Introduction
Resistance to chemotherapy is a major issue in the
treatment of cancer. Cancer cells exhibit intrinsic and acquired
resistance to anticancer agents, both resulting from various
genetic and epigenetic changes in the cells (1). Recent studies have revealed that
resistance to cancer chemotherapy occurs not only with conventional
chemotherapy, but also with targeted therapies such as gefitinib
(2) and imatinib (3). Thus, it is crucial to identify agents
that regulate chemotherapy resistance to promote effective clinical
outcomes. Cisplatin, cis-diamminedichloroplatinum (II) (CDDP), is a
vital anticancer agent commonly used in chemotherapy for various
human cancers. Previous studies have revealed that CDDP resistance
is correlated with reduced drug accumulation in the cells (4,5),
increased DNA repair 6), higher level of intracellular thiols such
as glutathione and metallothioneins (7,8), and
anti-apoptotic activity (9). These
findings suggest that a series of events contribute to acquired
CDDP resistance.
In the present study, we focused on members of
aldo-keto reductase (AKR) 1C family, AKR1C1, AKR1C2, AKR1C3, and
AKR1C4, as putative genes, which may be associated with CDDP
resistance. AKR1C is one of the AKR superfamily members and has 4
isoforms; AKR1C1, AKR1C2, AKR1C3, and AKR1C4. These are mapped on
chromosome 10p15-14 and share high sequence homology with each
other (10). These enzymes catalyze
steroids (11), prostaglandins
(12), and lipid aldehydes
(13); however, altered expression
profiles of AKR1C family members have been reported in some
malignant tumors. Upregulated AKR1C3 expression has been
demonstrated in breast cancer (14), prostate cancer (15), adenocarcinoma and squamous cell
carcinoma of the lung (16), and
squamous cell carcinoma of the head and neck (17). Previous studies suggested that
overexpression of AKR1C1 and AKR1C2 was closely associated with
platinum drug resistance in human cancers (18,19).
Thus, it is reasonable to conclude that AKR1Cs may regulate
chemotherapy resistance to anticancer agents and that controlling
AKR1C activity using its inhibitors may lead to favorable
therapeutic outcomes. The present study aimed to determine an
approach that suppresses the mechanism of anticancer drug
resistance by controlling AKR1C enzyme activities.
Materials and methods
Cell lines and cell culture
The cervical cancer cell line, HeLa and the
OSCC-derived cell line, Sa3 were used in the present study.
CDDP-resistant cells established from these cell lines, HeLa-R and
Sa3-R, were used (20). Cell lines
were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10%
fetal bovine serum and 50 U/ml of penicillin and streptomycin.
Ethics statement
The present study was approved by the Ethics
Committee of the Graduate School of Medicine, Chiba University
(approval number, 236) and performed according to the tenets of the
Declaration of Helsinki. All the animal experiments were performed
in accordance with the ethical standards of Canadian Council on
Animal Care (CCAC) and institutional guidelines.
RNA extraction and reverse
transcription
Total RNA was extracted using TRIzol Reagent
(Invitrogen, Carlsbad, CA, USA), according to the manufacturer's
instructions. The quality of the total RNA was determined using the
Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
cDNA was synthesized from total RNA using Ready-to-Go You-Prime
first-strand beads (GE Healthcare, Buckinghamshire, UK) and oligo
(dT) primer (Sigma Genosys, Ishikari, Japan) according to the
manufacturer's protocol.
Real-time qRT-PCR analysis
Real-time qRT-PCR was performed using
LightCycler® 480 Probes Master kit (Roche Diagnostics
GmbH, Mannheim, Germany) according to manufacturer's instructions.
PCR reactions were performed in the Light Cycler (Roche Diagnostics
GmbH) apparatus. Transcript amount was estimated using standard
curves and normalized against glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) transcripts determined in corresponding
samples. Nucleotide sequences of the specific primers for
AKR1C1, AKR1C2, AKR1C3, AKR1C4, and GAPDH are shown
in Table I.
 | Table I.Specific primers used in real-time
qRT-PCR. |
Table I.
Specific primers used in real-time
qRT-PCR.
| Gene | Forward | Reverse |
|---|
| AKR1C1 |
5′-catgcctgtcctgggattt-3′ |
5′-agaatcaatatggcggaagc-3′ |
| AKR1C2 |
5′-ttccatacagaaacttcttttccac-3′ |
5′-ggttaaccaatggcatgtga-3′ |
| AKR1C3 |
5′-cattggggtgtcaaacttca-3′ |
5′-ccggttgaaatacggatgac-3′ |
| AKR1C4 |
5′-tcggggtgtcaaacttcaa-3′ |
5′-gctctggttgaggtaaggatga-3′ |
| GAPDH |
5′-catctctgccccctctgctga-3′ |
5′-ggatgaccttgcccacagcct-3′ |
Small-interfering RNA and transfection
reagents
SMARTpool siRNA targeting AKR1Cs (siAKR1C1,
siAKR1C2, siAKR1C3, and siAKR1C4) (Dharmacon, Lafayette, CO, USA)
were used in gene silencing. Vehicle control and siCONTROL
nontargeting siRNA pool (D-001206-13-20; siNT) were used as
negative controls. cyclophilin B (siCONTROL cyclophilin B; siCyclo)
was used as a positive silencing control to ascertain the
transfection efficiency in each experiment. Cells were transfected
with siRNAs using DharmaFECT 1 siRNA transfection reagent
(Dharmacon).
3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium,
inner salt assay (MTS assay)
Cells (2000 cells/well) were seeded in triplicate in
a 96-well plate and cultured for 72 h. MTS reagent (Promega) was
then added and incubated for 4 h, and relative cell viability was
determined by recording the absorbance at 490 nm. Assays were
performed with or without anticancer agent [CDDP, 5-fluorouracil
(5-FU)] or non-steroidal anti-inflammatory drugs (NSAIDs). The
experiment was repeated 3 times.
Western blotting analysis
Expression of AKR1C family proteins was detected
using western blot analysis. Cells were pelleted and resuspended to
a concentration of 108 cells/ml in lysis buffer (150 mM
NaCl, 20 mM Tris-HCl, pH 7.4, containing 1% Triton X-100, 10%
glycerol, 5 mM EDTA, 10 mM NaF, 1 mM sodium orthovanadate, 100 U/ml
aprotinin, 10 mM iodoacetamide, and 25 µg/ml
p-nitrophenyl-p'-guanidinobenzoate) with a cocktail of proteinase
inhibitors (Roche Diagnostics GmbH). After centrifugation at 15,000
× g for 20 min, supernatants (cell lysates) were collected and
subjected to SDS-PAGE (4–12%) and transferred to nitrocellulose
membranes (Invitrogen). After blocking with Blocking One (Nacalai
Tesque, Kyoto, Japan), the membrane was incubated with each
antibody against AKR1C1, AKR1C2, AKR1C3, AKR1C4 (Sigma-Aldrich, St.
Louis, MO, USA), respectively. Then, the membrane was incubated
with the horseradish peroxidase-conjugated anti-mouse or
anti-rabbit IgG antibody (Promega, Madison, WI, USA). The signals
were detected using SuperSignal West Pico Chemiluminescent
substrate (Thermo) and were visualized using ATTO Light-Capture II
(Atto, Tokyo, Japan). Protein expression profiles of AKR1Cs were
normalized with the internal control, β-actin.
Xenograft
HeLa and HeLa-R cells were used in the xenograft
experiment. Cells (1×107) were dissolved in 200 µl of
phosphate buffered saline (PBS) and injected subcutaneously into
the backs of 6-week-old female athymic nude mice;
BALB/cAnNcrj-nu/nu (Charles River Japan Inc., Kanagawa, Japan).
When the transplanted tumor volume reached 100 mm3, the
mice were divided into 4 groups; control (n=5), anticancer agent
(CDDP or 5-FU) alone (n=5), mefenamic acid alone (n=5), and
anticancer agent plus mefenamic acid (n=5). CDDP (2 mg/kg) was
administered intraperitoneally once weekly for 4 weeks. 5-FU (7.5
mg/kg) was administered intraperitoneally five times a week for 3
weeks. The freeze-dried diet containing 0.0125% of mefenamic acid
was prepared and fed to the mice. In addition to tumor volume, body
weight of mice was monitored throughout the experiment period. Six
weeks after medication was initiated, tumor tissues were resected
and analyzed.
Statistical analysis
The average values and standard deviation were
analysed, and P-value was calculated using two-tailed, Student's
t-test in MTS assay and in xenograft experiment. For all tests,
α-level was 5% and the criterion for statistical significance was
P<0.05.
Results
Characterization of CDDP-resistant
cell lines
The cervical cancer cell line, HeLa (Japanese
Collection of Research Biosources Cell Bank, Osaka, Japan) and the
OSCC-derived cell line, Sa3 (RIKEN BioResource Center, Ibaraki,
Japan) were used in the present study. Moreover, CDDP-resistant
cells established from these cell lines, HeLa-R and Sa3-R, were
used (20). The sensitivity of
these cells to various concentrations of CDDP was determined using
MTS assay. Sa3-R and HeLa-R cells showed significantly higher
viable cell rates than the parent clones with the same
concentration of CDDP. The 50% inhibitory concentration
(IC50) values (µM) in the Sa3, Sa3-R, HeLa, and HeLa-R
cells were 1.4, 18.4, 1.0, and 12.9, respectively. CDDP-resistant
cell lines also showed drug resistance to 5-FU, suggesting that the
cell lines potentially had cross-resistance to anticancer reagents.
The IC50 values (µg/ml) in the Sa3, Sa3-R, HeLa, and
HeLa-R cells were 1.1, 38.3, 51.5, and 212.6, respectively. No
morphologic difference was observed between CDDP-resistant cell
lines and the parent cell lines. The CDDP-resistant cells as well
as the parent cell lines had proliferative ability.
Analysis of gene expression in
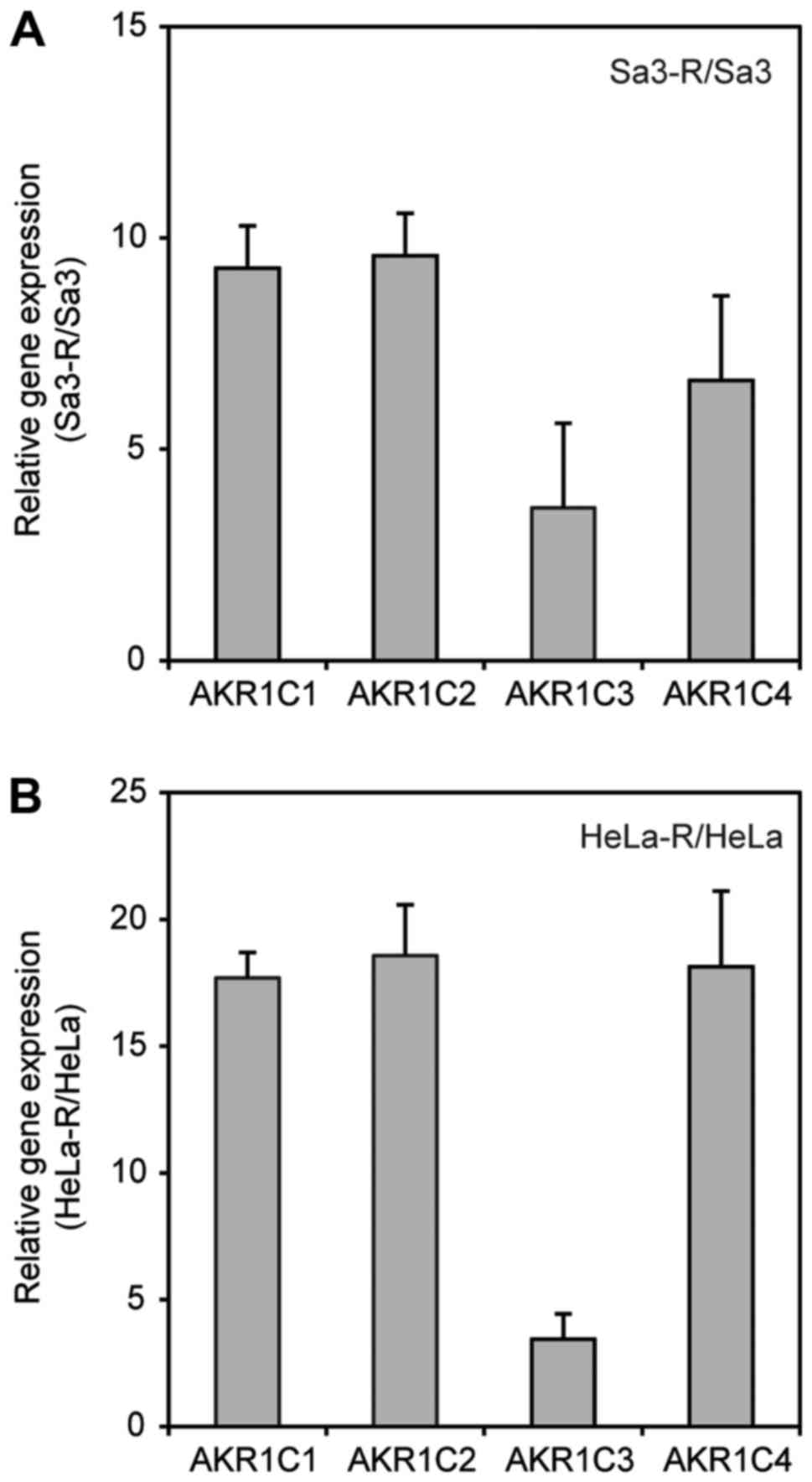
CDDP-resistant cells
Real-time quantitative reverse
transcriptase-polymerase chain reaction (qRT-PCR) analysis revealed
that aldo-keto reductase (AKR) 1C1, AKR1C2, AKR1C3, and
AKR1C4 were significantly upregulated in the CDDP-resistant
cell lines (Fig. 1). Data are
expressed as the average ± standard deviation (SD) of two
independent experiments with samples in triplicate. The results
indicated that AKR1C family members were associated with resistance
to CDDP; thus, the AKR1C family was subjected to further
investigation.
Functional analysis in
small-interfering RNA (siRNA)-transfected cells
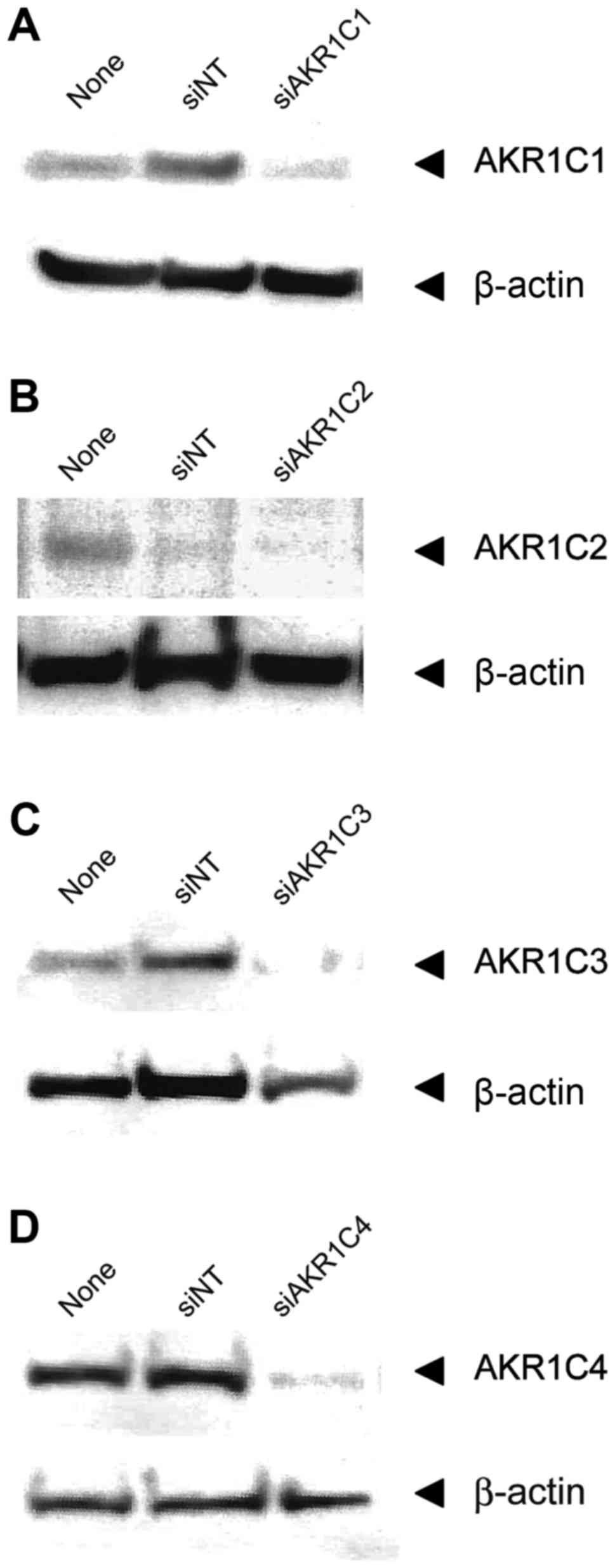
To determine whether AKR1C gene silencing
contributes to CDDP sensitivity, cells were transfected with
siRNAs. The AKR1C protein expression was examined by western blot
analysis in Sa3-R and HeLa-R cells 120 h after transfection with
siRNAs. AKR1C protein levels in non-targeting siRNA
(siNT)-transfected cells were comparable to those of AKR1Cs in
non-treated cells. AKR1C protein levels reduced significantly
compared to the siNT-transfected cells (Fig. 2). These transfected cells were
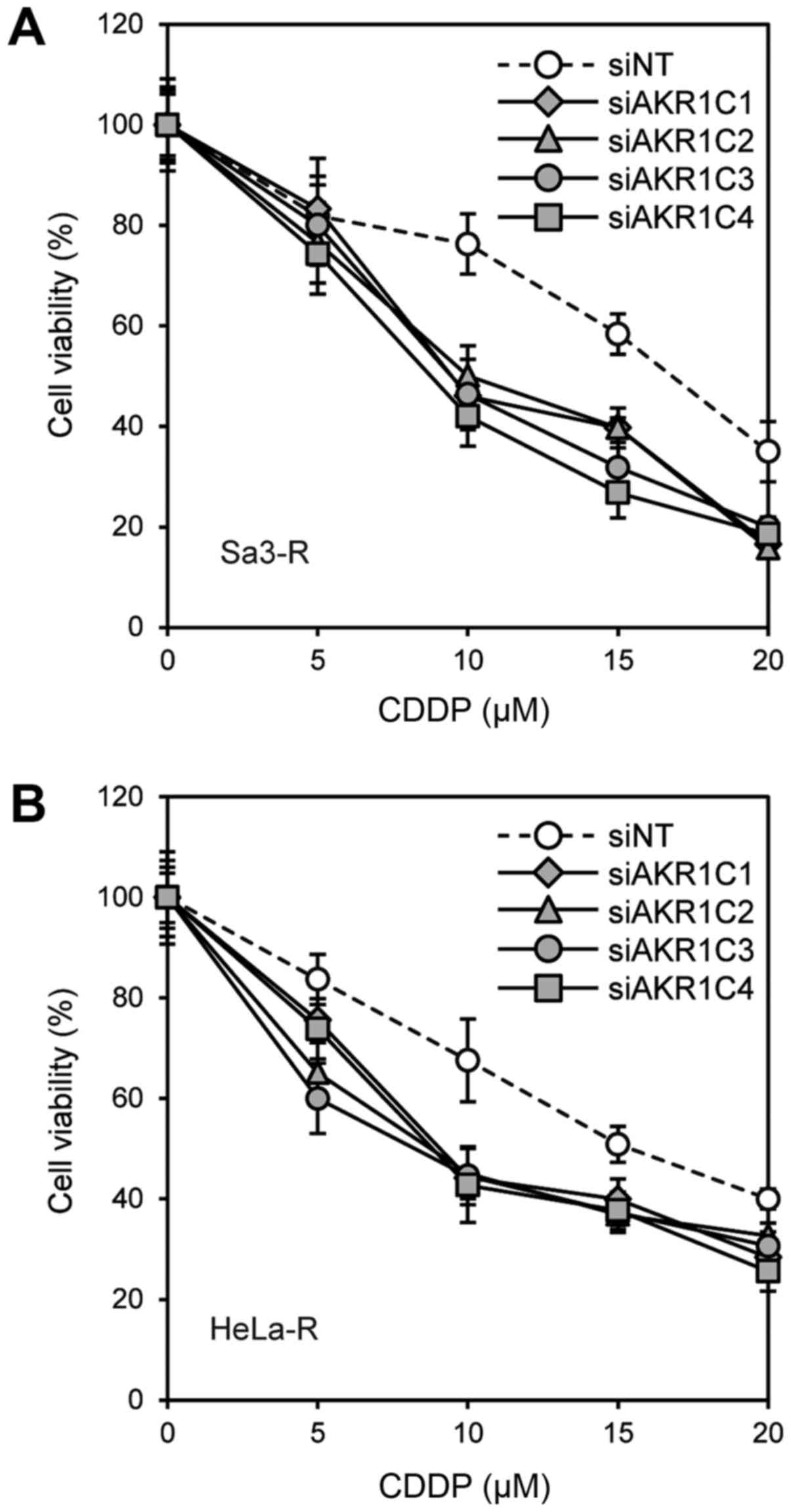
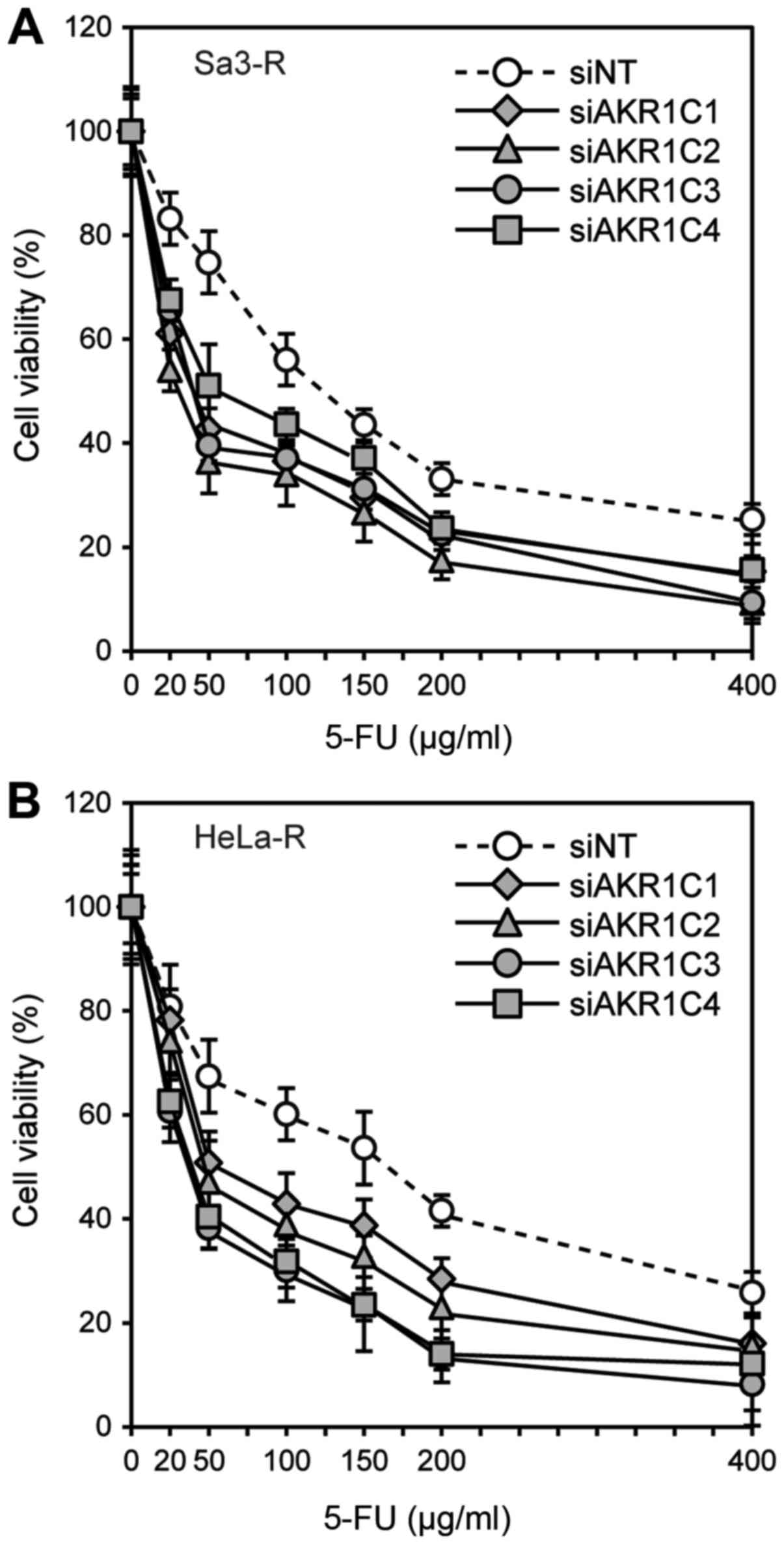
subjected to functional analysis to determine the effect of AKR1C
gene silencing on CDDP sensitivity. The sensitivities of these
cells to CDDP were determined by MTS assay. Increased sensitivity
to CDDP and 5-FU was observed in Sa3-R cells transfected with
siAKR1C1, siAKR1C2, siAKR1C3, and siAKR1C4, compared to that
observed in the corresponding cells treated with siNT (Figs. 3 and 4).
AKR1C inhibitor-induced enhancement of
CDDP sensitivity in vitro
Since mefenamic acid is an effective inhibitor of
AKR1Cs (21), mefenamic acid was
added, and the alteration of CDDP sensitivity in tumor cell lines
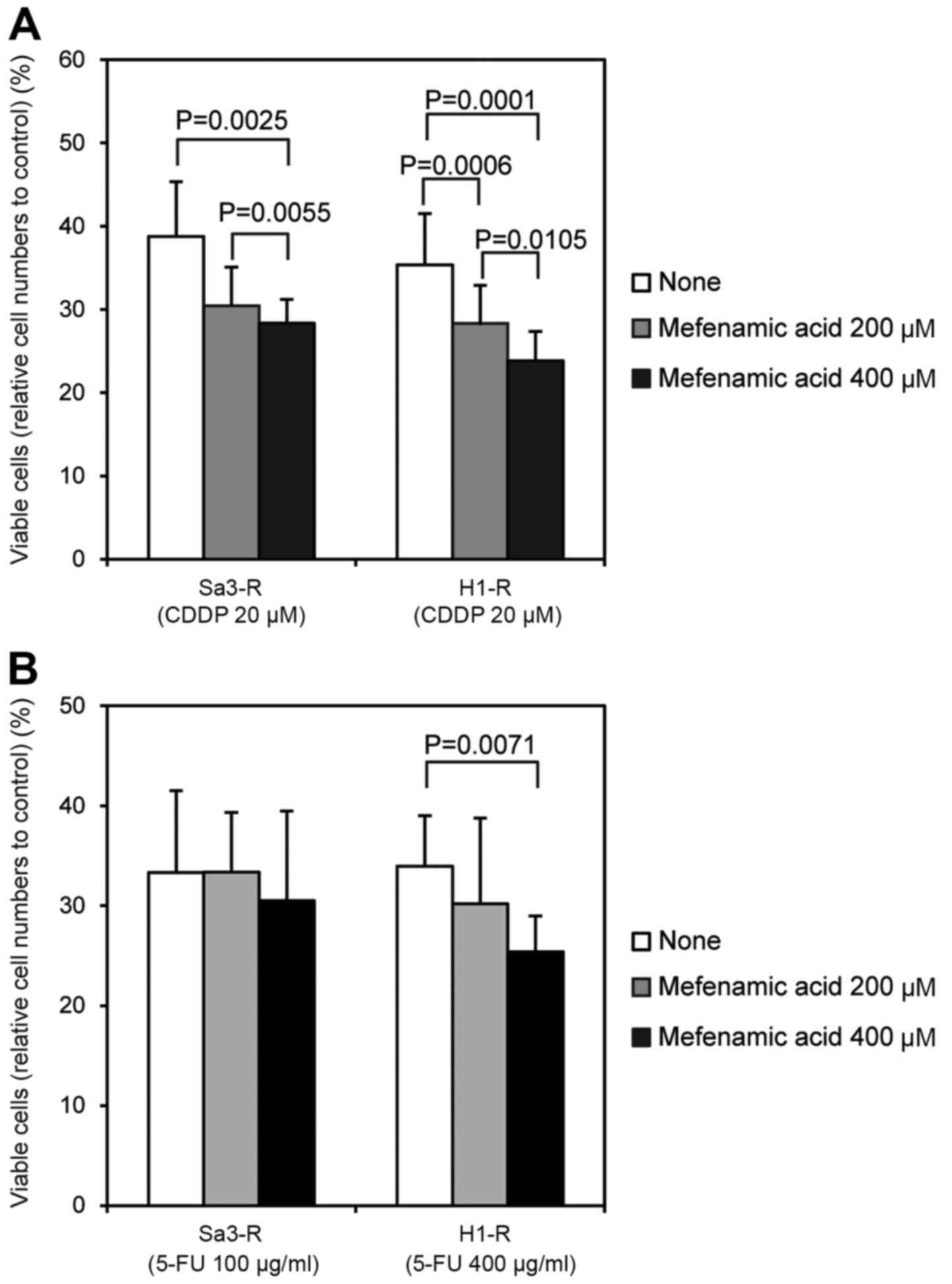
was determined by MTS assay. CDDP-resistant cells treated with
mefenamic acid (Sa3-R and HeLa-R) demonstrated restored sensitivity
to CDDP and 5-FU in a dose-dependent manner (Fig. 5). As a result, mefenamic acid was
used for further experiments in vivo.
Effect of mefenamic acid on CDDP
sensitivity in vivo
Mouse tumor xenografts were established to
investigate whether an AKR1C inhibitor enhances sensitivity to
CDDP. HeLa and HeLa-R cells were subcutaneously implanted into
female athymic nude mice, and the tumor volume was measured
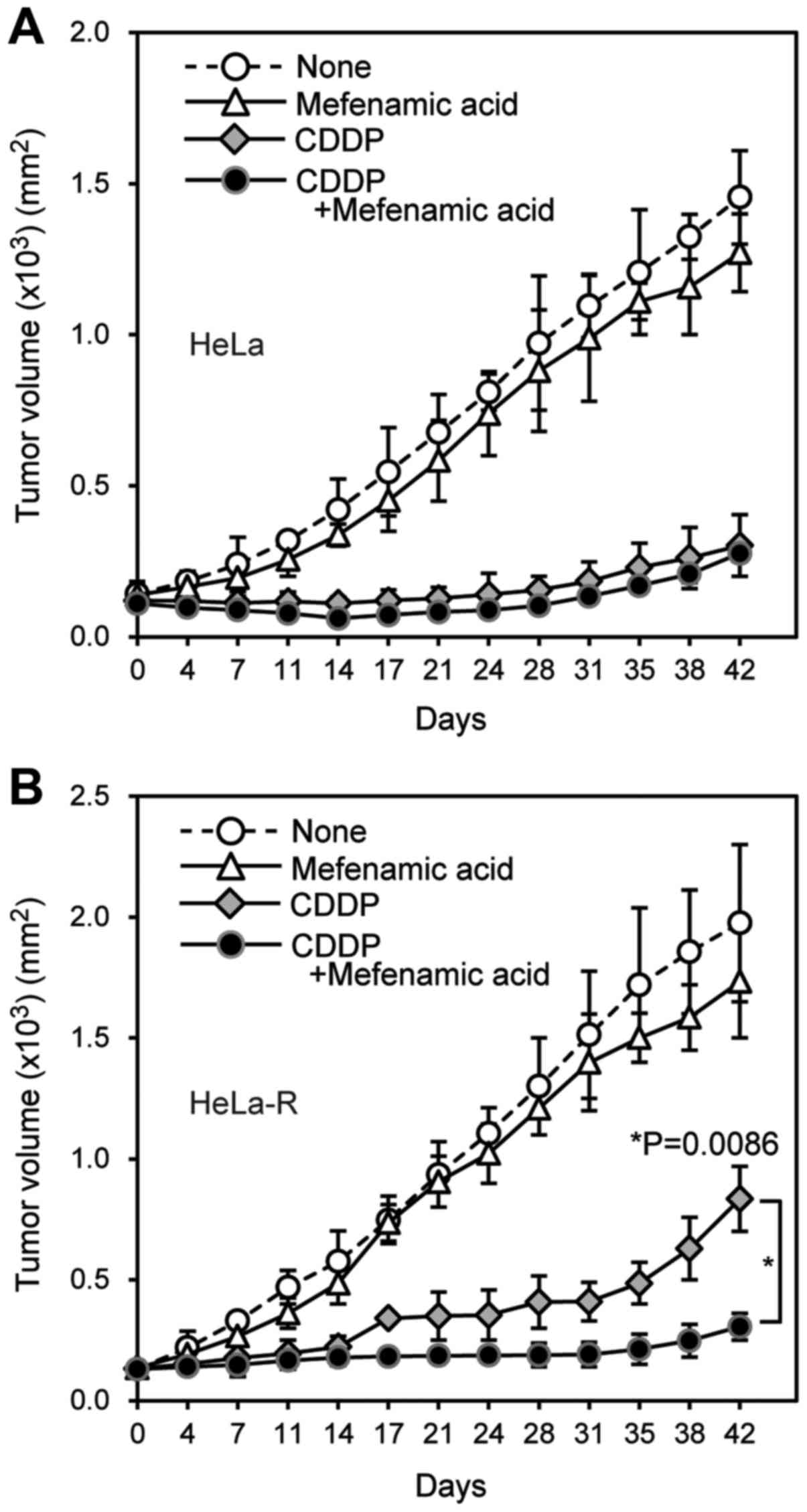
regularly until day 42. When CDDP was administered, tumor growth
was suppressed in HeLa cell transplanted mice, however, tumor
volume gradually increased in HeLa-R cell transplanted mice
probably due to the CDDP-resistant characteristic (Fig. 6). The increased tumor volume
significantly reduced in HeLa-R cell xenografts when mefenamic acid
was added to CDDP (Fig. 7).
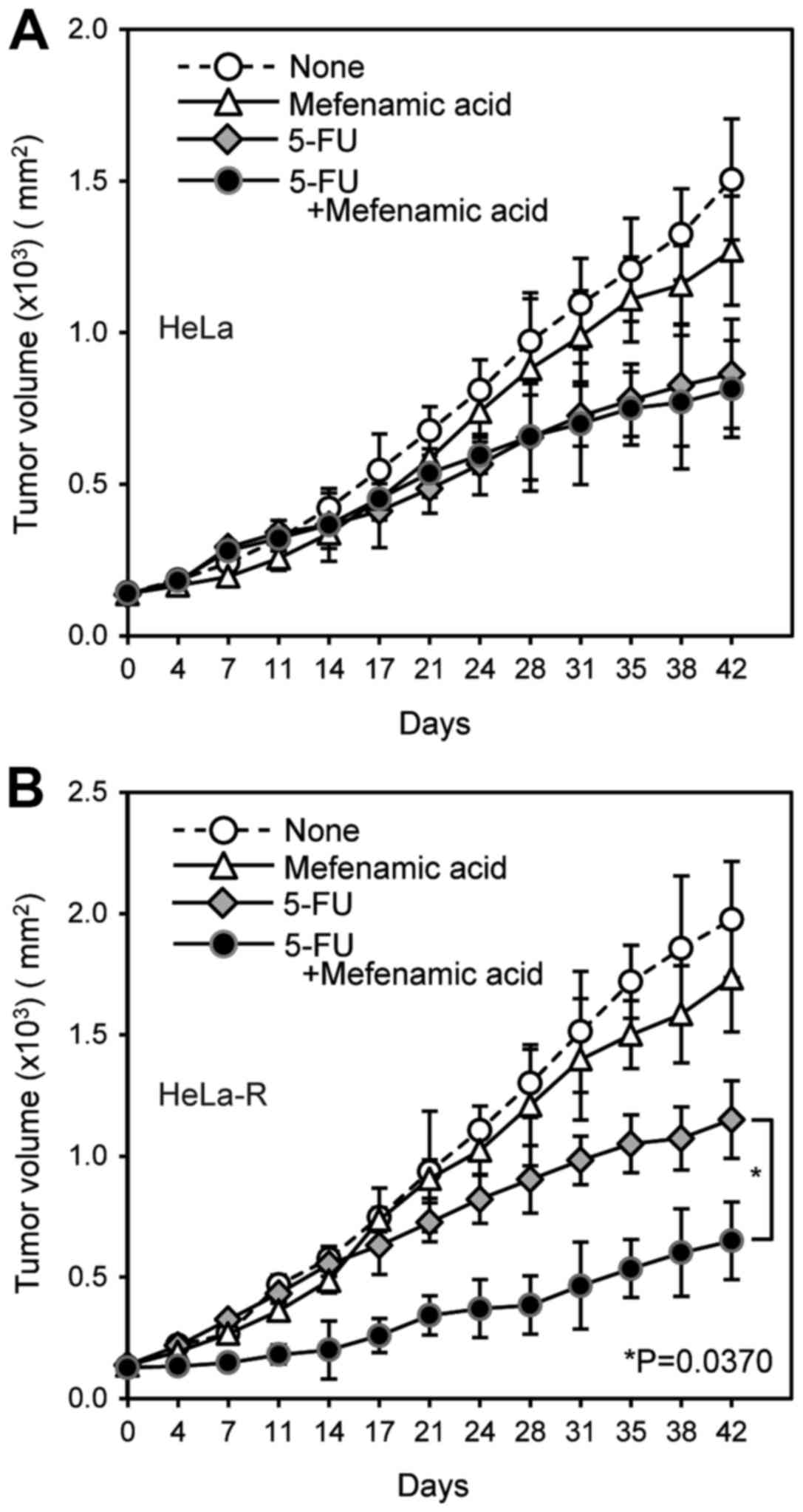
Similarly, in the 5-FU administered mice, tumor volume in the mice
treated with mefenamic acid was significantly smaller than without
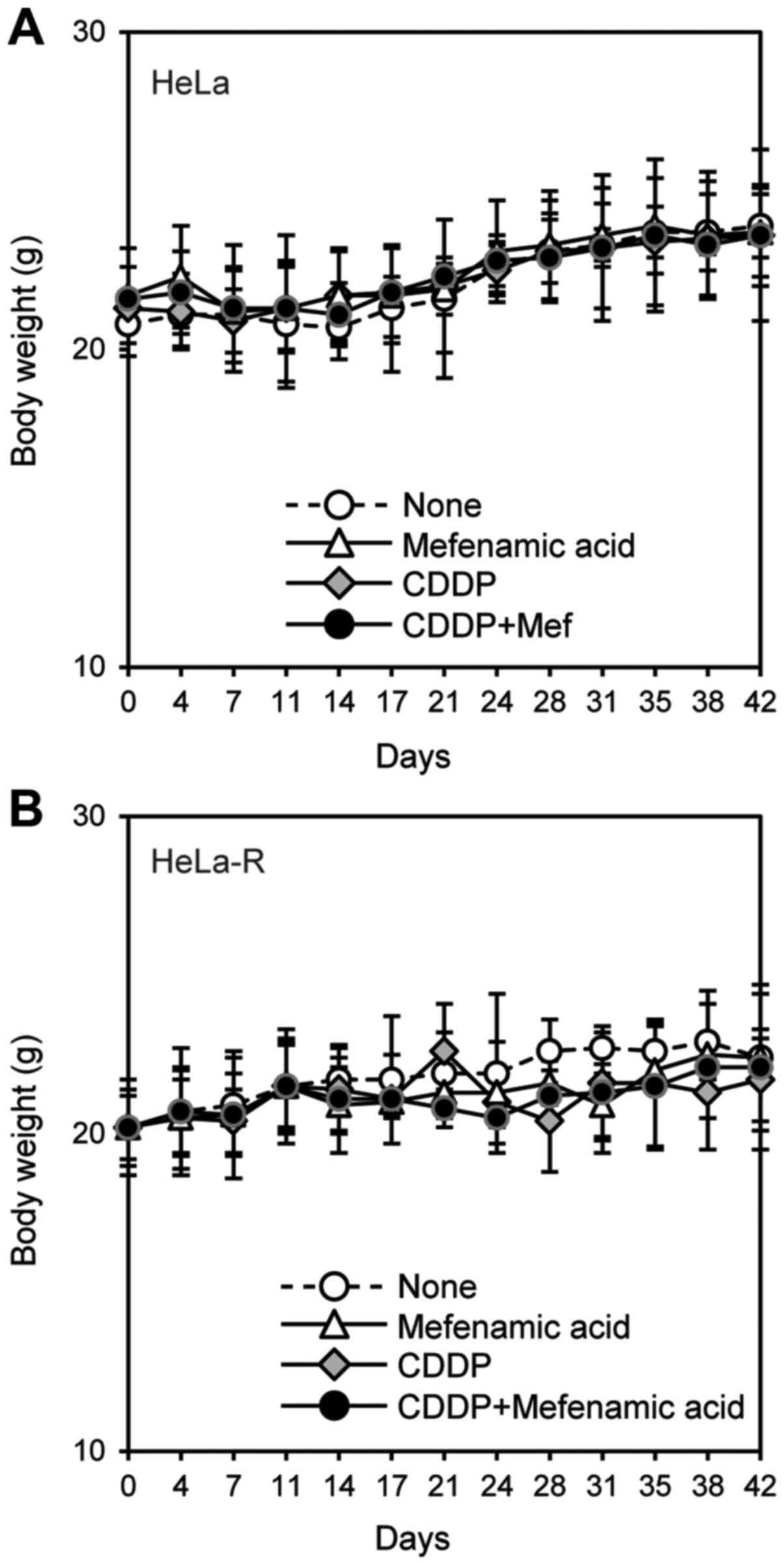
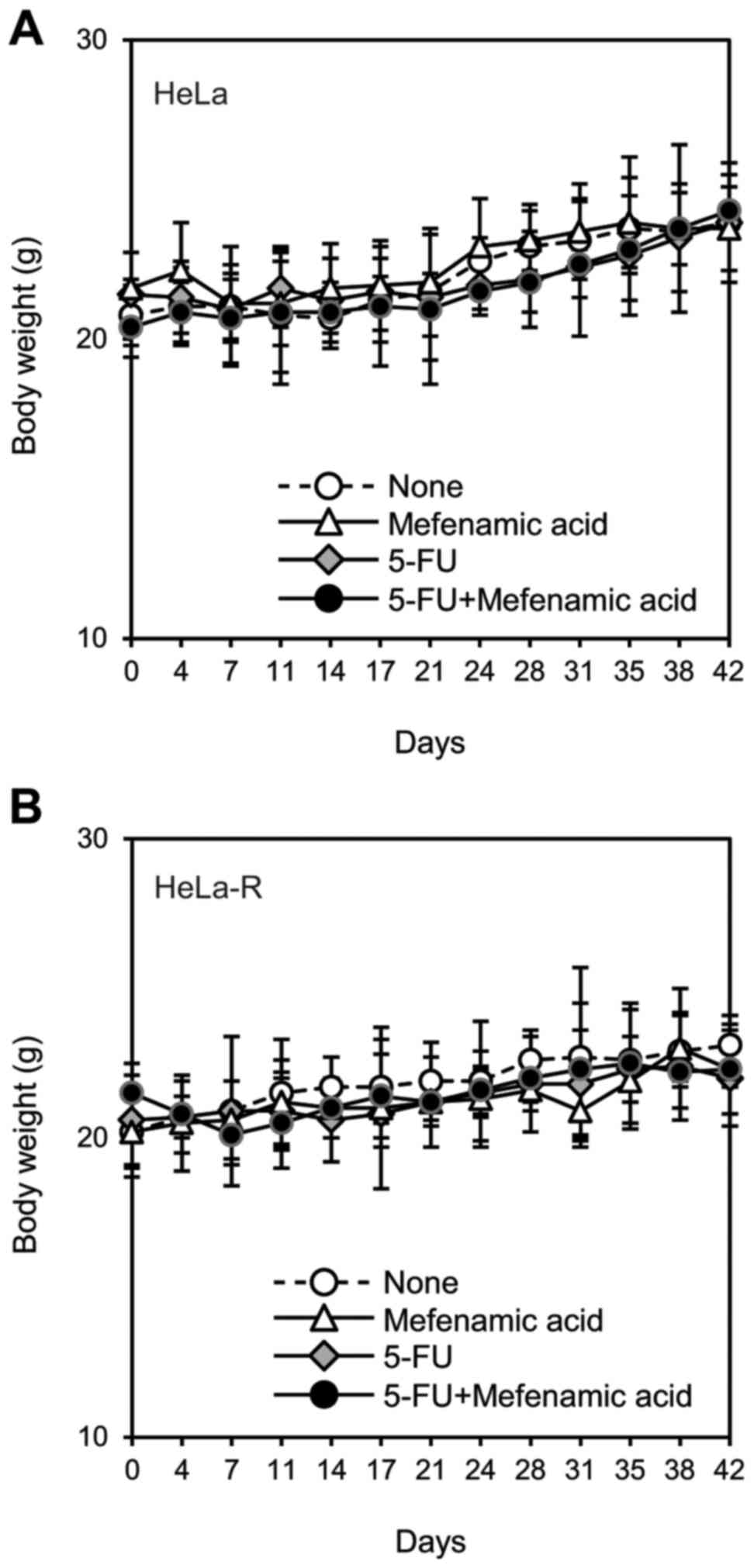
it (Fig. 8). Body weights of the
mice were also measured to examine the systemic effect induced by
these chemical reagents. The data revealed that the systemic
effects observed after combined administration of mefenamic acid
and anticancer agents were not different from that observed with
individual administration (Figs. 9
and 10).
Discussion
In the present study, we identified AKR1C1, AKR1C2,
AKR1C3, and AKR1C4 as putative genes, which may be associated with
anticancer drug resistance. Knockdown of AKR1Cs genes apparently
increased the cytotoxic effect of CDDP in CDDP-resistant cells,
suggesting that inhibition of AKR1Cs' activity can induce enhanced
CDDP sensitivity. Furthermore, administration of mefenamic acid, a
known inhibitor of AKR1C, increased sensitivity to CDDP and 5-FU,
in vitro and in vivo without mefenamic acid-induced
adverse effects.
The causes of resistance to anticancer medications
are multifactorial and involve numerous genetic and epigenetic
changes (14,18,22).
Cisplatin is converted to its active form by intracellular aquation
of one of two chloride-leaving groups and covalently binding to
purine DNA base leading to the formation of intrastrand or
interstrand crosslink adducts, which lead to cellular apoptosis
(23–25). This process of cisplatin activation
may be inhibited by the enzyme activity of AKR1C; however, the
mechanism by which this occurs is still unknown. Wang et al
(18) demonstrated that
interleukin-6, a pro-inflammatory cytokine, is crucial for
overexpression of AKR1C1 and AKR1C2 and for resistance to
anticancer drugs in NSCLC cells. Matsunaga et al reported
that knockdown of AKR1C1 and AKR1C3 and the use of their specific
inhibitors improved sensitivity to CDDP in human colon cancer
cells, and suggested that the underlying mechanism for CDDP
resistance is most likely due to aldehyde detoxification, resulting
from enhanced oxidative stress (26). Moreover, blockade of proteasome
leads to a compensatory upregulation of AKR1C1 and AKR1C3 in
CDDP-resistant cells (26). Novotna
et al reported that human AKR1C3 might mediate deactivation
of the anticancer drugs, oracin and doxorubicin, via carbonyl
reduction in hormone-dependent malignancies such as prostate and
breast cancers (27).
Previous studies suggested that formation of
reactive oxygen species (ROS) induces the CDDP toxicity (24,28,29).
Ebert et al reported that some proteasome inhibitors produce
mild oxidative stress, which activates nuclear factor-erythroid 2
related factor 2 (Nrf2)-related genes leading to AKR1C induction,
suggesting that proteasome inhibitors may elicit a protective
effect (30).
Noteworthy, the present study revealed that
overexpression of AKR1Cs is also associated with 5-FU drug
resistance. 5-FU has been used clinically since the late 1950s for
the treatment of various cancers (31). Both CDDP and 5-FU have specific
pharmacokinetics; the mechanism of CDDP involves covalent binding
to purine DNA bases, which primarily leads to cellular apoptosis
(22), whereas 5-FU is
enzymatically converted to the main active metabolites
fluorodeoxyuridine monophosphate (FdUMP), fluorodeoxyuridine
triphosphate (FdUTP), and fluorouridine triphosphate (FUTP). FUTP
and FdUTP lead to RNA and DNA damage, respectively, and FdUTP
induces DNA damage via inhibition of thymidylate synthase (TS)
(32). This suggests that AKR1Cs
may have various roles in anticancer drug resistance mechanisms,
such as drug uptake, DNA-damage recognition and repair, and
apoptosis.
Byrns et al reported that NSAIDs, sulindac,
mefenamic acid, arylpropionic acids, and indomethacin, inhibit
AKR1C enzyme activity (21,33). We investigated the inhibitory
activity of these NSAIDs and determined mefenamic acid to be the
most effective drug for this purpose, and we could not find the
significant altered expression of AKR1Cs induced by mefenamic acid
(data not shown). Both drug safety and effectiveness are equally
important parameters for clinical use of a drug. Mefenamic acid has
long been used as a medicinal agent and is deemed safe. Thus, the
use of mefenamic acid as an AKR1C inhibitor to enhance the effect
of chemotherapy is plausible.
In conclusion, the present study suggests that
AKR1C1, AKR1C2, AKR1C3, and AKR1C4 are closely associated with drug
resistance to both CDDP and 5FU, and that mefenamic acid, an
inhibitor of AKR1C, restores sensitivity through inhibition of
drug-resistance in human cancer cells. This implies that inhibition
of the AKR1C biological function may lead to an effective clinical
outcome by either overcoming anticancer drug resistance or reducing
adverse effects of concomitant medications.
References
|
1
|
Rebucci M and Michiels C: Molecular
aspects of cancer cell resistance to chemotherapy. Biochem
Pharmacol. 85:1219–1226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weisberg E, Manley PW, Cowan-Jacob SW,
Hochhaus A and Griffin JD: Second generation inhibitors of BCR-ABL
for the treatment of imatinib-resistant chronic myeloid leukaemia.
Nat Rev Cancer. 7:345–356. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gately DP and Howell SB: Cellular
accumulation of the anticancer agent cisplatin: A review. Br J
Cancer. 67:1171–1176. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Loh SY, Mistry P, Kelland LR, Abel G and
Harrap KR: Reduced drug accumulation as a major mechanism of
acquired resistance to cisplatin in a human ovarian carcinoma cell
line: Circumvention studies using novel platinum (II) and (IV)
ammine/amine complexes. Br J Cancer. 66:1109–1115. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Scanlon KJ, Kashani-Sabet M, Tone T and
Funato T: Cisplatin resistance in human cancers. Pharmacol Ther.
52:385–406. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hosking LK, Whelan RD, Shellard SA,
Bedford P and Hill BT: An evaluation of the role of glutathione and
its associated enzymes in the expression of differential
sensitivities to antitumour agents shown by a range of human tumour
cell lines. Biochem Pharmacol. 40:1833–1842. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Estrela JM, Ortega A and Obrador E:
Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci.
43:143–181. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Perego P, Giarola M, Righetti SC, Supino
R, Caserini C, Delia D, Pierotti MA, Miyashita T, Reed JC and
Zunino F: Association between cisplatin resistance and mutation of
p53 gene and reduced bax expression in ovarian carcinoma cell
systems. Cancer Res. 56:556–562. 1996.PubMed/NCBI
|
|
10
|
Penning TM, Burczynski ME, Jez JM, Hung
CF, Lin HK, Ma H, Moore M, Palackal N and Ratnam K: Human
3alpha-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the
aldo-keto reductase superfamily: Functional plasticity and tissue
distribution reveals roles in the inactivation and formation of
male and female sex hormones. Biochem J. 351:67–77. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schlegel BP, Pawlowski JE, Hu Y, Scolnick
DM, Covey DF and Penning TM: Secosteroid mechanism-based
inactivators and site-directed mutagenesis as probes for steroid
hormone recognition by 3 alpha-hydroxysteroid dehydrogenase.
Biochemistry. 33:10367–10374. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bohren KM, Bullock B, Wermuth B and Gabbay
KH: The aldo-keto reductase superfamily. cDNAs and deduced amino
acid sequences of human aldehyde and aldose reductases. J Biol
Chem. 264:9547–9551. 1989.PubMed/NCBI
|
|
13
|
Hyndman D, Bauman DR, Heredia VV and
Penning TM: The aldo-keto reductase superfamily homepage. Chem Biol
Interact. 143-144:621–631. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin HK, Steckelbroeck S, Fung KM, Jones AN
and Penning TM: Characterization of a monoclonal antibody for human
aldo-keto reductase AKR1C3 (type 2 3alpha-hydroxysteroid
dehydrogenase/type 5 17beta-hydroxysteroid dehydrogenase);
immunohistochemical detection in breast and prostate. Steroids.
69:795–801. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dozmorov MG, Azzarello JT, Wren JD, Fung
KM, Yang Q, Davis JS, Hurst RE, Culkin DJ, Penning TM and Lin HK:
Elevated AKR1C3 expression promotes prostate cancer cell survival
and prostate cell-mediated endothelial cell tube formation:
Implications for prostate cancer progression. BMC Cancer.
10:6722010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Miller VL, Lin HK, Murugan P, Fan M,
Penning TM, Brame LS, Yang Q and Fung KM: Aldo-keto reductase
family 1 member C3 (AKR1C3) is expressed in adenocarcinoma and
squamous cell carcinoma but not small cell carcinoma. Int J Clin
Exp Pathol. 5:278–289. 2012.PubMed/NCBI
|
|
17
|
Martinez I, Wang J, Hobson KF, Ferris RL
and Khan SA: Identification of differentially expressed genes in
HPV-positive and HPV-negative oropharyngeal squamous cell
carcinomas. Eur J Cancer. 43:415–432. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang HW, Lin CP, Chiu JH, Chow KC, Kuo KT,
Lin CS and Wang LS: Reversal of inflammation-associated dihydrodiol
dehydrogenases (AKR1C1 and AKR1C2) overexpression and drug
resistance in nonsmall cell lung cancer cells by wogonin and
chrysin. Int J Cancer. 120:2019–2027. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Deng HB, Adikari M, Parekh HK and Simpkins
H: Ubiquitous induction of resistance to platinum drugs in human
ovarian, cervical, germ-cell and lung carcinoma tumor cells
overexpressing isoforms 1 and 2 of dihydrodiol dehydrogenase.
Cancer Chemother Pharmacol. 54:301–307. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Negoro K, Yamano Y, Fushimi K, Saito K,
Nakatani K, Shiiba M, Yokoe H, Bukawa H, Uzawa K, Wada T, et al:
Establishment and characterization of a cisplatin-resistant cell
line, KB-R, derived from oral carcinoma cell line, KB. Int J Oncol.
30:1325–1332. 2007.PubMed/NCBI
|
|
21
|
Byrns MC, Steckelbroeck S and Penning TM:
An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a
selective inhibitor of aldo-keto reductase 1C3 (type 2 3alpha-HSD,
type 5 17beta-HSD, and prostaglandin F synthase), a potential
target for the treatment of hormone dependent and hormone
independent malignancies. Biochem Pharmacol. 75:484–493. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kelland L: The resurgence of
platinum-based cancer chemotherapy. Nat Rev Cancer. 7:573–584.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bauman DR, Rudnick SI, Szewczuk LM, Jin Y,
Gopishetty S and Penning TM: Development of nonsteroidal
anti-inflammatory drug analogs and steroid carboxylates selective
for human aldo-keto reductase isoforms: Potential antineoplastic
agents that work independently of cyclooxygenase isozymes. Mol
Pharmacol. 67:60–68. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Matsunaga T, Tsuji Y, Kaai K, Kohno S,
Hirayama R, Alpers DH, Komoda T and Hara A: Toxicity against
gastric cancer cells by combined treatment with 5-fluorouracil and
mitomycin c: Implication in oxidative stress. Cancer Chemother
Pharmacol. 66:517–526. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Szatrowski TP and Nathan CF: Production of
large amounts of hydrogen peroxide by human tumor cells. Cancer
Res. 51:794–798. 1991.PubMed/NCBI
|
|
26
|
Matsunaga T, Hojo A, Yamane Y, Endo S,
El-Kabbani O and Hara A: Pathophysiological roles of aldo-keto
reductases (AKR1C1 and AKR1C3) in development of cisplatin
resistance in human colon cancers. Chem Biol Interact. 202:234–242.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Novotna R, Wsol V, Xiong G and Maser E:
Inactivation of the anticancer drugs doxorubicin and oracin by
aldo-keto reductase (AKR) 1C3. Toxicol Lett. 181:1–6. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chirino YI and Pedraza-Chaverri J: Role of
oxidative and nitrosative stress in cisplatin-induced
nephrotoxicity. Exp Toxicol Pathol. 61:223–242. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang G, Reed E and Li QQ: Molecular basis
of cellular response to cisplatin chemotherapy in non-small cell
lung cancer (Review). Oncol Rep. 12:955–965. 2004.PubMed/NCBI
|
|
30
|
Ebert B, Kisiela M, Wsól V and Maser E:
Proteasome inhibitors MG-132 and bortezomib induce AKR1C1, AKR1C3,
AKR1B1, and AKR1B10 in human colon cancer cell lines SW-480 and
HT-29. Chem Biol Interact. 191:239–249. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Parker JB and Stivers JT: Dynamics of
uracil and 5-fluorouracil in DNA. Biochemistry. 50:612–617. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Longley DB, Harkin DP and Johnston PG:
5-fluorouracil: Mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Byrns MC, Jin Y and Penning TM: Inhibitors
of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3): Overview and
structural insights. J Steroid Biochem Mol Biol. 125:95–104. 2011.
View Article : Google Scholar : PubMed/NCBI
|