Introduction
Recently, various cancer driver gene mutations have
been identified in non-small cell lung cancers (NSCLC), including
epidermal growth factor receptor (EGFR), KRAS, HER2, and
BRAF mutations (1).
Personalized treatment based on this molecular genetic information
showed positive therapeutic effects (2). EGFR mutations, mostly exon 19
deletions and L858R point mutation in exon 21, are associated with
clinical benefits from EGFR tyrosine kinase inhibitors (EGFR-TKIs)
(3). Therefore, somatic mutational
analysis has become a routine part of clinical practice and is
indispensable for the treatment decisions in NSCLC clinical
settings. Though patients with EGFR mutations are initially
responsive to EGFR-TKIs, almost all of their tumors ultimately
acquire TKI resistance as a result of tumor heterogeneity or clonal
evolution (4,5). Various mechanisms of resistance to the
first generation EGFR-TKIs, including gefitinib and erlotinib, have
been identified, and the drugs to overcome resistance have been
developed. For example, the most frequently reported mechanism of
acquired resistance to EGFR-TKIs is a EGFR T790M point
mutation in exon 20, consisting of >50% of these cases (4,6).
Third-generation EGFR TKIs, including WZ4002, CO-1686, and AZD9291,
have been developed and potently appear to inhibit tumors with
EGFR activating mutations even in the presence of the T790M
mutation (7). Thus, the importance
of T790M mutation detection is increasing in NSCLC treatment.
However, the conventional method for mutation detection requires a
cancer cell biopsy, which is an invasive procedure and sometimes
difficult to perform repeatedly.
Circulating cell-free DNA (cfDNA) has been shown to
be released from apoptotic and necrotic cells into the circulation
and has been identified in plasma (8). Recent technological development has
demonstrated the potential of molecular genetic profiling from
cfDNA. cfDNA has received great attention as one of the critical
targets for non-invasive mutation detection (9). It enables repetitive biopsy and
monitoring of cancer status, though detection of cfDNA from tumor
cells in the circulation still remains challenging. The detection
of cfDNA from tumor cells requires a highly-sensitive assay,
because the majority of circulating cfDNA appears to be from normal
tissue (10). Droplet digital PCR
(ddPCR) is one of the digital PCR technologies and provides
highly-sensitive and absolute quantitative detection of target
genes (9). In ddPCR, template DNA
is partitioned into approximately 20,000 droplets in a single
reaction well, and then amplified within individual droplets. This
approach facilitates the detection and quantification of rare
mutants in a background of wild-type alleles, because rare mutant
alleles can avoid competition with wild-type ones. However, the
optimal method for detecting cfDNA remains debatable.
In this study, we evaluated the optimal method for
mutation analysis with EGFR T790M mutation in plasma using a ddPCR
system.
Materials and methods
Clinical samples and cell lines
This study was approved by the ethics committee of
Okayama University (receipt number: 2220). All patients enrolled in
the study provided written informed consent for the use of tissue
and blood sample. July 2014 through February 2015, we obtained 7-ml
peripheral blood samples using EDTA-2K tubes from two healthy
volunteers and 24 lung adenocarcinoma patients with EGFR
mutations. These patients had been treated with EGFR-TKI and their
EGFR T790M mutational status was examined through re-biopsy
as a part of clinical care. Clinical EGFR mutation tests in tissue
samples had been performed before entry into the study by an
commercial clinical laboratory via mutant-enriched PCR assay using
the peptide nucleic acid (PNA)-locked nucleic acids (LNA) PCR clamp
method (LSI Medience Corp., Tokyo, Japan) (11,12).
The approval of the Institutional Review Board and informed consent
from each patient were obtained. After drawing blood, samples were
immediately centrifuged at 2,330 × g for 10 min to collect plasma,
and the plasma samples were centrifuged at 16,000 × g for 10
additional min. The supernatants were collected, frozen, and stored
at −80°C. In addition, we used an immortalized normal human
bronchial epithelial cell line (HBEC-5KT) harboring the wild-type
EGFR gene and the NCI-H1975 cell line (H1975) harboring the
EGFR mutations L858R and T790M as negative and positive
controls, respectively (13,14).
HBEC-5KT and H1975 were kindly provided by Dr Adi F. Gazdar (The
University of Texas Southwestern Medical Center, Dallas, TX,
USA).
DNA extraction, quantification, and
fragmentation
After thawing the frozen plasma samples, DNA was
extracted using QIAamp Circulating Nucleic Acid kit (Qiagen, Venlo,
The Netherlands), and eluted in 20 µl of the kit's elution buffer.
Control DNA from cell lines was extracted using DNeasy Blood and
Tissue kit (Qiagen) and fragmented through sonication to obtain an
average size of 200 bp using Covaris M220 Focused-ultrasonicator
(Covaris Inc., Woburn, MA, USA) to closely mimic cfDNA in plasma
(15,16). DNA size distribution was analyzed
using Agilent 2200 TapeStation (Agilent Technologies, Santa Clara,
CA, USA). Genomic DNA was quantified by Qubit 2.0 Fluorometer and
Qubit dsDNA HS or BR assay kit (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA).
Primers and probes
The sequences of primers, probes, and blocking oligo
used in this study are shown in Table
I. S1 and AS1 primers and the M1 probe, which were designed in
a previous study, were used as a validated reference assay
(9). The primer melting
temperatures (Tm) were calculated using Primer3Plus (http://primer3plus.com/). Probes, including LNA, were
designed using IDT Biophysics software (http://biophysics.idtdna.com/) and synthesized by
IDT-MBL KK (Nagoya, Japan). The blocking oligo using PNA was
synthesized by Fasmac Co., Ltd. (Atsugi, Japan).
 | Table I.Sequences of primers and probes for
ddPCR. |
Table I.
Sequences of primers and probes for
ddPCR.
| Oligo name | Oligo sequenses 5′ to
3′ | Tm (°C) | Product size
(bp) | Match-mismatch Tm
difference (°C) |
|---|
| Primer S1 | GCCTGCTGGGCATCTG | 59.5 | 93 | NA |
| Primer AS1 |
TCTTTGTGTTCCCGGACATAGTC | 61.6 |
| NA |
| Primer S2 | CCTCACCTCCACCGTG | 57.8 | 43 | NA |
| Primer AS2 | CGAAGGGCATGAGCTG | 56.6 |
| NA |
| Probe M1 |
FAM/ATGAGCTGCATGATGAG/IABkFQ | 58.83 | NA | 5.57 |
| Probe M2 |
FAM/TCA+TC+A+T+GC+AGC/IABkFQ | 60.7 | NA | 11.41 |
| Blocking oligo | TCATCACGCAG (Peptide
nucleic acids) | 58.6 | NA | NA |
Droplet digital PCR assay for EGFR
T790M detection
ddPCR was performed using QX200 Droplet Digital PCR
system (Bio-Rad, Hercules, CA, USA). The final volume of the PCR
mixture was 20 µl, containing 10 µl of ddPCR Supermix for Probes
(No dUTP) (Bio-Rad), 1 µM of each primer, 0.25 µM of each probe,
200 µM of dNTP, and 5 µl of DNA (from approximately 2 ml of blood
sample or control DNA), with or without 5 µM of blocking oligo. The
following PCR conditions for ddPCR were used: 1) an initial
denaturation step at 95°C for 10 min followed by 2) 45 cycles at
94°C for 30 sec, and 3) 45 cycles at 57°C for 1 min. Each ramp rate
was 2°C per second. Annealing temperatures were optimized by
gradient PCR (data not shown). PCR products were then subjected to
analysis by the QX-200 droplet reader and QuantaSoft analysis
software (Bio-Rad). Assays were considered positive if >3
droplets were over the threshold fluorescence of 8,000. The
fluorescence threshold and cut off number for mutation detection
were determined by assessing these data using positive and negative
control DNA from cell lines.
Statistical analysis
The concentrations of target alleles were calculated
using QuantaSoft software 9.2.1 (Bio-Rad) based on Poisson
distribution.
Results
The size of circulating cell-free
DNA
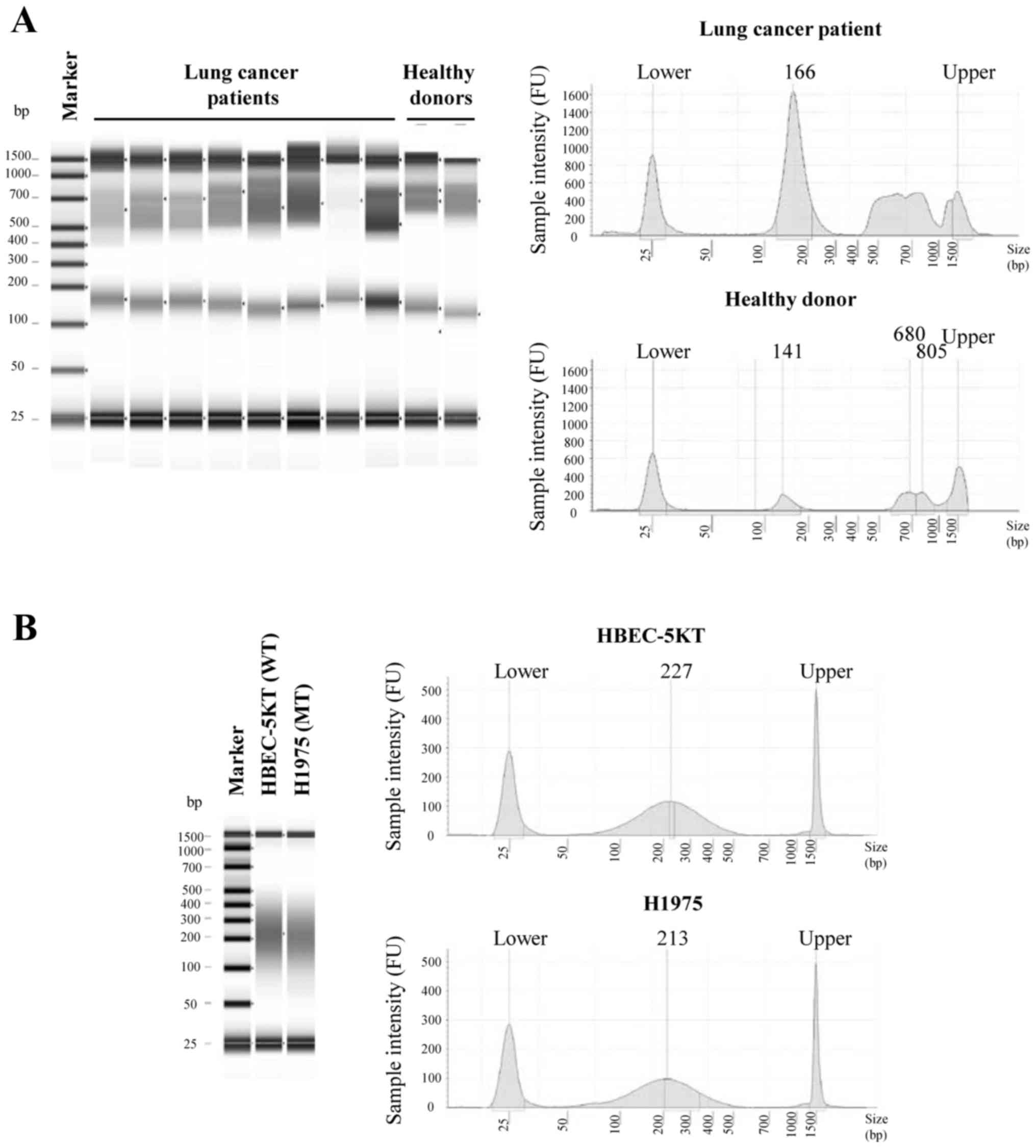
The size distribution of cfDNA extracted from
healthy donors and lung cancer patients was assessed (Fig. 1A). It showed similar distribution
patterns between healthy donors and lung cancer patients, and two
peaks were identified in both sets of patients. The first peak was
distributed from 128 to 168 bp (median, 158 bp), and the 2nd peak
was distributed from 526 to 797 bp (median: 681 bp). Next, we
prepared control cfDNA mimic samples through sonication to evaluate
the method of highly-sensitive detection with focus on cfDNA; the
size of the samples was then confirmed. After sonication, the
genomic DNA samples were fragmented to an average size of
approximately 200 bp (Fig. 1B).
The effect of LNA probe to suppress
the fluorescence intensity of negative droplets
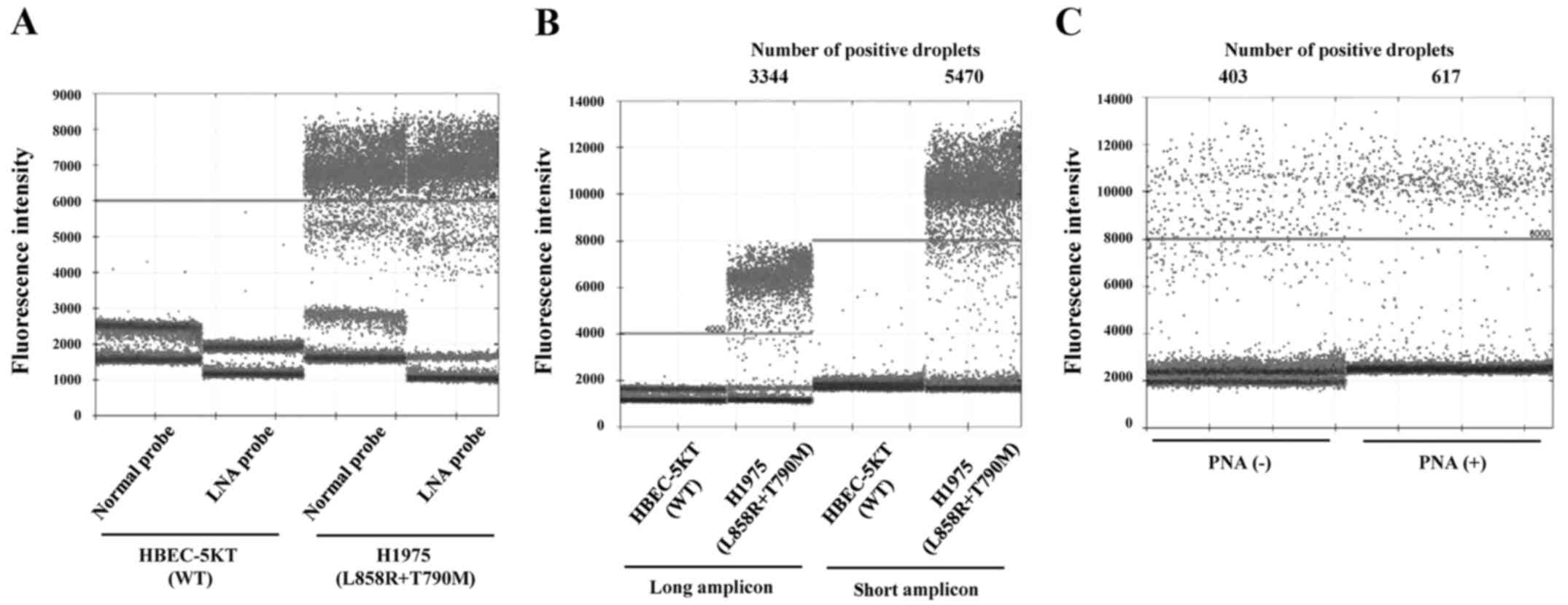
To examine the advantage of using the LNA probe,
ddPCR was performed using different probes consisting of a regular
oligonucleotide probe (M1) and an LNA probe (M2) and the same
primers (S1 and AS1). These probes were designed to display similar
Tms, and were based either on regular oligonucleotides or LNA. The
match-mismatch Tm difference of the regular oligonucleotides and
LNA probes were 5.6°C and 11.4°C, respectively. As shown in
Fig. 2A, the fluorescence intensity
of negative droplets was suppressed in the LNA probe group compared
with the regular oligonucleotide probe group in HBEC-5KT cells as
negative controls. In H1975 cells, the negative droplet suppression
by the LNA probe contributed to better separation between both
positive and negative droplet clusters. These results suggest that
the LNA probe displayed an improvement in mismatch discrimination,
which was thought to possibly result in better specificity.
Improvement in the detection of cfDNA
with short-size amplicon
Next, we assessed the amplicon size for better
detection of cfDNA. Since cfDNA is highly fragmented, we
hypothesized that ddPCR with a short-size amplicon improves the
detection efficiency of cfDNA. We designed new primers (S2 and
AS2), the amplicon size of which was 43 bp, whereas the amplicon
size of the validated reference primers (S1 and AS1) was 93 bp.
ddPCR was performed using either of the two primer sets and the
same LNA probe (M2). The template DNA from either HBEC-5KT or H1975
was fragmented through sonication to approximately 200 bp as
previously mentioned. The use of a short-size amplicon led to
approximately a 1.6-fold increase in the number of detected
positive droplets (Fig. 2B). On the
other hand, there were no significant differences in the number of
false positive droplets between the two primer sets. This approach
with short-size amplicon may be useful to increase cfDNA detection
sensitivity because cfDNA is highly fragmented.
Enrichment of the PCR efficiency of
targeted mutation alleles with PNA clamping method
Since plasma cfDNA contains abundant cfDNA released
from normal cells, a highly-sensitive method which enables
mutations to be selectively enriched in a large background of
wild-type alleles is preferable. We prepared a blocking oligo using
PNA to suppress PCR amplification of the wild-type alleles and 100
ng diluted DNA mixture with a mutant content of only 1% was
analyzed with or without the PNA clamp. The median positive droplet
fluorescence intensity was increased in the group with the PNA
clamp compared to the one without the clamp, which resulted in
better separation between positive and negative droplets clusters.
The use of the PNA clamp led to approximately a 1.5-fold increase
in the number of detected positive droplets (Fig. 2C). This approach appeared to enhance
PCR efficiency for T790M alleles by blocking the amplification of
wild-type alleles.
Detection limit and quantitative
ability of ddPCR for EGFR T790M detection
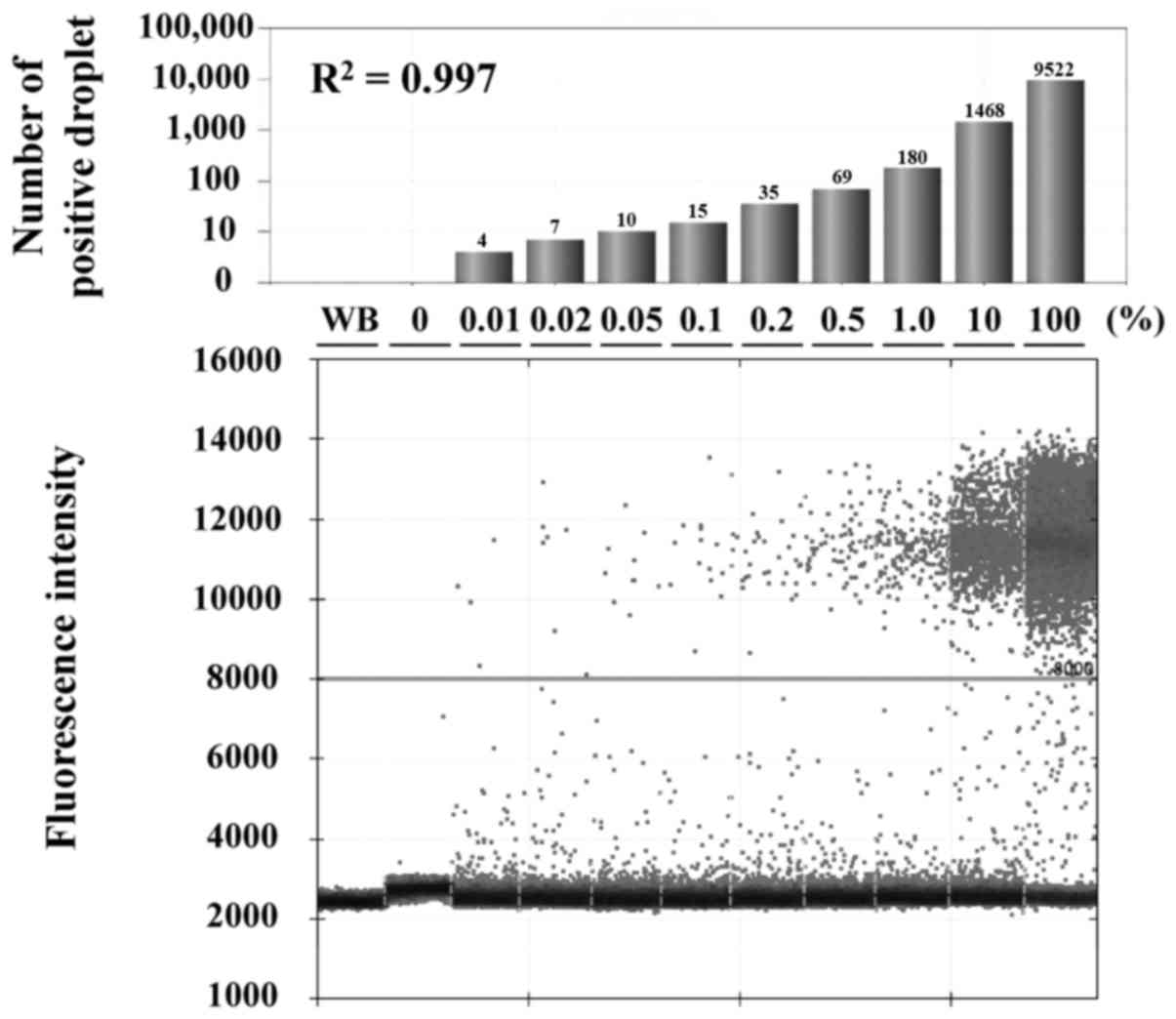
The detection limit and quantitative ability of this
assay were determined using serially diluted DNA mixtures of H1975
and HBEC-5KT. In total, 50 ng of template DNA was used for this
assay, and ddPCR was performed using LNA probe (M2), short amplicon
primers (S2 and AS2), and a PNA clamp. Results indicated that the
PNA-LNA-ddPCR clamp method can detect mutant alleles in the sample
with a mutant content of 0.01%, and the concentration of
detected-mutant alleles correlated with the applied control DNA
mixture concentrations in a linear fashion (R2=0.997)
(Fig. 3).
EGFR T790M detection and monitoring in
clinical plasma samples
We examined T790M mutation by ddPCR using the
PNA-LNA-ddPCR clamp method for clinical plasma samples. The patient
characteristics are shown in Table
II. All patients had EGFR-mutant NSCLC and had received
treatment with an EGFR-TKI, and 9 patients were positive for T790M
in tumor re-biopsy. Of the 59 clinical plasma samples, T790M
alleles were detected via ddPCR in 10 samples. As 21 of 59 plasma
samples were collected from 9 patients with T790M positive in
re-biopsy sample, the sensitivity of ddPCR was 42.8%. Among 10
positive samples in the ddPCR assay, nine were concordant with the
T790M mutational status of the re-biopsy samples, and one was
disconcordant. ddPCR specificity was determined as 97.3%. Results
are summarized in Table III. In
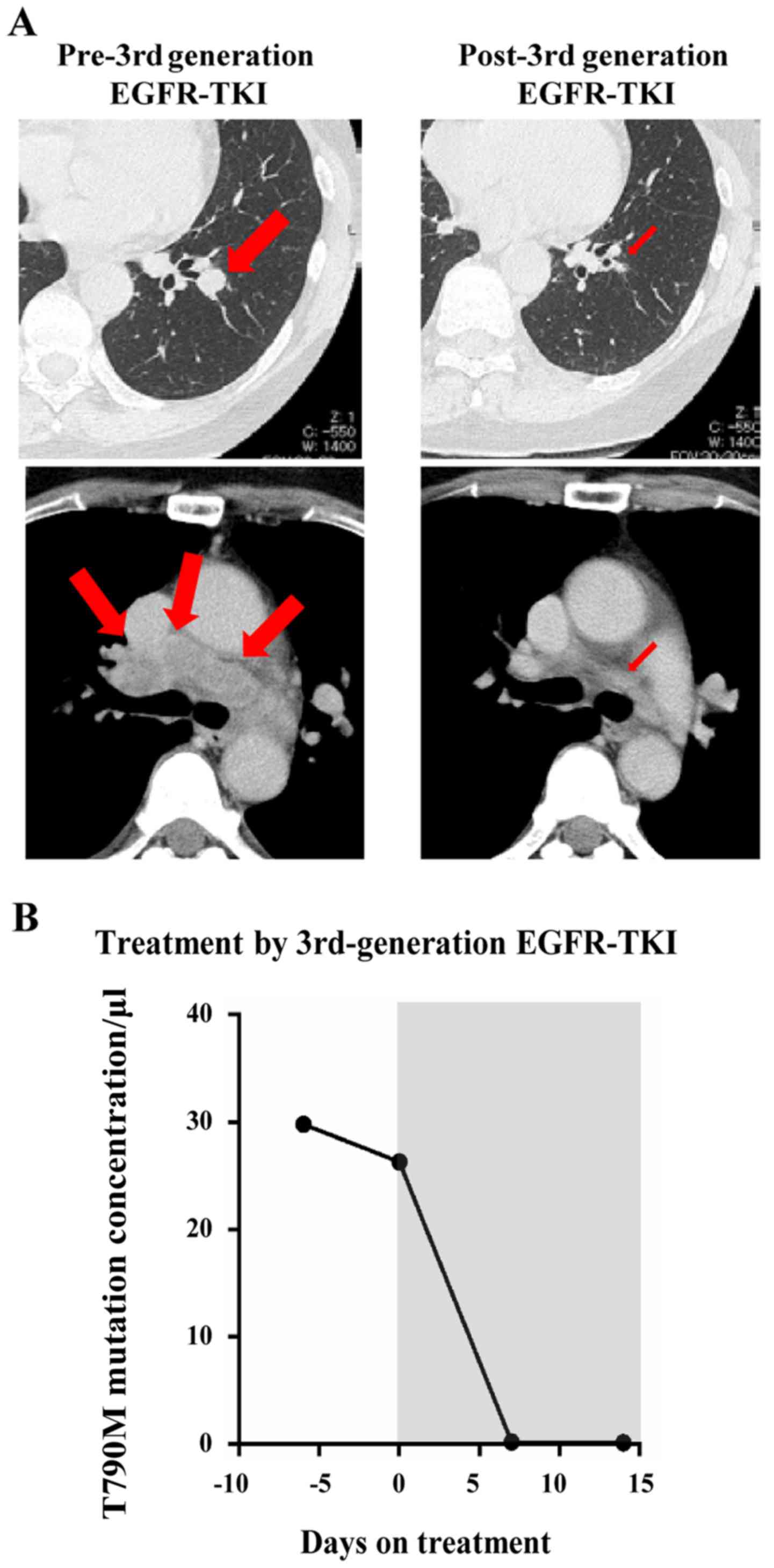
patients diagnosed as T790M-positive in plasma, one patient was
enrolled in this study before treatment with third-generation
EGFR-TKI after acquiring gefitinib resistance. We compared cfDNA
T790M mutant alleles in pre- versus post-treatment plasma (Fig. 4). It revealed that the number of
T790M alleles from plasma cfDNA rapidly decreased after
third-generation EGFR-TKI treatment. This decline in plasma cfDNA
was consistent with the computed tomography (CT) images. Six weeks
after the administration of third-generation EGFR-TKI treatment,
contrast-enhanced CT examination revealed that the primary tumor in
the left lower lobe of the lung and mediastinal lymph nodes had
significantly shrunk.
| Table II.Patient characteristics (n=24). |
Table II.
Patient characteristics (n=24).
| Subsets | n (%) |
|---|
| Median age, (range),
year | 67 (39–84) |
| Sex |
|
| Male | 7 (29.2%) |
|
Female | 17 (70.8%) |
| Smoking history |
|
|
Smoked | 9 (37.5%) |
|
Never-smoked | 15 (62.5%) |
| Drug-sensitive
EGFR mutation |
|
|
L858R | 7 (29.2%) |
| Exon 19
deletion | 16 (66.7%) |
|
G719A | 1 (4.2%) |
| Tissue T790M
mutation (PNA-LNA Clamp method) |
|
|
Positive | 9 (37.5%) |
|
Negative | 15 (62.5%) |
| Table III.Correlation of EGFR T790M
mutational status between cfDNA and rebiopsy tissue DNA. |
Table III.
Correlation of EGFR T790M
mutational status between cfDNA and rebiopsy tissue DNA.
|
| EGFR T790M
status in rebiopsy tissue-DNA (PNA-LNA PCR clamp method) |
|
|---|
|
|
|
|
|---|
|
| Positive | Negative | Sum |
|---|
| EGFR T790M
status in cfDNA (ddPCR) |
|
|
|
|
Positive | 9 | 1 | 10 |
|
Negative | 12 | 37 | 49 |
|
Sum | 21 | 38 | 59 |
Discussion
The emergence of a non-invasive mutational analysis
using cfDNA enables both repetitive disease assessment and
monitoring. However, mutation detection still remains challenging
because circulating bloodstream cfDNA derived from tumor tissue is
highly fragmented, yields small quantities, and presents at very
low allelic frequencies in abundant cfDNA released from normal
cells in nature (10,17). To overcome these difficulties, in
this study, we assessed the optimal approach using ddPCR for
mutation detection from plasma cfDNA.
In this study, the median cfDNA concentration from
lung cancer patients was 1.4 ng/µl, and the total amount in single
7 ml peripheral blood samples was only approximately 28 ng, which
equates to 8,400 copies of human haploid genomic equivalents.
Therefore, without consideration of copy number gain of mutant
alleles, the theoretical mutation detection limit for cfDNA in one
blood sample shows a sensitivity of 0.012%, indicating one mutant
allele in a background of 8,400 wild-type alleles.
Moreover, amplicon size is also a principal issue
for mutation analysis of cfDNA in plasma samples, because cfDNA has
been shown to be highly fragmented after nucleosomal cleavage in
apoptosis (17). As for cell-free
fetal DNA, Sikora et al reported that a real-time PCR assay
with the 50 bp amplicon was able to detect 1.6-fold more cell-free
fetal DNA than the conventional real-time PCR assay with a longer
amplicon (18). We confirmed that
the assay with the 42 bp amplicon was able to detect 1.6-fold more
fragmented template DNA when compared with the assay for the 93 bp
amplicon. Our results in this study were consistent with previous
results. This approach may be useful for all assays in which target
DNA is fragmented, including DNA from formalin-fixed
paraffin-embedded materials.
One of the advantages of digital PCR is the
partitioning of competing backgrounds of wild-type alleles, leading
to decrease in their PCR inhibitory effects and improvements in
detection sensitivity (19).
However, when DNA mixtures of positive and negative controls were
subjected to ddPCR assay, some components, possibly false
negatives, distributed between positive and negative clusters. The
fluorescent signal was higher than that of negative clusters, but
showed a weak signal when compared with positive clusters. We
hypothesized that one of the reasons for these false negative
droplets was PCR inhibitory effects because of a pool of wild-type
alleles. PNA is resistant to the 5′ nuclease activity of Taq DNA
polymerase, leading to a PNA oligo with a superior clamp primer for
inhibiting PCR amplification of wild-type sequences. Nagai et
al reported that an EGFR mutation detection system using
PNA-clamping PCR with a melting curve analysis can detect
EGFR mutations in the presence of 100- to 1,000-fold
background of wild-type EGFR (11). In this study, we showed that the PNA
clamping method in ddPCR also enriched the PCR efficiency of
targeted mutation alleles. These strategies resulted in
highly-sensitive ddPCR assay that enables detection of one mutant
allele in the presence of 10,000 background wild-type alleles. A
potential disadvantage of the PNA clamping PCR is that the mutant
frequency cannot be measured because of the suppression of a
wild-type allele control by PNA clamping. However, ddPCR can
provide absolute quantitation of target sequences in the loaded DNA
sample, and the levels of plasma DNA concentrations in cancer
patients is known to be associated with tumor burden and malignant
progression (15). Therefore, the
number of mutant fragments in the loaded DNA sample are also deemed
to represent the tumor biology, same as mutant frequency.
Of the 59 clinical plasma samples, T790M alleles in
10 samples were successfully detected by ddPCR. The sensitivity of
ddPCR was 42.8%, and the specificity was 97.3% when referred to
that of diagnostic tissue biopsies determined by clinical routine
analysis using the PNA-LNA PCR clamp method. In 12 plasma samples,
our assay could not identify the T790M mutation that had been
detected in the corresponding biopsy samples, and in one plasma
sample, the T790M mutation was identified only in the plasma
sample. However, the limitation of this study can be identified.
The plasma samples were obtained at different times from the tissue
re-biopsy tests. The median time interval between plasma collection
and re-biopsy test was 12.5 months. Therefore, we have to consider
these different timings of sample collections and heterogeneity of
tumor cells, and the discrepancy between the two results could be
because of biology and not technological capability. Every mutation
may not necessarily be represented in a patient's circulation
system. Furthermore, the amount of template DNA is also a critical
factor for detection of rare mutations from blood samples. Even
though the assay can achieve a better sensitivity of 0.01%, one in
10,000 alleles, it is impossible to maximize its ability without
applying >10,000 alleles. However, in practice, it is often
difficult to apply 10,000 alleles because of the limitation of
blood sampling volume. Recently, a BRAF mutation detection
system using cfDNA from urine samples was reported (20,21).
In these studies, 60–120 ml of urine was collected for a urinary
cfDNA assay. Urine sampling is less invasive and more suitable for
repeated and large volume sampling than blood, which may solve the
problem with small template DNA amounts.
In conclusion, we showed that the use of a
short-size amplicon, LNA probe, and PNA blocker improve the
sensitivity of a mutation detection for cfDNA samples. ddPCR is a
promising method for non-invasive T790M mutation detection.
Acknowledgements
The authors thank Dr Mizuki Morita (Biorepository
Research and Networking, Okayama University Graduate School of
Medicine, Dentistry and Pharmaceutical Sciences, Okayama, Japan),
Ms. Fumiko Isobe (Department of Thoracic, Breast and
Endocrinological Surgery, Okayama University Graduate School of
Medicine, Dentistry and Pharmaceutical Sciences, Okayama, Japan),
Dr Takehiro Matsubara, Ms. Yuko Hanafusa and Ms. Yayoi Kubota
(Biobank of Okayama University Hospital, Okayama, Japan) for their
technical support.
Glossary
Abbreviations
Abbreviations:
|
EGFR
|
epidermal growth factor receptor
|
|
ddPCR
|
droplet digital PCR
|
|
LNA
|
locked nucleic acids
|
|
PNA
|
peptide nucleic acids
|
References
|
1
|
Collisson EA, Campbell JD, Brooks AN,
Berger AH, Lee W, Chmielecki J, Beer DG, Cope L, Creighton CJ,
Danilova L, et al: Cancer Genome Atlas Research Network:
Comprehensive molecular profiling of lung adenocarcinoma. Nature.
511:543–550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kris MG, Johnson BE, Berry LD, Kwiatkowski
DJ, Iafrate AJ, Wistuba II, Varella-Garcia M, Franklin WA, Aronson
SL, Su PF, et al: Using multiplexed assays of oncogenic drivers in
lung cancers to select targeted drugs. JAMA. 311:1998–2006. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mok TS, Wu YL, Thongprasert S, Yang CH,
Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et
al: Gefitinib or carboplatin-paclitaxel in pulmonary
adenocarcinoma. N Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sequist LV, Waltman BA, Dias-Santagata D,
Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger
S, Cosper AK, et al: Genotypic and histological evolution of lung
cancers acquiring resistance to EGFR inhibitors. Sci Transl Med.
3:75ra262011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Soucheray M, Capelletti M, Pulido I, Kuang
Y, Paweletz CP, Becker JH, Kikuchi E, Xu C, Patel TB, Al-Shahrour
F, et al: Intratumoral heterogeneity in EGFR-Mutant NSCLC results
in divergent resistance mechanisms in response to EGFR tyrosine
kinase inhibition. Cancer Res. 75:4372–4383. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kobayashi S, Boggon TJ, Dayaram T, Jänne
PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos
B: EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cross DA, Ashton SE, Ghiorghiu S, Eberlein
C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ,
et al: AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated
resistance to EGFR inhibitors in lung cancer. Cancer Discov.
4:1046–1061. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Crowley E, Di Nicolantonio F, Loupakis F
and Bardelli A: Liquid biopsy: Monitoring cancer-genetics in the
blood. Nat Rev Clin Oncol. 10:472–484. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Oxnard GR, Paweletz CP, Kuang Y, Mach SL,
O'Connell A, Messineo MM, Luke JJ, Butaney M, Kirschmeier P,
Jackman DM, et al: Noninvasive detection of response and resistance
in EGFR-mutant lung cancer using quantitative next-generation
genotyping of cell-free plasma DNA. Clin Cancer Res. 20:1698–1705.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Diaz LA Jr and Bardelli A: Liquid
biopsies: Genotyping circulating tumor DNA. J Clin Oncol.
32:579–586. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nagai Y, Miyazawa H, Huqun Tanaka T,
Udagawa K, Kato M, Fukuyama S, Yokote A, Kobayashi K, Kanazawa M,
et al: Genetic heterogeneity of the epidermal growth factor
receptor in non-small cell lung cancer cell lines revealed by a
rapid and sensitive detection system, the peptide nucleic
acid-locked nucleic acid PCR clamp. Cancer Res. 65:7276–7282. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tanaka T, Nagai Y, Miyazawa H, Koyama N,
Matsuoka S, Sutani A, Huqun Udagawa K, Murayama Y, Nagata M, et al:
Reliability of the peptide nucleic acid-locked nucleic acid
polymerase chain reaction clamp-based test for epidermal growth
factor receptor mutations integrated into the clinical practice for
non-small cell lung cancers. Cancer Sci. 98:246–252. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ramirez RD, Sheridan S, Girard L, Sato M,
Kim Y, Pollack J, Peyton M, Zou Y, Kurie JM, Dimaio JM, et al:
Immortalization of human bronchial epithelial cells in the absence
of viral oncoproteins. Cancer Res. 64:9027–9034. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gandhi J, Zhang J, Xie Y, Soh J,
Shigematsu H, Zhang W, Yamamoto H, Peyton M, Girard L, Lockwood WW,
et al: Alterations in genes of the EGFR signaling pathway and their
relationship to EGFR tyrosine kinase inhibitor sensitivity in lung
cancer cell lines. PLoS One. 4:e45762009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schwarzenbach H, Hoon DS and Pantel K:
Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev
Cancer. 11:426–437. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Giacona MB, Ruben GC, Iczkowski KA, Roos
TB, Porter DM and Sorenson GD: Cell-free DNA in human blood plasma:
Length measurements in patients with pancreatic cancer and healthy
controls. Pancreas. 17:89–97. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Heitzer E, Ulz P and Geigl JB: Circulating
tumor DNA as a liquid biopsy for cancer. Clin Chem. 61:112–123.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sikora A, Zimmermann BG, Rusterholz C,
Birri D, Kolla V, Lapaire O, Hoesli I, Kiefer V, Jackson L and Hahn
S: Detection of increased amounts of cell-free fetal DNA with short
PCR amplicons. Clin Chem. 56:136–138. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weisenberger DJ, Trinh BN, Campan M,
Sharma S, Long TI, Ananthnarayan S, Liang G, Esteva FJ, Hortobagyi
GN, McCormick F, et al: DNA methylation analysis by digital
bisulfite genomic sequencing and digital MethyLight. Nucleic Acids
Res. 36:4689–4698. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hyman DM, Diamond EL, Vibat CR, Hassaine
L, Poole JC, Patel M, Holley VR, Cabrilo G, Lu TT, Arcila ME, et
al: Prospective blinded study of BRAFV600E mutation detection in
cell-free DNA of patients with systemic histiocytic disorders.
Cancer Discov. 5:64–71. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Janku F, Vibat CR, Kosco K, Holley VR,
Cabrilo G, Meric-Bernstam F, Stepanek VM, Lin PP, Leppin L,
Hassaine L, et al: BRAF V600E mutations in urine and plasma
cell-free DNA from patients with Erdheim-Chester disease.
Oncotarget. 5:3607–3610. 2014. View Article : Google Scholar : PubMed/NCBI
|