used as novel diagnostic tools for
high-grade glioma. We disclose the preferential regulation of
PAX6-5a expression by miR-183, while the canonical PAX6 expression
is more strongly affected by both miR-182 and miR-96.
Mechanistically, we demonstrate that the tumor suppressive
potential of the increased PAX6/PAX6-5a ‘dose’, could be a result
of down regulation of SPHK1 and CTNND2, two key factors for glioma
progression, leading to decreased proliferation and increase death
of GBM cells, in vitro.
Materials and methods
RNA extraction and qRT-PCR
Total RNA was purified from cell cultures using
TRIzol reagent according to manufacturer's instructions
(Invitrogen). For mature miRNA expression analysis, cDNA was
synthesized using the TaqMan MicroRNA Reverse Transcription kit
(Applied Biosystems) and 50–100 ng of total RNA along with miRNA
specific stem loop primer, supplied with the TaqMan microRNA Assays
kit (Applied Biosystems). The assays used were: hsa-miR-183
(002269; Applied Biosystems), hsa-miR-96 (000186; Applied
Biosystems) and hsa-miR-182 (002334; Applied Biosystems). qPCR was
performed with TaqMan PCR Master Mix according to the
manufacturer's instructions and U6 snRNA (001973; Applied
Biosystems) or snoRNA202 TaqMan (001232; Applied Biosystems) probes
were used as endogenous controls. For gene expression analysis,
cDNA was synthesized using the QuantiTect Reverse Transcription kit
(Qiagen) according to manufacturer's instructions and 100–150 ng of
total RNA. qPCR was performed with GoTaq® qPCR Master
Mix (Promega). Primer sequences of target genes were as follows;
Pax6-forward: 5′-gaatcagagaagacaggcca-3′; Pax6-reverse:
5′-gtgtaggtatcataactccg-3′; Pax6(5a)-forward: 5′-gagtgaatcag
ctcggtgg-3′; Pax6(5a)-reverse 1: 5′-ggacttttgcatctgcatgg-3′;
Pax6(5a)-Reverse 2: 5′-cgttttgattgtccagcac-3′; β-actin-Forward:
5′-tccttcctgggcatggagt-3′; β-actin-Reverse: 5′-aaagccatgc
caatctcatc-3′. Also the following Quantitect primer assays were
purchased from Qiagen and used according to manufacturer's advice:
Hs_PDGFRA_1_SG (QT00012719), Hs_SPHK1_1_SG (QT01011927),
Hs_PDGFRB_1_SG (QT00082327), Hs_CTNND1_1_SG (QT00033831),
Hs_PAX6_1_SG (QT00071169), Hs_PDGFA_1_SG (QT01664488), Hs_TAM1_1_SG
(QT01009568), Hs_GADPH_1_SG (QT00079247), Hs_PDFGB_1_SG
(QT00001260), Hs_ACTB_1_SG (QT00095431), Hs_NF1_1_SG (QT00065016).
In all cases, qPCR was performed on a Realplex2 Mastercycler
(Eppendorf).
miRNA expression vectors, mimics and
inhibitors
The pre-miR-182, pre-miR-183 and pre-miR-96
sequences were amplified by PCR from human genomic DNA, using the
following oligos; miR-182-For: 5′-ctgtgggaagagcgccctc-3′;
miR-182-Rev: 5′-caccgagaagaggtcgacttc-3′; miR-183-For:
5′-gagctggtgaggagggttgc-3′; miR-183-Rev: 5′-ccaagcagatg
gtactggaac-3′; miR-96-For: 5′-gcaggctggagagtgtgactc-3′; miR-96-Rev:
5′-caggcagtgaaaggtgatctg-3′. The products were purified and cloned
into the pCIG2 expression vector (CMV-EGFP) upstream of the EGFP
sequence. The expression of mature miRNA was verified by qRT-PCR,
with TaqMan MicroRNA assays. The following commercial miRNA mimics
and inhibitors were purchased from Qiagen and were used according
to the manufacturer's advice; miScript miRNA mimics:
Syn-hsa-miR-183 (MSY0000261), Syn-hsa-miR-182 (MSY0000259),
Syn-hsa-miR-96-5p (MSY0000095). miScript miRNA inhibitors:
anti-hsa-96-5p (MIN0000095), anti-hsa-miR-183-5p (MIN0000261) and
anti-hsa-miR-182-5p (MIN0000259).
Immunocytofluorescence (ICF)
For ICF, the cells were seeded on
Lab-Tek®II Chamber Slide™ system (Nalge Nunc
International) chambers. The cells were transfected and remained in
culture for 24–72 h, after which, they were fixed in 4%
praraformaldehyde (w/v) for 7 min in room temperature (RT).
Following washes in PBS, the cells were treated with PBS containing
10% normal goat serum (NGS) (blocking buffer) for 1 hour in RT and
incubated with the primary antibodies overnight (O/N) at 4°C. The
antibodies were diluted as follows in blocking buffer; chicken
anti-GFP (1:500; Abcam: ab13970), rabbit anti-δ-catenin (1:50;
Santa Cruz Biotechnology Inc. (H-160) sc-33553), rabbit anti-SPHK1
antibody (1:100; Abcam: ab71700), rabbit anti-Pax6 (1:300; Covance:
PRB-278P-100), rabbit anti-Ki67 (1:100; Vector Laboratories, Enzo
Life Sciences GmbH, Lörrach, Germany VP-RM04), rabbit
anti-phospho-Histone H3 (1:500; Abcam: ab5176), rabbit anti-PDGF BB
(1:300; Abcam: ab23914). Primary antibodies were detected with
appropriate secondary antibodies conjugated to Alexa Fluor
fluorophores (1:500; Invitrogen), and cells were counterstained
with Vectashield mounting medium containing
4′,6′diamidino-2-phenylindole (DAPI) (Vector Laboratories, Enzo
Life Sciences GmbH, Lörrach, Germany) to label the cell nuclei.
Five randomly selected microscopic fields were selected and
pictured for each condition and the positive cells counted. The
images were acquired using an Olympus XC30 camera attached to an
Olympus BX60 fluorescence microscope and they were processed with
Microsoft's power point format-picture command for corrections in
brightness/contrast.
Luciferase reporter assays
The 3′UTR of Pax6, was cloned into the pGL3-promoter
reporter vector (Promega, GmbH, Mannheim, Germany). Constructs
carrying mutations in the potential targeting sites were also
generated and used as controls. Transfections were performed in
24-well plates using Lipofectamine 2000 (Invitrogen Life
Technologies GmbH, Darmstadt, Germany) and the following vector DNA
concentrations per well: 0.4ng Pax6 3′UTR construct (or Pax6
3′UTR-mutated construct or pGL3 empty plasmid), 0.4 ng miRNA
expression plasmid, 0.2 ng Renilla Luciferase plasmid (Promega,
GmbH, Mannheim, Germany). Dual-Luciferase Reporter Assay (Promega,
GmbH, Mannheim, Germany) was carried out 48 h after transfection
and Firefly luciferase activity (F) was normalized to Renilla
luciferase reporter activity (F/R). The value (F/R) of the miRNA
expression plasmid + pGL3 empty vector transfected cells, was set
to 1.
Cell culture and transfections
U118, U87 and U251 glioma cell lines as well as HeLa
and P19 cells were purchased from Cell Lines Services, and were
grown in Dulbecco's modified Eagle's medium (DMEM) (Gibco),
supplemented with 10% Fetal Bovine Serum (FBS) (Gibco). Transient
transfections of cells were performed using Lipofectamine LTX with
PLUS reagent (Invitrogen) and endotoxin-free DNA plasmids (Qiagen
kit) or commercially purchased (Qiagen) miRNA mimics and inhibitors
(see miRNA expression vectors, mimics and inhibitors section of
Materials and methods), in 96-well plates, 24-well plates and
Lab-Tek II Chamber Slide system chanbers (Nalge Nunc International)
according to the supplier's guidelines.
Human glioma samples
The collection of the glioma tumor specimens was
done in collaboration with the Medical University, Varna, Bulgaria.
(Department of Anatomy, Histology and Embryology - Head, Professor
A. Tonchev; The Neurosurgery Clinic - Director, Dr Y. Enchev;
Department of Neurology - Dr A. Kaprelyan). In total 22 GBM and 7
low-grade glioma tumors were analyzed in Max Planck Institute for
Biophysical Chemistry, Göttingen, Germany. The RNA extraction and
qRT-PCR analyses were done as described in previous sections. The
collection and use of tissues were approved by the Ethics Committee
of the Medical University of Varna.
Cell culture assays
To perform wound-healing assay, U87MG cells were
seeded in 24-well plates and transfected with miRNA expression
plasmids, miRNA inhibitors, control GFP-expressing plasmid, PAX6 or
PAX6-5a expression plasmids. When the cells reached confluence, a
wound was gently made through the central axis of the plate, by
scraping the cell monolayer with a micropipette 200 µl tip. The
cultures were washed with serum free (SF) media to remove floating
cells and placed back in the incubator. Pictures of the scratches
were taken at selected time points, in five randomly selected
microscopic fields for each condition and time point, using a
digital camera Prog/Res/3012 from Kotron Elektronik coupled to a
Leitz Labovert microscope (Leica). The images were processed with
Microsoft power point format-picture command, for brightness and
contrast corrections. The same filter was applied in every picture
as to make the cells clearer and to correct any background
coloration.
The invasion assays were performed using the BD
Biocoat™ kit (BD Matrigel Invasion Chamber 24-well plate 8.0
Micron; BD Biosciences; Heidelberg, Germany) as instructed by the
supplier. The invaded cells on the lower membrane surface were
fixed in 100% methanol for 10 min, air-dried, stained with DAPI and
counted under an Olympus BX60 fluorescence microscope. In the cell
survival assays U87 cells were seeded in 24-well plates and allowed
to reach 80% confluence in media with serum, followed by washes in
SF-media. The cells were then transfected with either miRNA or
PAX6/PAX6-5a expression plasmids and allowed to grow in SF-media
for 72 h in total. Pictures were taken at defined time points with
a digital camera Prog/Res/3012 from Kotron Elektronik coupled to a
Leitz Labovert microscope (Leica).
Western blot (WB) analysis
WB analyses were done as previously described
(36). The following primary and
secondary antibody dilutions were used: rabbit anti-Pax6 polyclonal
antibody (1:500; Covence: PRB-278P-100), rabbit anti-δ-catenin
(1:200; Santa Cruz Biotechnology Inc.: (H-160) sc-33553), rabbit
anti-PDGF BB (1:500; Abcam: ab23914), rabbit anti-β-actin
polyclonal antibody (1:500, Abcam: ab8227). β-actin served as
loading control and PageRuler pre-stained protein ladders
(Fermentas) were utilized for the identification of the correct
sized proteins.
Caspase 3/7 assays
Cell death measurement studies were performed using
a Caspase-Glo® 3/7 Assay System from Promega, according
to the manufacturer's instructions. The luminescence was measured
in a Centro LB 960 Luminometer (Berthold Technologies).
Ago2 immunoprecipitation of
targets
The Ago2 IP experiments were done exactly as
previously described (30), using
P19 cells seeded in 24-well plates. The immunoprecipitated RNA was
analyzed by qRT-PCR using gene-specific primers and normalized to
U6 RNA. GAPDH served as negative control. The analysis was done as
follows: GFP control vector, pull-down/input (A); miRNA,
pull-down/input (B); fold enrichment = B/A.
Algorithm tools/Target prediction
databases
The following algorithms were applied to find miRNAs
that target PAX6: Targetscan (http://www.targetscan.org), DIANAmT (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=microT_CDS/index),
miRWalk (http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/),
PICTAR 5 (http://pictar.mdc-berlin.de), RNA22
(http://cbcsrv.watson.ibm.com/rna22.html). For the
analysis of the miR-183-96-182 promoter for putative PAX6 binding
sites, we used the RVISTA 2.0 database (http://rvista.dcode.org).
Results
Upregulation of the miR-183-96-182
cluster correlates with reduced level of PAX6 and PAX6-5a and
increased glioma progression
Although high expression of PAX6 has been
assumed to slow down the GBM progression (20), the specific role of each of the
PAX6 isoforms has not been approached so far. In order to
determine whether the expression of PAX6/PAX6-5a and
miR-183-96-182 cluster correlates at different stages of glioma
progression, we utilized qRT-PCR (∆∆Ct relative quantification) to
analyze primary human glioma tissue specimens (29 samples in total)
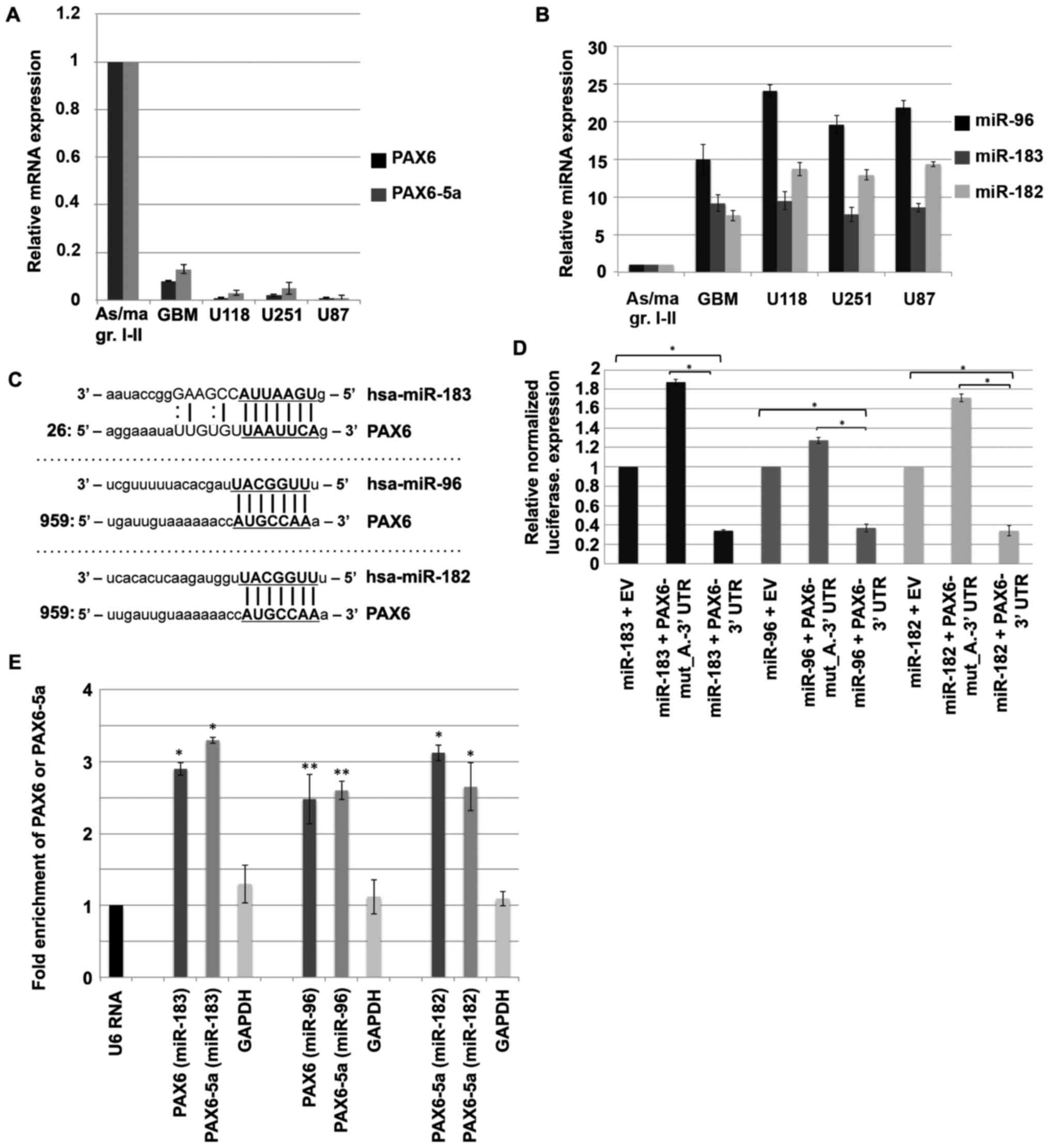
and glioma cell lines (U118, U251 and U87). Fig. 1A shows the relative expression
levels of both PAX6 isoforms that are significantly lower
(7.7–12.5-fold down, respectively) in GBM (22 samples of primary
GBM, grade IV) compared to astrocytomas (grades I and II; 7 samples
in total). In the tested four high-grade glioma cell lines,
PAX6/PAX6-5a level was markedly downregulated compared to
low-grade glioma tumors. On the contrary, all members of the
miR-183-96-182 cluster were highly upregulated in GBM, as well as
in high-grade glioma cell lines (Fig.
1B). miR-96 appears the most abundantly expressed miRNA in
glioblastoma tumors, showing more than 14-fold upregulation
compared to low-grade astrocytoma (Fig.
1B).
PAX6 is a direct target of
miR-183-96-182 cluster
These observations prompted us to inquire more into
the possible regulatory relationship between the PAX6 isoforms and
the microRNA cluster. Using several target prediction programs (see
Materials and methods) we examined whether the 3′-UTR of PAX6
contains putative sites for miR-183, miR-96 and miR-182
interactions. All programs predict one binding site for each
miR-182 and miR-96 in the 3′UTR region of the PAX6 gene.
Both sites, are eptamers (7mer-1A) and similar for the two miRNAs
because their seed sequences are alike (Fig. 1C). MiR-183 complementarity is
predicted by two algorithms (miRanda and miRWalk).
To validate direct binding and targeting by
miR-183-96-182, we constructed firefly luciferase reporter plasmids
containing either wild-type or mutated 3′UTR of PAX6. In
order to disrupt the predicted binding, plasmids containing
mutations within the miR-183, miR-96 and miR-182 seed-binding
regions in the 3′UTR of PAX6 were designed and generated.
Reporter activities were quantified and normalized to non-targeted
Renilla luciferase activity. As shown in Fig. 1D, cotransfections of miRNA
expression plasmids together with wild-type
PAX6-3′UTR-reporter in HeLa cells, resulted in pronounce
reduction of the relative luciferase activity, approximately
2.9-fold by either miR-182 or miR-183 and 2.7-fold by miR-96.
Mutations (Table I) of the
predicted specific miRNA recognition elements completely restored
luciferase expression (Fig. 1D). To
further confirm the direct regulation of PAX6 and
PAX6-5a by miR-182-96-183 cluster, we utilized AGO2
immunoprecipitation of targets (AGO2-IP) as previously described
(30). The analysis of the results
shows that miR-182/AGO2, miR-183/AGO2 and miR-96/AGO2 complexes
associate selectively with the PAX6 or PAX6-5a
transcripts (Fig. 1E). The levels
of both PAX6 and PAX6-5a mRNA were significantly
higher (more than 2.5-fold) than control transcripts (U6 RNA and
GAPDH mRNA) (Fig. 1E),
suggesting that both PAX6 and PAX6-5a are targeted by the clustered
miRNAs.
 | Table I.Primers used for PAX6-3′UTR
mutagenesis. |
Table I.
Primers used for PAX6-3′UTR
mutagenesis.
| Primers | Sequence |
|---|
| PAX6 mutant_A
3′UTR | Forward:
ATTGTCTAGAGGAAAGGAAATATTGTGTTAGCTCAG |
|
| Reverse:
GCTTTCTAGATCATGACCAACACAGATCAAAC |
| PAX6 mutant_B
3′UTR | Forward:
CAGGTCTCTAGACCATGTGAAAGCCTTTGTATTTCC |
|
| Reverse:
GTAACTTCTAGACTGACAATGGAAATCTGCCCAG |
Frequently, miRNAs and TFs participate in
feed-forward and feedback loop regulatory networks (37,38). A
search for putative binding sites of PAX6 in the promoter of
miR-183-96-182 in silico, identifies TFs PAX6 and BACH2 as
candidate regulators of the miR-183-96-182 cluster (based on highly
conserved sequences between species and known TF binding motifs
within these regions, the RVISTA database) (data not shown).
miR-183 preferentially regulates
PAX6-5a expression in glioma cells
To further investigate whether PAX6 and
PAX6-5a are specifically targeted by the miRNA cluster we
used qRT-PCR and WB, to compare PAX6/PAX6-5a expression in
glioma cell lines (U87, U118, U251) transfected with microRNA
mimics or inhibitors. All glioma cell lines used have, minimal
levels of both PAX6 and PAX6-5a (Fig.
1A). Since the results were similar for all cell lines, only
those obtained from U87 cells are shown, unless otherwise
specified.
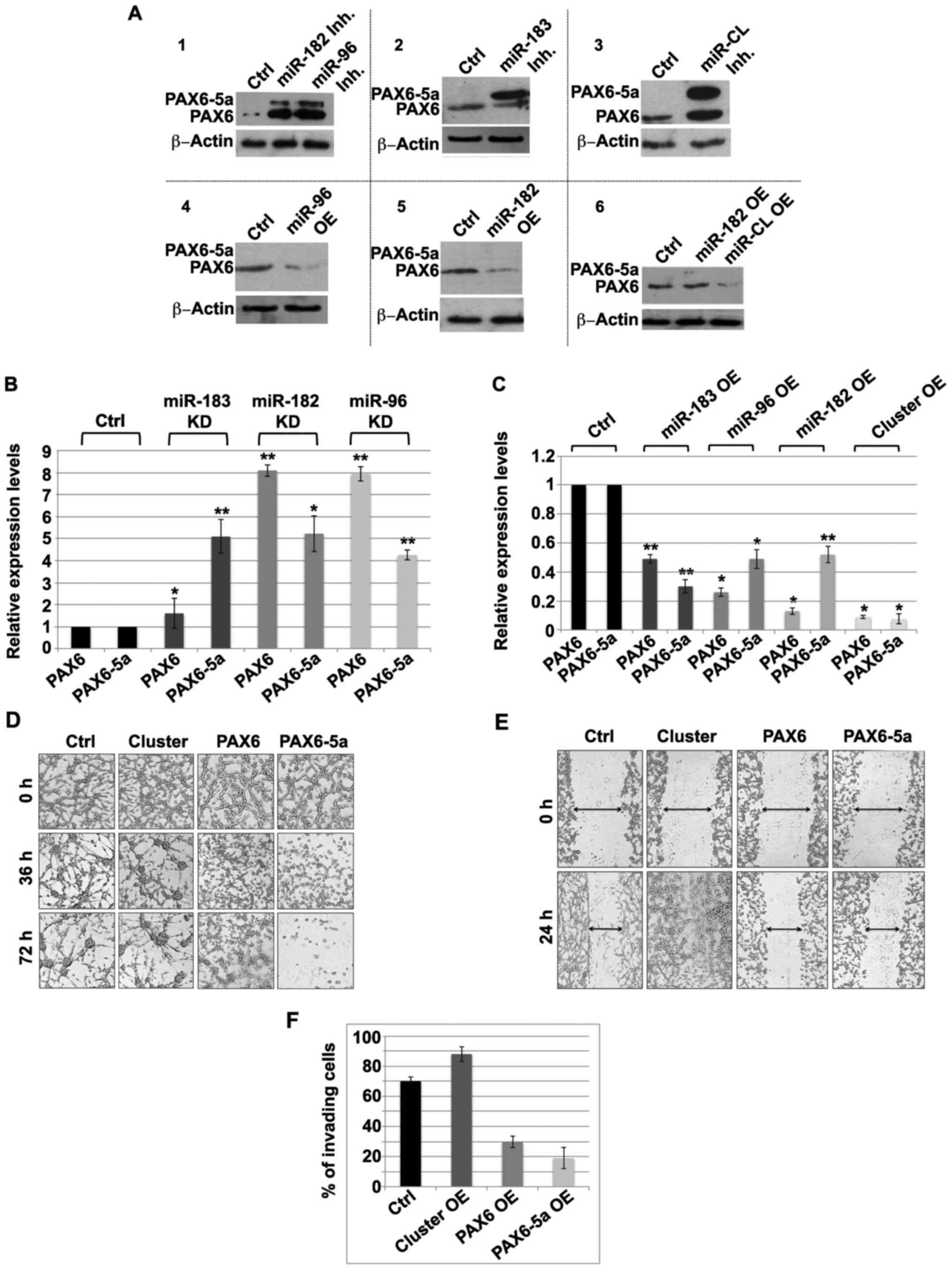
Noteworthy, inhibition of miR-183 alone results in
significant increase of the PAX6-5a protein levels while PAX6 is
only insignificantly affected (Fig.
2A2). On the other hand, inhibition of either miR-182 or miR-96
affects the expression of both PAX6 and PAX6-5a. Noteworthy, the
regulatory relationship between the two PAX6 isoforms is by itself
complex. Despite slight fluctuations during embryogenesis, under
physiological conditions, PAX6 is maintained at higher levels than
PAX6-5a. In addition, PAX6 directly and positively autoregulates
itself and PAX6-5a, while PAX6-5a regulates PAX6 expression in a
dose-dependent manner (39–41). Accordingly, increase of PAX6 levels
would also result in an increase of the PAX6-5a, while higher
PAX6-5a, as is the case when miR-183 is inhibited (Fig. 2A2), results in decrease or no-change
of the PAX6 levels. However, inhibition of the whole miRNA cluster
leads to an increase of both PAX6 and PAX6-5a protein levels
(Fig. 2A3).
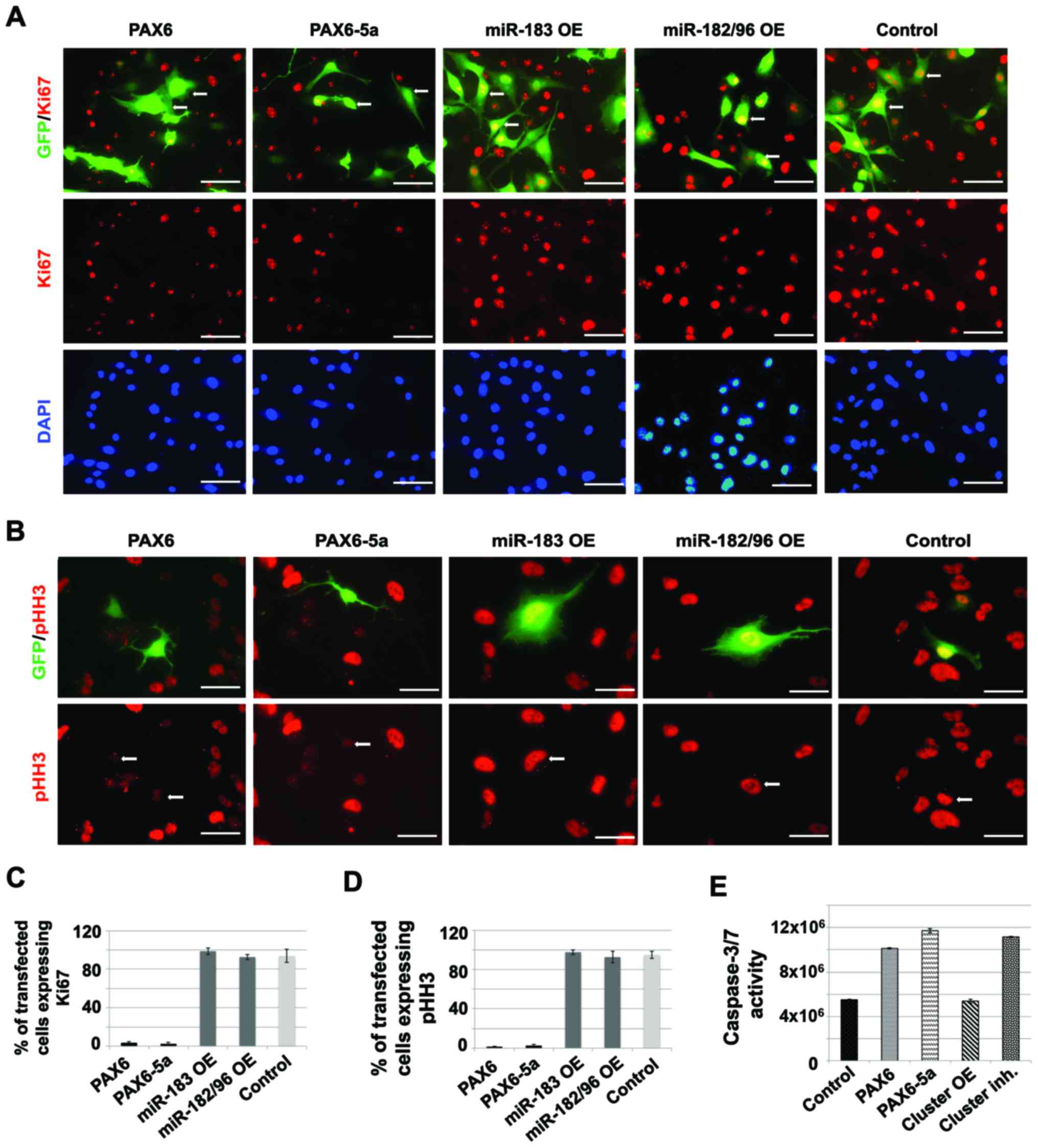
 | Figure 2.miR-183-96-182 cluster negatively
regulates the expression of both PAX6 and PAX6-5a
isoforms and affects tumorigenicity of glioma cell lines. (A)
Western blotting analysis of the PAX6 and PAX6-5a protein levels in
glioma cell line U87MG, after inhibition (Inh.) or overexpression
(OE) of single components of the miR-183-96-182 cluster or the
whole cluster (miR-CL). (B) qRT-PCR analysis of PAX6 and
PAX6-5a mRNA, in U87 cells, after miR-183, miR-96 and
miR-182 knockdown (KD) using specific miRNA inhibitors (details are
shown in Materials and methods) (*P<0.01; **P<0.05; Error
bars show SEM from 4 independent transfections) or (C) after OE of
miR-183, miR-96, miR-182 or the cluster (*P<0.01; **P<0.05;
Error bars show SEM from 4 independent transfections). (D) Time
course (0–72 h) of U87MG cell survival potential following
simultaneous transfection with miR-182 miR-96 and miR-183
(Cluster), PAX6 and PAX6-5a. Note that cells
transfected with the miRNAs (cluster) proliferate faster and start
to form large clusters, similarly to the control native cells,
while the cells transfected with either PAX6 isoform fail to form
clusters and become apoptotic. The latter is especially prominent
after transfection with the PAX6-5a expression plasmid
(representative images from 1 out of 3 independent experiments).
(E) Time course (0–24 h) of U87MG glioma cell migration in a
monolayer-based wound-healing assay (scratching assay), after OE of
the miR-183-96-182 cluster (Cluster), PAX6 and
PAX6-5a. Transfection of either PAX6 or PAX6-5a markedly
reduces the migratory potential of the glioma cells, while the
opposite is evident after miRNA cluster OE. The gap is indicated by
double arrows. (representative images from 1 out of 3 independent
experiments). (F) The PAX6 or PAX6-5a OE U87MG cells become less
invasive compared to high miR-183-96-182 cluster expression
conditions, as indicated by Matrigel invasion assays (details in
Materials and methods). (Cluster: miR-182 mimic, miR-96 mimic and
miR-183 mimic simultaneous transfection. Control: non-transfected
U87MG cells. Values represent average percentage of invading cells
from 3 independent experiments. Error bars show SEM.
P<0.05). |
Consistently, after overexpression (OE) of miR-182,
miR-96, or the whole miRNA cluster, the level of PAX6 is reduced
(Fig. 2A4, A5 and A6), while
miR-183 OE does not seem to significantly affect PAX6 level
(Fig. 2A6). Notably, in both cases,
PAX6-5a, which is expressed at lower levels in the native U87 cell
line, is not detectable. Similar results were obtained at the mRNA
level, where knockdown (KD) of any of the miR-183-96-182 members,
results in increased expression levels of both PAX6 and PAX6-5a
(Fig. 2B), while the opposite is
obvious following OE of any of the miRNAs (Fig. 2C). At the mRNA level it is also
evident that miR-183 has a greater impact on the expression levels
of PAX6-5a, while PAX6 is only mildly increased (Fig. 2B and C). On the contrary, miR-182
and miR-96, both have more influence on the expression levels of
the PAX6 isoform (Fig. 2B and
C).
Overexpression of miR-183-96-182
promotes glioma cell migration, survival and proliferation in
glioma cell lines
Next, we transfected glioma cell lines U87, U118,
U251 with either mimics specific for each component of the cluster,
or with PAX6 and PAX6-5a expression constructs and examined the
effect on the survival and migration of the cells. Since the
results were similar for all cell lines, only those obtained from
U87 cells are shown, unless otherwise specified. First, we used a
simple cell viability assay to assess the effect the whole miRNA
cluster on the survival of U87 cells, compared to PAX6 or PAX6-5a
OE. As shown in Fig. 2D the miRNA
cluster promoted the survival of glioma cells, indicated by the
formation of large clumps from 36 h onwards. On the contrary, PAX6
OE caused decreased cell viability, while the effect of the OE of
PAX6-5a isoform was even more prominent (Fig. 2D).
The migratory potential of glioma cells, was further
evaluated by wound healing (scratch) assays after OE of the whole
miRNA cluster. PAX6 and PAX6-5a OE markedly slowed down the
migration. This is evident by the closure of the wound only by
approximately 30% in 24 h, closely resembling the control
situation. In the latter, the closure was more prominent (38%) with
few cells already migrated in the middle of the wound (Fig. 2E). In order to circumvent the
assumed cell migration that might partially be the result of cell
proliferation, we limited the experiment to 24 h. Nevertheless,
further future experiments are required in order to assess absolute
rates of total cell proliferation and determine net migration
independent of proliferation.
Lastly, we examined the invasive ability of glioma
cells by applying matrigel transwell assays. We found that upon
miR-cluster OE, the cells become more invasive (more than 80% of
the transfected cells have migrated through the matrigel), whilst
upon PAX6 or PAX6-5a OE, 30% and 24% of the cells
invaded, respectively (Fig. 2F).
Overall, these findings indicate that U87 glioma cells acquire
higher tumorigenic aptitude after transfection with miR-183, miR-96
and miR-182 mimic oligonucleotides, whereas PAX6 and PAX6-5a push
them towards a less motile, non-invasive and non-viable track.
In order to evaluate whether the restrained
survival, migration and invasion of the glioma cells after
PAX6/PAX6-5a OE is due to aberrant proliferation, we performed
immunostainings with Ki67, which marks cells at any stage of
proliferation and pHH3, as a specific mitotic marker, using U87
cells. Our analysis shows a reduced number of Ki67 and pHH3
positive cells after transfection with PAX6 OE (3.4±1.55%
Ki67+ cells; 1.4±0.23% pHH3+ cells) or
PAX6-5a OE constructs (2.34±1.77% KI67+; 2.5±0.98%
pHH3+ cells) (Fig. 3).
On the contrary, transfection with mimics for miR-183 or miR-182/96
results in significantly increased proliferation rates (miR-183
transfection: 98.7±3.66% Ki67+ and 97.5±2.14%
pHH3+ cells; miR-182/96 transfection: 92.6±2.75%
Ki67+ and 92.5±5.67% pHH3+ cells); control
GFP transfection: 93.7±6.88% Ki67+ and 94.78±3.66%
pHH3+ cells) (Fig.
3).
Finally, we examined whether the reduction of U87
cell proliferation rate after PAX6/PAX6-5a OE is also associated
with increased cell death. To test this, we utilized a generic
enzymatic assay that measures caspase-3 and −7 activity. After OE
of PAX6 or PAX6-5a, we observed a 1.9- and 2.2-fold activation of
these effector caspases, respectively. At the same time, inhibition
of the miR-cluster resulted in 2-fold higher enzyme activity,
compared to the controls. These findings suggest that
miR-183-96-182 regulate glioma growth and proliferation by
inhibiting PAX6 and PAX6-5a, which otherwise, would make the cells
less viable.
PAX6 and PAX6-5a specifically regulate
a number genes involved in GBM progression
From our literature search we assorted at least 8
putative candidates [(SPHK1, CTNND2 (δ-catenin), PDGF-A,
PDGF-B, PDGFR-α, PDGFR-β, Neurofibromatosis 1 (NF1), and
T-cell lymphoma invasion and metastasis 1 (TIAM1)] that
could be downstream known or putative interaction partners of PAX6
and PAX6-5a during glioma progression. Using qRT-PCRs assays and
∆∆Ct quantification, we found that the expression of PAX6
and PAX6-5a (Fig. 4A) is
highly downregulated in 22 high-grade glioma tumors, while at the
same time the expression of SPHK1 is upregulated by
3.27±0.049. SPHK1, an oncogenic enzyme known to contribute to the
acquisition of the apoptosis resistance of the glioma cells, has
recently been suggested as a direct downstream target of PAX6-5a
(42–44), but such an interaction has not been
validated so far. On the other hand, the central nervous system
specific protein CTNND2 is known to be regulated by PAX6 and
PAX6-5a, in a dose- and isoform-sensitive fashion (45). Of note, high levels of CTNND2
correlate with positive patient outcome, while inactivation of
CTNND2 is a key genetic alteration driving the aggressive
mesenchymal phenotype of GBM (46).
In agreement with previous data (45), in our 22 GBM samples we observed a
decreased expression of CTNND2 accompanied by a low PAX6 and
PAX6-5a expression level. Finally, we found that the
relative expression of PDGF-B and the corresponding receptor
PDGFR-β was highly diminished by 9.09±0.00154 and
4.34±0.0038-fold, respectively, during glioma progression.
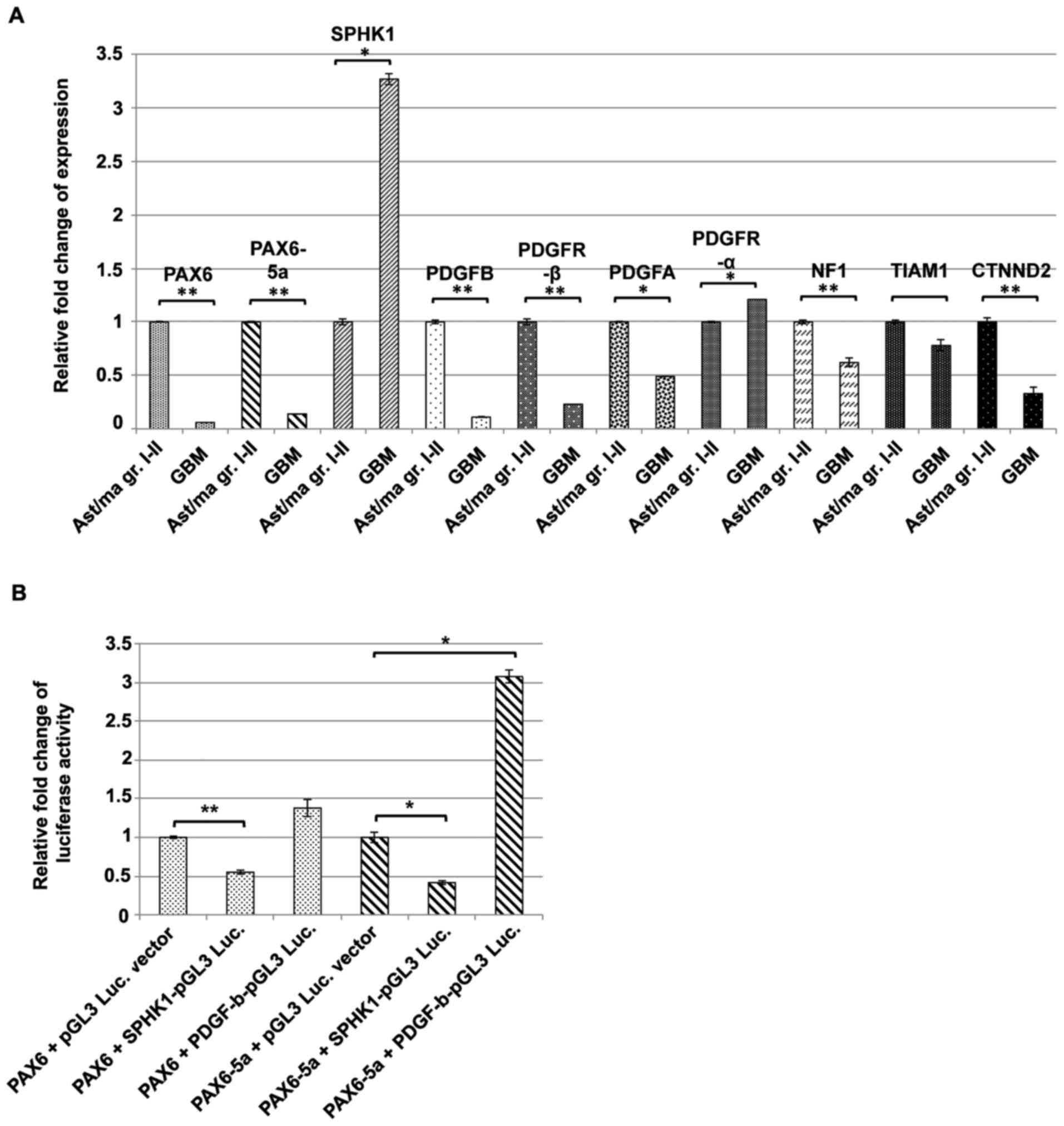 | Figure 4.Deregulation of putative PAX6/PAX6-5a
target genes in GBM in vivo. (A) Comparative qRT-PCR
analysis of the expression levels of PAX6, PAX6-5a, SPHK1, PDGF-B,
PDGFR-β, PDGF-A, PDGFR-α, NF1, TIAM1 and CTNND2, from biopsies of
diffuse fibrillary astrocytoma (stages I–II; 7 samples) and GBM
(stages III–IV; 22 samples). The analysis indicates significant
(**P<0.05) downregulation of PAX6, PAX6-5a, PDGF-B,
PDGFR-β and CTNND2, while SPHK1 shows significant
(*P<0.01) upregulation. PDGF-A, PDGFR-α, NF1 and TIAM1 did not
show any significant changes in this pool of samples. (Error bars
show SEM from 22 GBM samples and 7 low-grade glioma samples). (B)
Investigation of possible binding of PAX6 or PAX6-5a on the SPHK1
and PDGF-B promoters with luciferase reporter assays in NIH 3T3
cells indicates that SPHK1 is negatively regulated by both PAX6
isoforms although more prominently by PAX6-5a (*P<0.01;
**P<0.05). On the contrary, PDGF-B is positively regulated
exclusively by PAX6-5a, while PAX6 exerts only a minor effect
(*P<0.01) (Error bars show SEM from 4 independent
transfections). |
Intrigued to further explore the connection of
PAX6 isoforms with SPHK1, and PDGF-B, we
constructed firefly luciferase reporters containing the putative
promoter region of SPHK1 and PDGF-B and we
co-transfected each construct with either PAX6 or
PAX6-5a OE plasmids in HeLa cells. Transfections with either
PAX6 + pGL3-luciferase or PAX6-5a + pGL3-luciferase empty vector
were used as control. The luciferase activity was reduced by
0.56±0.096-fold when PAX6 was co-transfected with
SPHK1/PGL3-Luciferase construct and 0.46±0.0225-fold upon PAX6-5a
co-transfection (Fig. 4B)
suggesting that PAX6-5a tends to exert a slightly stronger effect.
Co-transfection of PDGF-B/PGL3-Luciferase construct with PAX6
resulted in minor but not significant upregulation
(1.46±0.112-fold) of the luciferase activity. However, when
co-transfected with PAX6-5a, the luciferase reporter activity was
upregulated by more than 3±0.089-fold, suggesting a specific
positive regulation of PDGF-B by the PAX6-5a isoform. Together, all
the above findings show that PAX6 and PAX6-5a could possibly
regulate at least 3 key downstream effectors with important roles
in glioma progression. However, additional ChIP experiments are
required in order to absolutely verify this.
Specific regulation of PDGF-B, SPHK1
and CTNND2 by different PAX6 isoforms in glioma cell lines
We used glioma cells lines (U87, U118 and U251) in
order to determine how PAX6 and PAX6-5a regulate PDGF-B,
SPHK1 and CTNND2. Since the results were similar for all
cell lines, only those obtained from U87 cells are shown, unless
otherwise specified.
We overexpressed either PAX6 or
PAX6-5a using specific for each isoform constructs, in U87MG
cells (similar results were also obtained from U251 and U118 cell
lines - data not shown). As shown in Fig. 5A and B, OE of PAX6 results in minor
(2.12±0.39-fold) upregulation of PDGF-B, whereas PAX6-5a OE yields
a much stronger effect, causing a 7.59±0.52-fold upregulation of
the PDGF-B levels. Although previously we were able to show
(Fig. 4B) that PAX6-5a and not PAX6
is the actual regulator of PDGF-B, it is expected that PAX6 OE
would result in a similar, but milder to PAX6-5a OE effect, due to
the reported regulation of the PAX6-5a by PAX6 (44,45,75).
In our experiments we also observed upregulation (4.6±0.54-fold) of
PAX6-5a under the PAX6 OE conditions, explaining the slight
upregulation of PDGF-B by PAX6 OE. The effect, naturally becomes
more prominent after higher (10.6±0.64-fold) PAX6-5a OE as seen in
Fig. 5B. On the other hand, we did
not witness ‘strong’ upregulation of PAX6 after PAX6-5a OE, meaning
that PAX6 probably regulates PAX6-5a but not necessarily vice
versa, or that the PAX6-5a isoform regulates negatively PAX6 after
a certain OE level. The latter has been previously reported
(45). Our WB analysis shown in
Fig. 5C is in accordance with the
above.
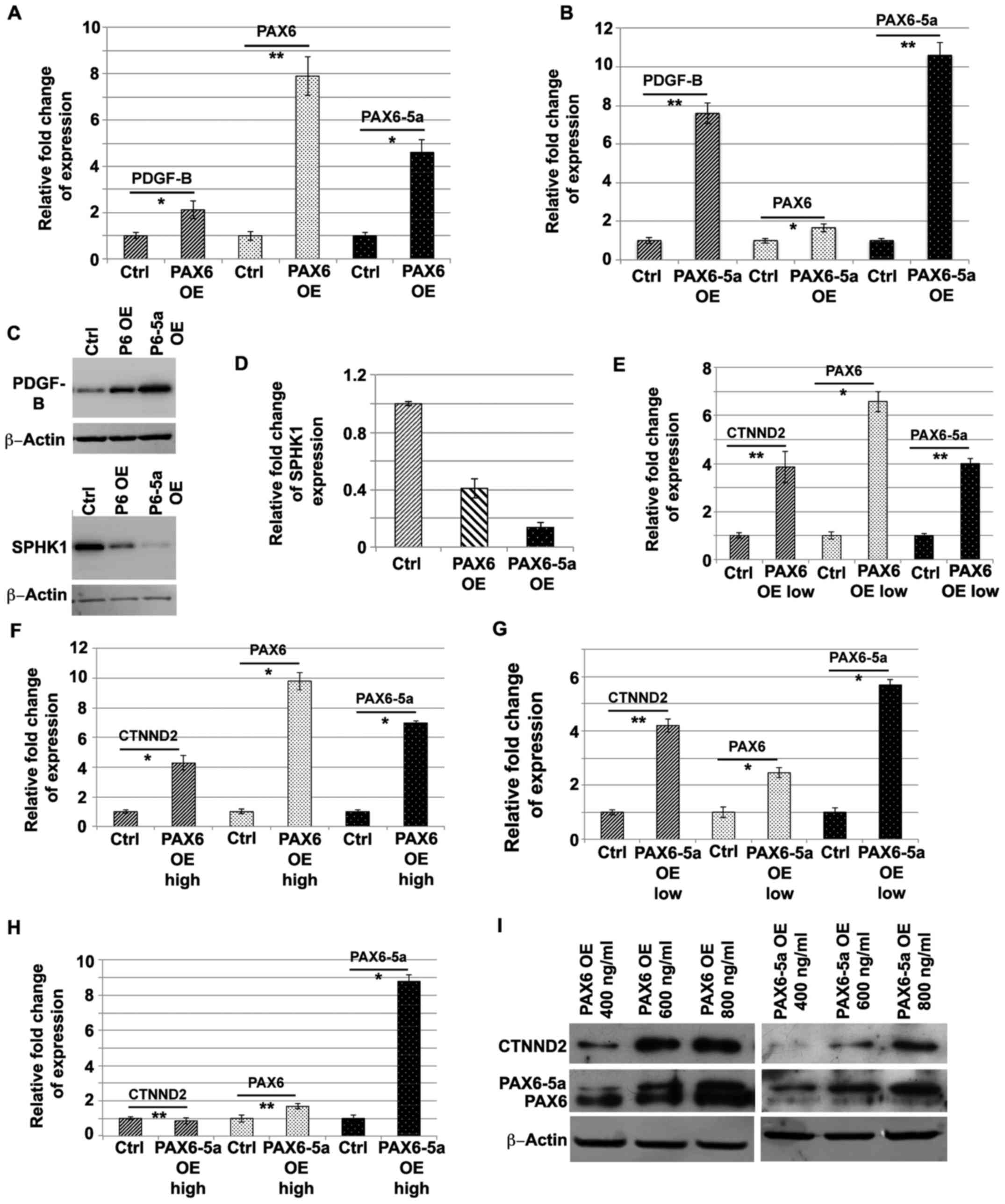 | Figure 5.Regulation of PAX6 and PAX6-5a target
genes in glioma cell lines. (A and B) qRT-PCR and (C) western blot
analysis of the expression levels of PDGF-B, PAX6 and PAX6-5a in
U87MG glioma cells, after OE of PAX6 or PAX6-5a. Evidently, PDGF-B
is highly upregulated in the PAX6-5a OE cells (B and C), while a
mild upregulation is also observed when PAX6 is overexpressed,
possibly resulting from the observed simultaneous increase of
PAX6-5a levels due to PAX6 OE. (C) Western blot analysis of SPHK1
after Pax6 or PAX6-5a OE. (D) qRT-PCR analysis of the levels of
SPHK1 under this conditions, shows decreased SPHK1 expression after
expression of both PAX6 isoforms, however most prominently under
PAX6-5a overexpression (P<0.05). (E-I) qRT-PCR and western blot
analysis of CTNDD2 expression after PAX6 or PAX6-5a OE, shows
positive regulation of CTNND2 by PAX6-5a. This is evident only
under moderate (<6-fold increase) PAX6-5a OE conditions. When
PAX6-5a is expressed at higher levels (>8-fold), CTNDD2 is
either diminished or not affected, compared to the control
situation. PAX6 OE also results in a mild but significant
upregulation of CTNDD2, however the effect could possibly result
from the observed simultaneous increase in PAX6-5a levels
(*P<0.01; **P<0.05; Error bars represent SEM from 3–4
independent experiments). |
We examined the expression of SPHK1 level,
after OE of PAX6 or PAX6-5a and we found a 2.4- and
7.7-fold reduction of its activity, respectively, suggesting that
Pax6-5a exerts even a slightly greater regulatory potential as
compared to PAX6 (Fig. 5D). Similar
results were obtained at the protein level as shown on Fig. 5C, where OE of either PAX6 isoform,
results in diminished SPHK1 protein levels. The greater reduction
achieved after PAX6-5a OE, points towards a theoretically greater
regulatory potential of this isoform to SPHK1.
The investigation of the regulatory relationship
between the PAX6 isoforms and CTNDD2, proved more difficult
because of the complex regulatory feedback loop between these genes
(45). When PAX6 is
‘moderately’ overexpressed (less than 7-fold), then PAX6-5a
and CTNND2 are also ‘moderately’ upregulated (3.9±0.21 and
3.85±0.65-fold, respectively) (Fig.
5E). Higher OE of PAX6 (9.8±0.59-fold) results in higher
expression (6.98±0.16-fold) of PAX6-5a, however
CTNND2 levels are only very mildly up (4.28±0.48-fold)
compare to the moderate PAX6 OE situation (Fig. 5F). One possibility could be that
PAX6 alone is not sufficient to promote higher upregulation
of CTNND2 or alternatively, that PAX6-5a is the main
regulator of CTNND2 and the upregulation seen under
PAX6 OE conditions is due to the simultaneous upregulation
of PAX6-5a. For clarification, we overexpressed PAX6-5a at
‘moderate’ or ‘high’ levels in U87 cells. A ‘moderate’ OE of
PAX6-5a (5.6±0.19-fold) results in only ‘mild’ upregulation of
PAX6 (2.45±0.19-fold), while CTNND2 is upregulated by
4.2±0.23-fold (Fig. 5G).
Nevertheless, a higher (8.78±0.39-fold) OE of PAX6-5a
results in insignificant (1.68±0.14-fold) upregulation of PAX6, but
even more interestingly the expression of CTNDD2 is borderline
downregulated (no change). This finding suggests that the levels of
PAX6-5a as well as the PAX6/PAX6-5a ratio, is
important for the passage from positive to negative regulation of
CTNND2 (Fig. 5H). The results from
WB analyses follow the same pattern described above under three
different concentrations of PAX6 or PAX6-5a expression plasmid
transfection (Fig. 5I).
Overexpression of either CTNND2 or
SPHK1, in glioma cells, disrupts proliferation and promotes cell
death
As shown in Fig. 3,
OE of either PAX6 or PAX6-5a in glioma cell lines
leads to decreased proliferation and increased apoptosis. This is
in accordance with previous studies showing that conditional
activation of the two isoforms during corticogenesis in
vivo, results in defects in cell cycle progression (inhibition
of proliferation) and in the acquisition of apoptotic or neuronal
cell fate of the progenitors (11).
In order to verify whether this is the result of the de-regulation
of the downstream targets CTNND2 and of PAX6/PAX6-5a, we
overexpressed them in U87 glioma cells and 48 h later we conducted
immunostainings with Ki67.
As shown in Fig. 6A and
B, in the control (GFP-vector) situation (where PAX6 and
PAX6-5a are already expressed at insignificant levels due to their
downregulation by enhanced expression of the miR-183-96-182
cluster) the GFP+ cells expressing Ki67 reach almost
100%. Under CTNND2-GFP OE, less than half (32.8±2.6%) of the
GFP+ cells express Ki67. Similarly, when SPHK1 is
overexpressed, then the fraction of
SPHK1-GFP+/Ki67+ cells become only 40±1.89%).
Caspase 3/7 activity measurement also points towards a significant
increase of cell death when CTNND2 is overexpressed; however, SPHK1
OE leads to only a mild increase of caspase 3/7 activity (Fig. 6C). Thus, although less than half of
the SPHK1/GFP+ cells are proliferating, only a fraction
of the non-proliferating cells dies. On the other hand, PDGF-B OE,
results in no significant change in the proliferative or apoptotic
status of the glioma cells (Fig.
6A-C). The latter is somewhat expected, since U87 glioma cells
already express high PDGF-B levels.
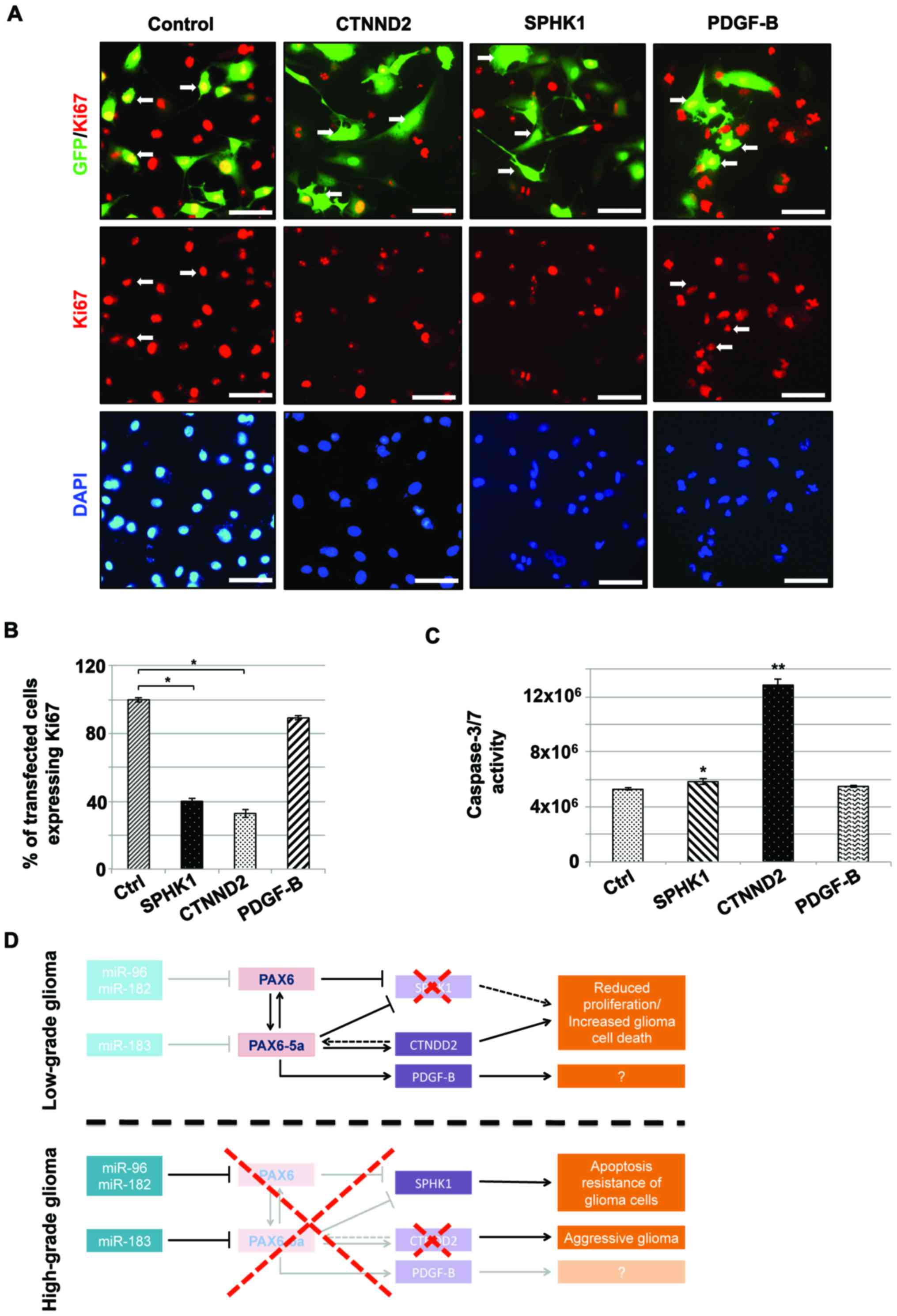 | Figure 6.The PAX6 and PAX6-5a target genes
CTNND2, SPHK1 and PDGF-B inhibit proliferation of glioma cells. (A)
Representative immunofluorescence images of glioma cells following
transfection of pCIG2-GFP control vector, CTNND2-GFP, SPHK1-GFP and
PDGF-B-GFP, showing differences in the proliferation, as indicated
by Ki67 immunostaining (red). (B) Counting of
GFP+/Ki67+ glioma cells, under the same
conditions, specifies a significantly (*P<0.01) decreased cell
proliferation after OE of CTNND2-GFP and SPHK1-GFP, while
PDGF-B-GFP transfection displays no significant changes (Error bars
show SEM from 3 independent transfections). (C) Measurement of
caspase 3/7 activity of those transfected U87MG glioma cells,
indicates increased rates of cell death after CTNND2-GFP
(*P<0.01) and SPHK1-GFP (**P<0.05) OE (Error bars show SEM
from 3 independent transfections) (Scale bars: 100 µm). (D)
Simplified schematic illustration of our proposed theory of
PAX6/PAX6-5a regulation in glioma. The miR-183-96-182 cluster
downregulates TFs PAX6 and PAX6-5a, while glioma progresses from
lower (I–II) to higher grades (III–IV). While all miRNAs of the
cluster have the capacity to regulate both PAX6 isoforms, however,
there is a strong tendency for higher specificity between miR-183
and PAX6-5a and miR-96-182 and PAX6. In low-grade glioma, the
expression levels of miR-183-96-182 (faint blocks) are extremely
low and thus not enough to control PAX6/PAX6-5a. As a result both
isoforms are available in high quantities. PAX6/PAX6-5a block (red
dashed X) the expression of its putative downstream target SPHK1,
resulting in decreased glioma cell proliferation-increased cell
death. Simultaneously, PAX6-5a induces CTNND2, which has
anti-oncogenic function, to be expressed at higher levels. PDGF-B
is also induced by PAX6-5a but the effect that this interaction has
on glioma progression remains elusive. In high-grade glioma (GBM),
the miR-183-96-182 are highly expressed. As a result both PAX6 and
PAX6-5a isoforms are strongly downregulated and can no longer
control the expression of their downstream effectors. SPHK1 is no
longer blocked and its levels increase significantly leading to
apoptosis resistance of the glioma cells. CTNND2 expression
diminishes, resulting in aggressive glioma phenotype. PDGF-B is
also reduced but as before, we have no significant data as to what
effect this might have for the glioma progression. (−-----┤,
indicates inhibition or downregulation; black arrows indicate
induction or upregulation; dashed black arrows indicate milder
effect). |
Discussion
Understanding the complex molecular mechanisms
underlying cellular processes in cancer, is of great importance in
order to decipher how tumors develop and progress. In this study,
we unveiled the existence of cross-regulatory interactions between
the two, active in vertebrate CNS, PAX6 isoforms,
PAX6 and PAX6-5a, with the miR183-96-182 cluster that
appears to play an essential role in glioma progression. Moreover,
we found that the expression levels of miR-183-96-182 and
PAX6/PAX6-5a could be valuable prognostic markers for disease
outcome in human probands.
The results from our qRT-PCR analysis from 29 human
glioma tumor biopsies and 3 glioma cell lines, revealed a
significantly great reduction of both PAX6 isoform expression
levels, as glioma progresses from low- to high-grade. On the
contrary, members of the miR-183-96-182 cluster showed the exact
opposite expression pattern, thus indicating a possible regulatory
relationship between the miRNAs and TFs PAX6/PAX6-5a. Frequently
miRNAs and TFs participate in feed-forward and feedback loop
regulatory networks (37,38) so that cellular processes are
fine-tuned and temporally controlled. Indicated by luciferase
reporter assays and bioinformatics analyses (data not shown), a
feedback positive regulation of miR-183-96-182 by TFs PAX6/PAX6-5a
might also exist, adding further complexity to the regulation of
glioma tumor progression. However, this remains to be extensively
investigated. Importantly, we found that different members of the
miR-183-96-182 cluster regulate specifically the expression of
either PAX6 or PAX6-5a. To our knowledge, such a preferential
regulation of one or the other isoform of a TF by members of the
same polycistronic, paralogous miRNA cluster, is a novel
observation, concerning the basic mechanisms governing miRNA
function.
Multiple studies have shown the involvement of the
canonical PAX6 isoform in NSC proliferation, neurogenesis,
generation of specific types of neurons, and changes in spatial
pattern (15,16,21,47).
Measured by migration, invasion and survival assays, the OE of
either PAX6 or PAX6-5a in high-grade glioma cells leads to
decreased tumorigenicity, as opposed to the OE of miR-183-96-182,
disclosing the important role of both PAX6 isoforms in restricting
glioma progression. Our results are in accordance with previous
findings reporting that PAX6-deficient cultured astrocytes display
higher migration potential, which along with other capacities that
they acquire, they resemble highly malignant glioma cells (48). Importantly, we were able to show
that the expression of both PAX6 isoforms has a marked effect on
glioma cell proliferation and death, however PAX6-5a exerts a more
intense influence in reversing tumorigenicity. These findings,
demonstrate for the first time that PAX6-5a may actually be the key
isoform functioning in the early stages of glioma development to
inhibit cell proliferation and promote glioma cell death.
Although high level of PAX6 has been reported to
correlate with a better patient prognosis, the involved mechanism
is still unknown (19,20). The performed qRT-PCR experiments on
human tumor biopsies, resulted in the identification of at least 3
important candidates acting downstream of PAX6/PAX6-5a in glioma
development, namely SPHK1, CTNND2 and PDGF-B.
Notably, while SPHK1 was negatively regulated by both PAX6
isoforms, PDGF-B was positively affected only by PAX6-5a. A
complex cross-regulatory interaction of PAX6-5a and CTNND2, is
already known (45), however we
showed that a similar feedback regulation also occurs in gliomas.
Intriguingly, our analysis indicates that the upregulation of
CTNND2 by PAX6-5a and the simultaneous down-modulation of SPHK1 by
both PAX6 isoforms, results in reduced glioma cell proliferation
and elevated cell death, thus providing a molecular explanation of
how these genes are controlled in relation to glioma
progression.
To our knowledge, a specific, if any, association
between PDGF-B and PAX6-5a has not been demonstrated. We found that
PDGF-B is specifically upregulated by PAX6-5a. Nevertheless in
glioma cells, high PDGF-B expression does not result in decreased
proliferation/increased cell death. Previous studies suggested that
overexpression of PDGF and corresponding receptors plays a role in
development of CNS tumors (4,5). In an
in vivo model PDGF-B induced oligodendroglioma, PAX6 and the
bHLH TF OLIG2, antagonize each other, where high PAX6 expression
results in downregulation of OLIG2 and a less malignant phenotype
(49). What is interesting though
is that mice overexpressing PDGF-B alone do not form tumors,
whereas they do so only when they are crossed with mice bearing
mutations in other oncogenes (e.g. Tp53 null mice) (5).
The present results highlight a novel aspect for
direct and specific regulation of the expression of the two PAX6
isoforms by the members of the miR-183-96-182 cluster. We show that
high expression levels of both PAX6 and PAX6-5a result in reduced
tumorigenicity of glioma cells, more prominently for isoform
PAX6-5a, which is due to deregulated proliferation and increased
cell death. Given the specific direct interaction of members of
miR183-96-182 cluster with PAX6 and PAX6-5a, our findings argue
towards the use of the clustered miRNAs, as well as the two PAX6
isoforms, as negative or positive markers, respectively, for glioma
progression. Another line of presented evidence suggests that
SPHK1, CTNND2 and PDGF-B could be PAX6/PAX6-5a
downstream target genes. We herein show that elevated expression of
CTNND2 or SPHK1 in glioma cells results in a similar
to PAX6/PAX6-5a OE phenotype, leading to reduced cell growth in
both cases and increased cell death in the case of CTNND2.
Together, our findings suggest that the molecular interaction
between PAX6 and PAX6-5a with specific members of the
miR-183-96-182 cluster plays an essential role in progression of
glioma, in glioma cell lines, delivering their anti-tumorigenic
effect, through the regulation of specific downstream pathways.
Acknowledgements
The authors wish to thank Silke Schlot and Martina
Daniel for excellent technical assistance. The study was funded by
Max Planck Gesellschaft and Cluster of Excellence and DFG Research
Center for Nanoscale Microscopy and Molecular Physiology of the
Brain (CNMPB).
References
|
1
|
Furnari FB, Fenton T, Bachoo RM, Mukasa A,
Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al:
Malignant astrocytic glioma: Genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Adamson C, Kanu OO, Mehta AI, Di C, Li N,
Mattox AK and Bigner DD: Glioblastoma multiforme: A review of where
we have been and where we are going. Expert Opin Investig Drugs.
18:1061–1083. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siebzehnrubl FA, Reynolds BA, Vescovi A,
Steindler DA and Deleyrolle LP: The origins of glioma: E Pluribus
Unum? Glia. 59:1135–1147. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nazarenko I, Hede SM, He X, Hedrén A,
Thompson J, Lindström MS and Nistér M: PDGF and PDGF receptors in
glioma. Ups J Med Sci. 117:99–112. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Koul D: PTEN signaling pathways in
glioblastoma. Cancer Biol Ther. 7:1321–1325. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang SI, Puc J, Li J, Bruce JN, Cairns P,
Sidransky D and Parsons R: Somatic mutations of PTEN in
glioblastoma multiforme. Cancer Res. 57:4183–4186. 1997.PubMed/NCBI
|
|
8
|
Chang JY, Hu Y, Siegel E, Stanley L and
Zhou YH: PAX6 increases glioma cell susceptibility to detachment
and oxidative stress. J Neurooncol. 84:9–19. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jacques TS, Swales A, Brzozowski MJ,
Henriquez NV, Linehan JM, Mirzadeh Z, O'Malley C, Naumann H,
Alvarez-Buylla A and Brandner S: Combinations of genetic mutations
in the adult neural stem cell compartment determine brain tumour
phenotypes. EMBO J. 29:222–235. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sanai N, Alvarez-Buylla A and Berger MS:
Neural stem cells and the origin of gliomas. N Engl J Med.
353:811–822. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Berger J, Berger S, Tuoc TC, D'Amelio M,
Cecconi F, Gorski JA, Jones KR, Gruss P and Stoykova A: Conditional
activation of Pax6 in the developing cortex of transgenic mice
causes progenitor apoptosis. Development. 134:1311–1322. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Götz M, Stoykova A and Gruss P: Pax6
controls radial glia differentiation in the cerebral cortex.
Neuron. 21:1031–1044. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Heins N, Malatesta P, Cecconi F, Nakafuku
M, Tucker KL, Hack MA, Chapouton P, Barde YA and Götz M: Glial
cells generate neurons: The role of the transcription factor Pax6.
Nat Neurosci. 5:308–315. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Osumi N, Shinohara H, Numayama-Tsuruta K
and Maekawa M: Concise review: Pax6 transcription factor
contributes to both embryonic and adult neurogenesis as a
multifunctional regulator. Stem Cells. 26:1663–1672. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Quinn JC, Molinek M, Martynoga BS, Zaki
PA, Faedo A, Bulfone A, Hevner RF, West JD and Price DJ: Pax6
controls cerebral cortical cell number by regulating exit from the
cell cycle and specifies cortical cell identity by a cell
autonomous mechanism. Dev Biol. 302:50–65. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Warren N, Caric D, Pratt T, Clausen JA,
Asavaritikrai P, Mason JO, Hill RE and Price DJ: The transcription
factor, Pax6, is required for cell proliferation and
differentiation in the developing cerebral cortex. Cereb Cortex.
9:627–635. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Haubst N, Berger J, Radjendirane V, Graw
J, Favor J, Saunders GF, Stoykova A and Götz M: Molecular
dissection of Pax6 function: The specific roles of the paired
domain and homeodomain in brain development. Development.
131:6131–6140. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shimizu N, Watanabe H, Kubota J, Wu J,
Saito R, Yokoi T, Era T, Iwatsubo T, Watanabe T, Nishina S, et al:
Pax6-5a promotes neuronal differentiation of murine embryonic stem
cells. Biol Pharm Bull. 32:999–1003. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou YH, Tan F, Hess KR and Yung WK: The
expression of PAX6, PTEN, vascular endothelial growth factor, and
epidermal growth factor receptor in gliomas: Relationship to tumor
grade and survival. Clin Cancer Res. 9:3369–3375. 2003.PubMed/NCBI
|
|
20
|
Zhou YH, Wu X, Tan F, Shi YX, Glass T, Liu
TJ, Wathen K, Hess KR, Gumin J, Lang F, et al: PAX6 suppresses
growth of human glioblastoma cells. J Neurooncol. 71:223–229. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sansom SN, Griffiths DS, Faedo A, Kleinjan
DJ, Ruan Y, Smith J, van Heyningen V, Rubenstein JL and Livesey FJ:
The level of the transcription factor Pax6 is essential for
controlling the balance between neural stem cell self-renewal and
neurogenesis. PLoS Genet. 5:e10005112009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Garzon R, Calin GA and Croce CM: MicroRNAs
in cancer. Annu Rev Med. 60:167–179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lavon I, Zrihan D, Granit A, Einstein O,
Fainstein N, Cohen MA, Cohen MA, Zelikovitch B, Shoshan Y, Spektor
S, et al: Gliomas display a microRNA expression profile reminiscent
of neural precursor cells. Neuro Oncol. 12:422–433. 2010.PubMed/NCBI
|
|
25
|
Li H, Kloosterman W and Fekete DM:
MicroRNA-183 family members regulate sensorineural fates in the
inner ear. J Neurosci. 30:3254–3263. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu S, Witmer PD, Lumayag S, Kovacs B and
Valle D: MicroRNA (miRNA) transcriptome of mouse retina and
identification of a sensory organ-specific miRNA cluster. J Biol
Chem. 282:25053–25066. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fendler A, Jung M, Stephan C, Erbersdobler
A, Jung K and Yousef GM: The antiapoptotic function of miR-96 in
prostate cancer by inhibition of FOXO1. PLoS One. 8:e808072013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu Z, Liu J, Segura MF, Shao C, Lee P,
Gong Y, Hernando E and Wei JJ: MiR-182 overexpression in
tumourigenesis of high-grade serous ovarian carcinoma. J Pathol.
228:204–215. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mi D, Carr CB, Georgala PA, Huang YT,
Manuel MN, Jeanes E, Niisato E, Sansom SN, Livesey FJ, Theil T, et
al: Pax6 exerts regional control of cortical progenitor
proliferation via direct repression of Cdk6 and hypophosphorylation
of pRb. Neuron. 78:269–284. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Moskwa P, Buffa FM, Pan Y, Panchakshari R,
Gottipati P, Muschel RJ, Beech J, Kulshrestha R, Abdelmohsen K,
Weinstock DM, et al: miR-182-mediated downregulation of BRCA1
impacts DNA repair and sensitivity to PARP inhibitors. Mol Cell.
41:210–220. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Song L, Liu L, Wu Z, Li Y, Ying Z, Lin C,
Wu J, Hu B, Cheng SY, Li M, et al: TGF-β induces miR-182 to sustain
NF-κB activation in glioma subsets. J Clin Invest. 122:3563–3578.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weeraratne SD, Amani V, Teider N,
Pierre-Francois J, Winter D, Kye MJ, Sengupta S, Archer T, Remke M,
Bai AH, et al: Pleiotropic effects of miR-183~96~182 converge to
regulate cell survival, proliferation and migration in
medulloblastoma. Acta Neuropathol. 123:539–552. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jiang L, Mao P, Song L, Wu J, Huang J, Lin
C, Yuan J, Qu L, Cheng SY and Li J: miR-182 as a prognostic marker
for glioma progression and patient survival. Am J Pathol.
177:29–38. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tanaka H, Sasayama T, Tanaka K, Nakamizo
S, Nishihara M, Mizukawa K, Kohta M, Koyama J, Miyake S, Taniguchi
M, et al: MicroRNA-183 upregulates HIF-1α by targeting isocitrate
dehydrogenase 2 (IDH2) in glioma cells. J Neurooncol. 111:273–283.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tang H, Bian Y, Tu C, Wang Z, Yu Z, Liu Q,
Xu G, Wu M and Li G: The miR-183/96/182 cluster regulates oxidative
apoptosis and sensitizes cells to chemotherapy in gliomas. Curr
Cancer Drug Targets. 13:221–231. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Paul V, Tonchev AB, Henningfeld KA,
Pavlakis E, Rust B, Pieler T and Stoykova A: Scratch2 modulates
neurogenesis and cell migration through antagonism of bHLH proteins
in the developing neocortex. Cereb Cortex. 24:754–772. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tsang J, Zhu J and van Oudenaarden A:
MicroRNA-mediated feedback and feedforward loops are recurrent
network motifs in mammals. Mol Cell. 26:753–767. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang HM, Kuang S, Xiong X, Gao T, Liu C
and Guo AY: Transcription factor and microRNA co-regulatory loops:
Important regulatory motifs in biological processes and diseases.
Brief Bioinform. 16:45–58. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Aota S, Nakajima N, Sakamoto R, Watanabe
S, Ibaraki N and Okazaki K: Pax6 autoregulation mediated by direct
interaction of Pax6 protein with the head surface ectoderm-specific
enhancer of the mouse Pax6 gene. Dev Biol. 257:1–13. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pinson J, Mason JO, Simpson TI and Price
DJ: Regulation of the Pax6: Pax6(5a) mRNA ratio in the developing
mammalian brain. BMC Dev Biol. 5:13–17. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pinson J, Simpson TI, Mason JO and Price
DJ: Positive autoregulation of the transcription factor Pax6 in
response to increased levels of either of its major isoforms, Pax6
or Pax6(5a), in cultured cells. BMC Dev Biol. 6:25–34. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Guan H, Song L, Cai J, Huang Y, Wu J, Yuan
J, Li J and Li M: Sphingosine kinase 1 regulates the Akt/FOXO3a/Bim
pathway and contributes to apoptosis resistance in glioma cells.
PLoS One. 6:e199462011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kiselev Y, Eriksen TE, Forsdahl S, Nguyen
LH and Mikkola I: 3T3 cell lines stably expressing Pax6 or Pax6(5a)
- a new tool used for identification of common and isoform specific
target genes. PLoS One. 7:e319152012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang H, Li W, Sun S, Yu S, Zhang M and
Zou F: Inhibition of sphingosine kinase 1 suppresses proliferation
of glioma cells under hypoxia by attenuating activity of
extracellular signal-regulated kinase. Cell Prolif. 45:167–175.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang J, Lu JP, Suter DM, Krause KH, Fini
ME, Chen B and Lu Q: Isoform- and dose-sensitive feedback
interactions between paired box 6 gene and delta-catenin in cell
differentiation and death. Exp Cell Res. 316:1070–1081. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Frattini V, Trifonov V, Chan JM, Castano
A, Lia M, Abate F, Keir ST, Ji AX, Zoppoli P, Niola F, et al: The
integrated landscape of driver genomic alterations in glioblastoma.
Nat Genet. 45:1141–1149. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Estivill-Torrus G, Pearson H, van
Heyningen V, Price DJ and Rashbass P: Pax6 is required to regulate
the cell cycle and the rate of progression from symmetrical to
asymmetrical division in mammalian cortical progenitors.
Development. 129:455–466. 2002.PubMed/NCBI
|
|
48
|
Sakurai K and Osumi N: The
neurogenesis-controlling factor, Pax6, inhibits proliferation and
promotes maturation in murine astrocytes. J Neurosci. 28:4604–4612.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Appolloni I, Calzolari F, Barilari M,
Terrile M, Daga A and Malatesta P: Antagonistic modulation of
gliomagenesis by Pax6 and Olig2 in PDGF-induced oligodendroglioma.
Int J Cancer. 131:E1078–E1087. 2012. View Article : Google Scholar : PubMed/NCBI
|