Introduction
Triazoles, including miconazole (MIC), ketoconazole
(KT) and fluconazole (FT), are used as fungicides in agriculture
and as antifungal drugs in humans (1,2).
Topical MIC is efficacious for the treatment of most mycoses
(3–5). MIC buccal tablets have recently been
approved by the US Food and Drug Administration for the treatment
of oropharyngeal candidiasis in human immunodeficiency virus
(HIV)-infected patients (6). MIC
alters the fungal cell membrane, and exerts its therapeutic effect
mainly by inhibiting fungal ergosterol biosynthesis (7). Additionally, MIC inhibits a variety of
cytochrome P-450-dependent enzymes (8).
Recent evidence suggests that triazoles exhibit
antiproliferative effects, and are modified on their 1,2,4-triazole
nucleus to generate different types of anticancer agents (9). MIC has also been shown to be an
effective anticancer drug. For example, MIC was found to inhibit
the proliferation of human acute myelogenous leukemia (10) and breast and bladder cancer cells
(11), and to induce apoptosis
through G0/G1 cell cycle arrest in some types of human colorectal
cancer cell lines (12). Other
studies have revealed that MIC may inhibit melanogenesis in B16
cells and that MIC may be useful in the treatment of
hyperpigmentation disorders, such as ephelis and melasma (13). Furthermore, 10–100 µM MIC was found
to decrease cell proliferation rates in human osteosarcoma cells in
a concentration-dependent manner and to increase intracellular
Ca2+ levels in these cells, mainly from the endoplasmic
reticulum, independent of phospholipase C activity and external
Ca2+ influx (14).
Although these findings demonstrate the antitumor
activity of MIC, the mechanism underlying the effect of MIC on
bladder cancer remains unknown. In the present study, we evaluated
the effect of MIC on bladder cancer cell apoptosis and
characterized its underlying molecular mechanism.
Materials and methods
Cell culture and reagents
Human bladder cancer cell lines T24 (p53-mutant) and
TSGH-8301 (wild-type p53) were purchased from the Bioresource
Collection and Research Center (BCRC; Hsinchu, Taiwan). The J82
(p53-mutant) cell line was purchased from the American Type Culture
Collection (ATCC; Rockville, MD, USA). Peripheral blood mononuclear
cells (PBMCs) were obtained from healthy donors. All of these cells
were cultured in RPMI medium supplemented with 10% fetal bovine
serum (FBS) (Gibco, Gaithersburg, MD, USA) and 1%
antibiotic-antimycotic solution. Cells were incubated at 37°C with
5% CO2. Miconazole nitrate was purchased from
Sigma-Aldrich (St. Louis, MO, USA). MIC was prepared in dimethyl
sulfoxide (DMSO) to a final concentration of 0.2 mmol/l.
Cytotoxicity assay
The cytotoxicity of MIC in T24, and TSGH-8301 cells
and PBMCs was determined using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay (Sigma-Aldrich), which was performed as previously described
(14). The
3-(4,5-dimethylthiazol-2-yl)-5-
(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS)
assay (Promega, Madison, WI, USA) was performed according to the
manufacturer's instructions. Cells were seeded on 96-well plates
for 24 h, and then treated with pifithrin-α (PFT-α), NAC or caspase
inhibitors with and without MIC at 50 µM. After incubation for 24
h, 20 µl of CellTiter 96 Aqueous One Solution Reagent (Promega) was
added to each well and incubated for 1–4 h followed by absorbance
reading at 490 nm.
Flow cytometry
Cells (1×106) were treated with MIC for
24 h. All cells were collected after trypsinization followed by
fixation and permeabilization with 70% ethanol at −20°C overnight.
After washing with ice-cold phosphate-buffered saline (PBS), the
cells were incubated with propidium iodide solution (PI: 0.2 mg/ml,
RNase: 20 µg/ml, 0.1% Triton X-100) for 30 min at room temperature,
in the dark. Flow cytometry was performed using WinMDI v2.9
(Scripps Research Institute, La Jolla, CA, USA). Ten thousand
events/sample were counted for triplicate experiments.
DNA fragmentation analysis
Cells were treated with various concentrations of
MIC. At indicated time points, the cells were collected by 0.25%
(v/v) trypsin digestion, followed by centrifugation at 2,000 × g
for 10 min. Cells were lysed in 800 µl of lysis buffer (50 mM Tris,
pH 8.0, 10 mM EDTA and 0.3% Triton X-100), and treated with RNase
(0.1 mg/ml) followed by proteinase K. The extracted DNA was
electrophoresed on 2% agarose gels and stained with ethidium
bromide (15).
Western blotting
Equal amounts of protein (measured by Bradford
assay) were loaded on 10–15% sodium dodecyl sulfate polyacrylamide
gels, transferred to polyvinylidene fluoride (PVDF) membranes, and
blocked with 5% non-fat milk in Tris-buffered saline and Tween-20
(TBST) buffer (20 mM Tris-HCl, 120 mM NaCl, 0.1% Tween-20). The
membranes were incubated with antibodies against TNF-related
apoptosis-inducing ligand (TRAIL), death receptors (DR4 and DR5)
(1:1,000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), DR5
(1:10,000; Abcam, Cambridge, MA, USA), β-actin, Bcl-2, Bax,
caspase-3, −8 and −9, poly(ADP-ribose) polymerase (PARP) (1:1,000;
Cell Signaling Technology, Boston, MA, USA), cytochrome c (1
µg/ml; from BD Pharmingen, San Diego, CA, USA), Smac/DIABLO, cyclin
D1, cyclin-dependent kinase 2 (CDK2), CDK4 (1:1,000; GeneTex,
Hsinchu City, Taiwan), p53, p21, p27 and cyclin E1 (1:1,000;
Epitomics, Burlingame, CA, USA) at 4°C overnight. After washing,
the blots were incubated with horseradish peroxidase-labeled
secondary antibodies for 1 h, and visualized using enhanced
chemiluminescence.
Mitochondrial membrane potential
assay
Cells were seeded onto 10-cm dishes and treated with
various concentrations of MIC for 24 h, followed by staining with 5
µM JC-1 (Invitrogen, Carlsbad, CA, USA) for 30 min at 37°C.
Fluorescence was monitored using a plate reader at wavelengths of
490 nm (excitation)/540 nm (emission) and 540 nm (excitation)/590
nm (emission). Mitochondrial membrane potential (Δψm) changes were
indicated by the changes in the ratio of 590 nm (red) to 540 nm
(green) fluorescence.
Reactive oxygen species detection
Intracellular reactive oxygen species (ROS) were
determined using the fluorescent superoxide indicator
dihydroethidium (DHE; Setareh Biotech, LLC, Eugene, OR, USA)
(16–19). T24 and TSGH-8301 cells were treated
with 50 µM MIC for 8, 16 or 24 h, and then incubated with 2 µM DHE
in serum-free medium, at 37°C for 15 min, and then analyzed by flow
cytometry.
Statistical analyses
Statistical comparisons were performed using
unpaired, two-tailed Student's t-tests. p-values of <0.05 were
considered to indicate a statistically significant result.
Results
MIC is cytotoxic to T24, J82 and
TSGH-8301 cells
We determined the cytotoxic effect of various
concentrations of MIC (Fig. 1A) on
T24, J82 and TSGH-8301 cells using the MTT assay. The cell
viability of both T24 (p53 mutant) and TSGH-8301 (p53
wild-type) cells was lower than that of the J82 bladder cancer
cells after treatment with 50 µM MIC. We therefore chose T24 and
TSGH-8301 cells for further assays. MIC exhibited cytotoxic effects
in a concentration- and time-dependent manner (Fig. 1B and C). After MIC treatment for 24,
48 and 72 h, the IC50 values were estimated to be
47.5±0.5, 42.0±1.8 and 37.3±0.3 µM, respectively, in the T24 cells;
and 45.9±0.4, 32.2±0.8 and 29.4±0.2 µM, respectively, in the
TSGH-8301 cells. Treatment of PBMCs with 100 µM MIC for 24 h showed
a lower cytotoxic effect (85% cell-survival rate) than that noted
in the bladder cancer cells (Fig.
1B). Thus, human bladder cancer cells were more sensitive than
PBMCs to the cytotoxic effect of MIC. Furthermore, the
morphological changes in MIC-treated cells at 24 h were examined
using a phase-contrast microscope. Cell proliferation in the T24
and TSGH-8301 cells was inhibited with 25 µM MIC, but the cells
were mostly alive (Fig. 1D). At
concentrations of ≥50 µM, MIC induced cell death with marked
morphologic changes, such as shrinkage, rounding and floating of
the cells, indicating that MIC at higher concentrations induced
apoptotic cell death.
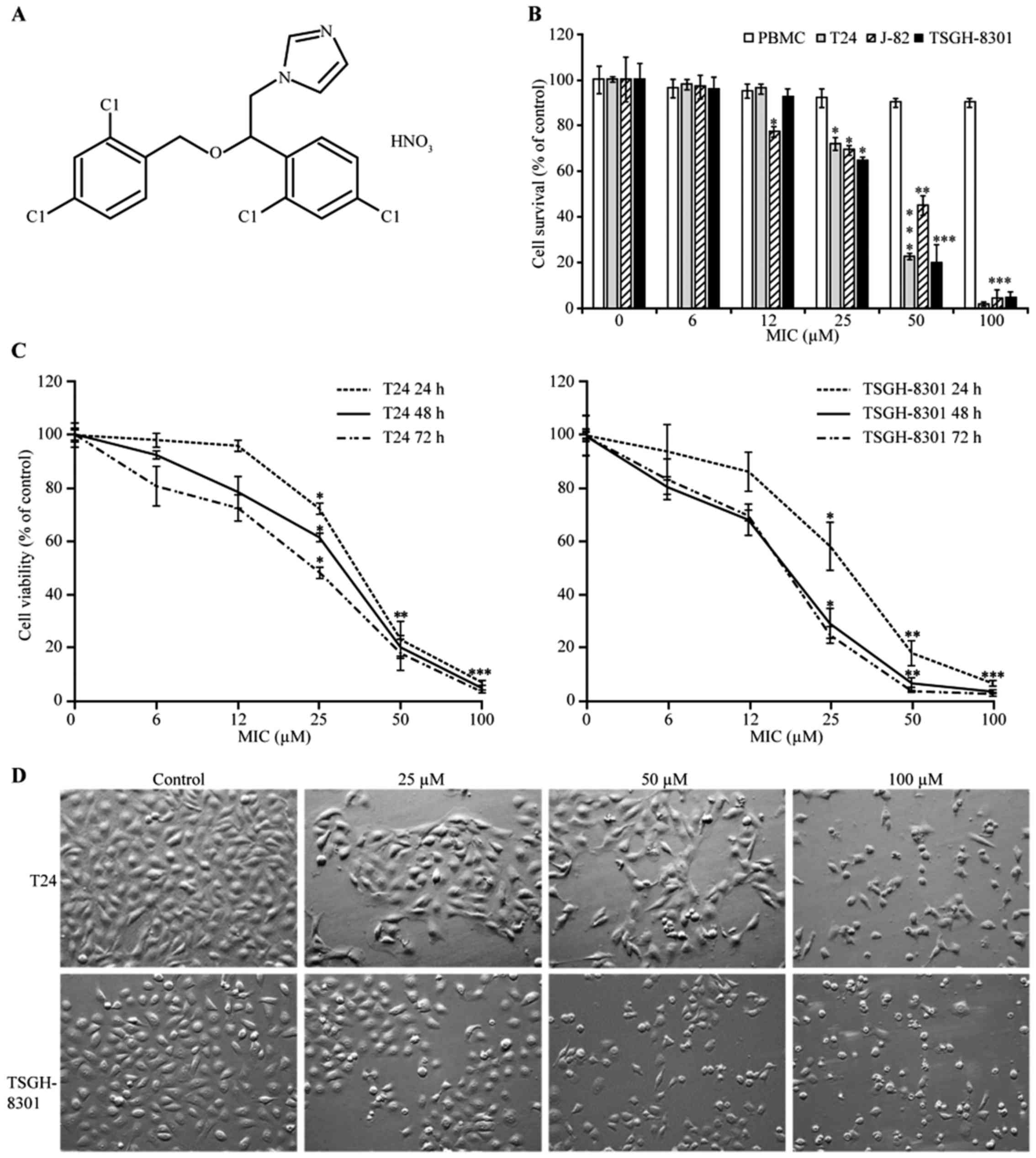 | Figure 1.Cytotoxic effects of miconazole (MIC)
on human T24, J82 and TSGH-8301 bladder cancer and normal human
peripheral blood mononuclear cells (PBMCs). (A) The chemical
structure of MIC. (B) T24, J82 and TSGH-8301 cells, and PBMCs were
seeded at a density of 2×104 cells/well, and then
treated with MIC (6.25, 12.5, 25, 50 or 100 µM) or a vehicle
control for 24 h. The MTT assay (described in the ‘Materials and
methods’ section) was used to quantify cell viability. (C) T24 and
TSGH-8301 cells were also treated with the same concentrations of
MIC for 24, 48 or 72 h, and then cell viability was assessed with
the MTT assay. (B and C) Data points and error bars represent the
mean ± SD of three experiments, respectively. Statistical
significance: *p<0.05, **p<0.01, ***p<0.001 as compared
with the control. (D) Effect of MIC on the morphology of T24 and
TSGH-8301 cells. T24 and TSGH-8301 cells were treated with 25, 50
or 100 µM MIC for 24 h. Cells were viewed by a phase contrast
microscopy and photographed at a magnification of ×200. |
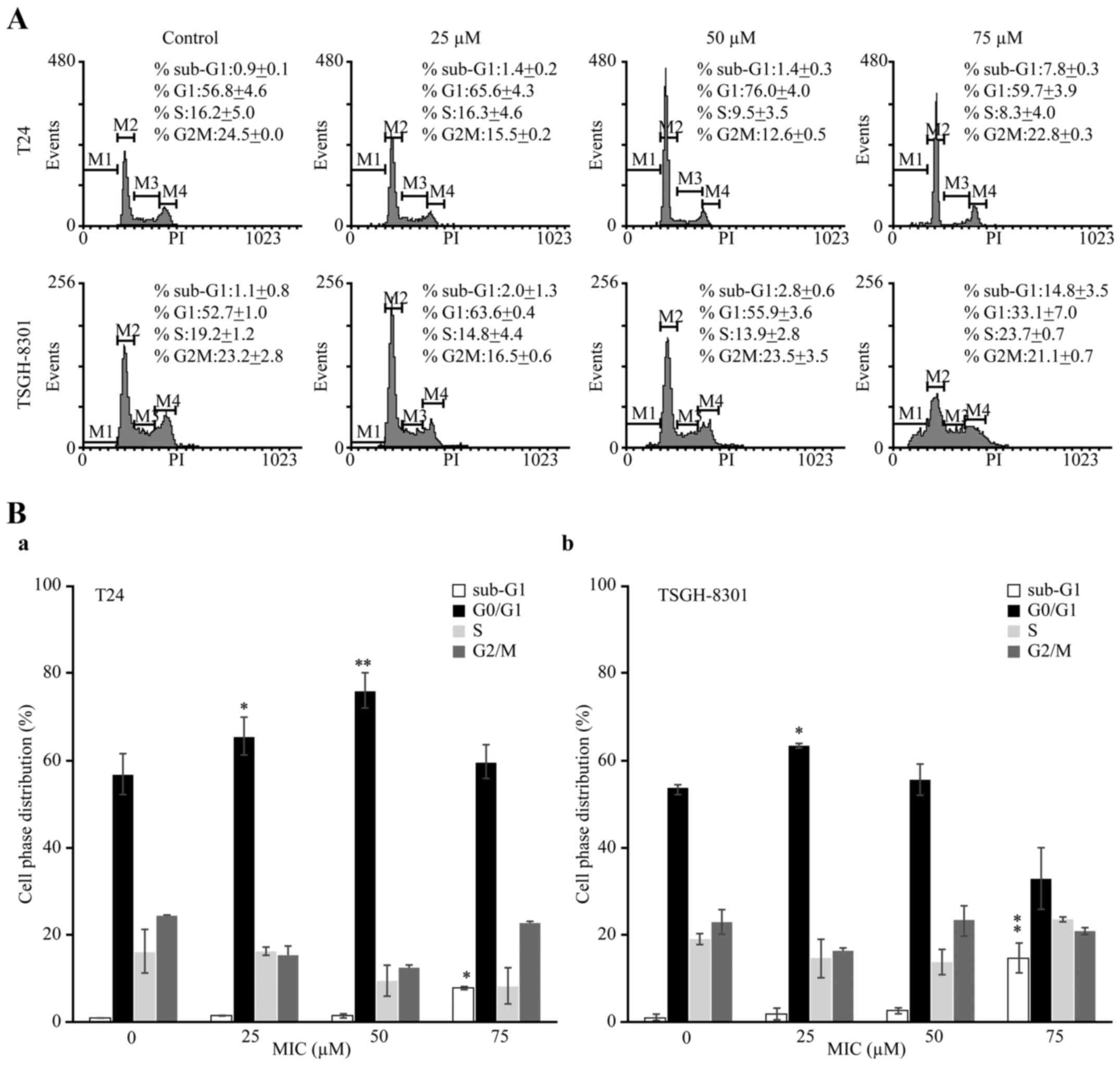
MIC induces cell cycle arrest and
apoptosis in a p53-dependent manner in T24 and TSGH-8301 cells
To elucidate the mechanism underlying MIC-induced
cell death, we analyzed the cell cycle phase distribution in the
T24 and TSGH-8301 cells treated with various MIC concentrations for
24 h. The percentage of cells in the sub-G1 phase, indicating cell
death, in the T24 and TSGH-8301 cells treated with 75 µM MIC
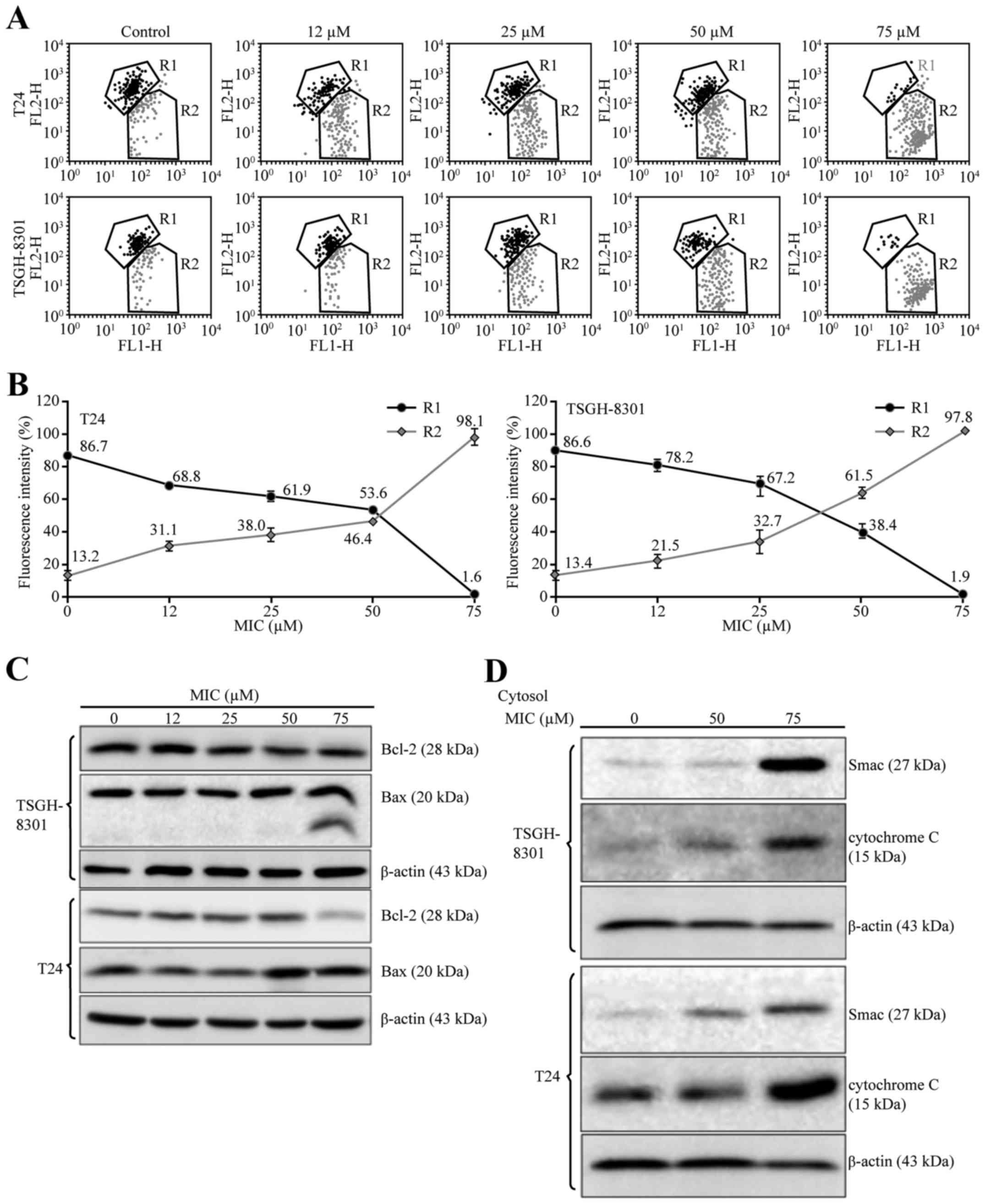
(7.8±0.3 and 14.8±3.5%, respectively; Fig. 2A and B) was significantly higher
than the percentages in the untreated (control) cells (0.9±0.1 and
1.1±0.8%, respectively). Additionally, the percentages of T24 cells
in G0/G1 transition after treatment with 25 and 50 µM MIC (65.6±4.3
and 76.0±4.0%, respectively) were significantly higher than that in
the untreated cells (56.8±4.6%; Table
I). Similarly, the percentage (63.6±0.4%) of TSGH-8301 cells in
G0/G1 transition after treatment with 25 µM MIC was significantly
higher than that noted in the untreated cells (52.7±1.0%; Table I). Thus, MIC caused G0/G1 cell cycle
arrest in both cell lines.
 | Table I.Cell cycle phase distribution of T24
and TSGH-8301 cells following MIC treatment. |
Table I.
Cell cycle phase distribution of T24
and TSGH-8301 cells following MIC treatment.
|
|
| Cell cycle phase
distribution |
|---|
|
|
|
|
|---|
| Cell lines | MIC (µM) | Sub-G1 (%) | G1 (%) | S (%) | G2/M (%) |
|---|
| T24 |
0 |
0.9±0.1 | 56.8±4.6 | 16.2±5.0 | 24.5±0.0 |
|
| 25 |
1.4±0.2 |
65.6±4.3a | 16.3±4.6 | 15.5±0.2 |
|
| 50 |
1.4±0.3 |
76.0±4.0a, b |
9.5±3.5 | 12.6±0.5 |
|
| 100 |
7.8±0.3a | 59.7±3.9 |
8.3±4.0 | 22.8±0.3 |
| TSGH-8301 |
0 |
1.1±0.8 | 52.7±1.0 | 19.2±1.2 | 23.2±2.8 |
|
| 25 |
2.0±1.3 |
63.6±0.4a | 14.8±4.4 | 16.5±0.6 |
|
| 50 |
2.8±0.6 | 55.9±3.6 | 13.9±2.8 | 23.5±3.5 |
|
| 100 |
14.8±3.5a, b | 33.1±7.0 | 23.7±0.7 | 21.1±0.7 |
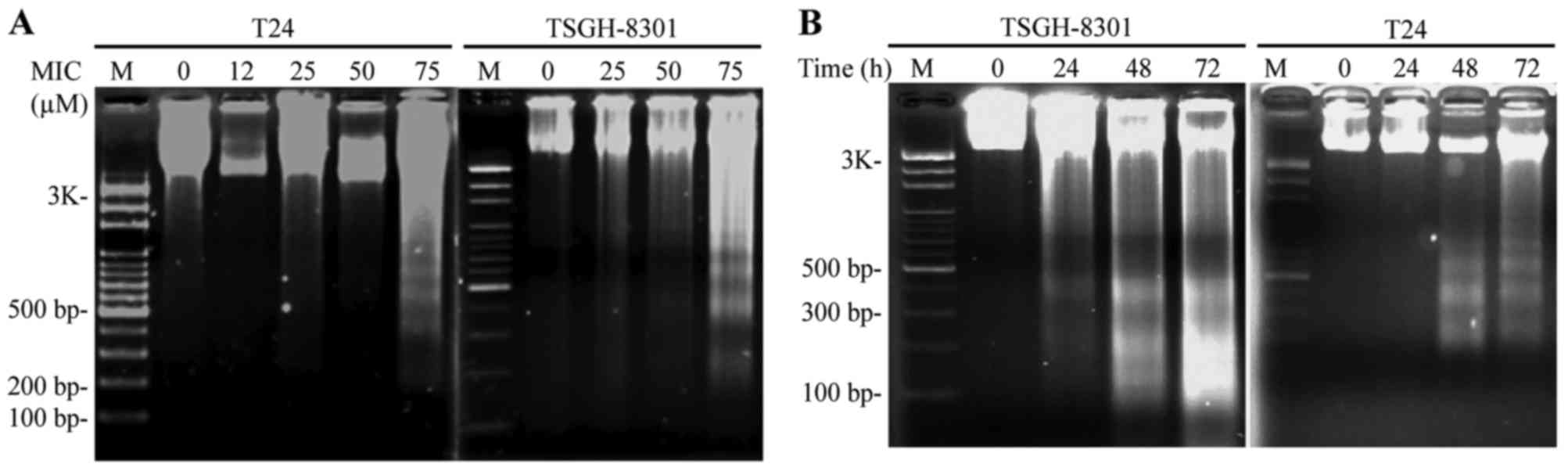
To further investigate the type of
cell death caused by MIC, we assessed DNA fragmentation in both
types of cells
MIC-treated cells exhibited significant
internucleosomal DNA degradation, and elicited a DNA fragment
ladder on 2% gels, which was significantly increased in the T24 and
TSGH-8301 cells treated with 75 µM MIC for 24 h (Fig. 3A) and 50 µM MIC for 24, 48 and 72 h
(Fig. 3B) relative to the control.
Importantly, TSGH-8301 cells exhibited more DNA fragmentation than
T24 cells after 50 µM MIC treatment for 24 h (Fig. 3B). This indicated that the TSGH-8301
cells, expressing wild-type p53, were more sensitive to MIC-induced
apoptosis than the T24 cells, which express p53 with an in-frame
deletion of tyrosine 126.
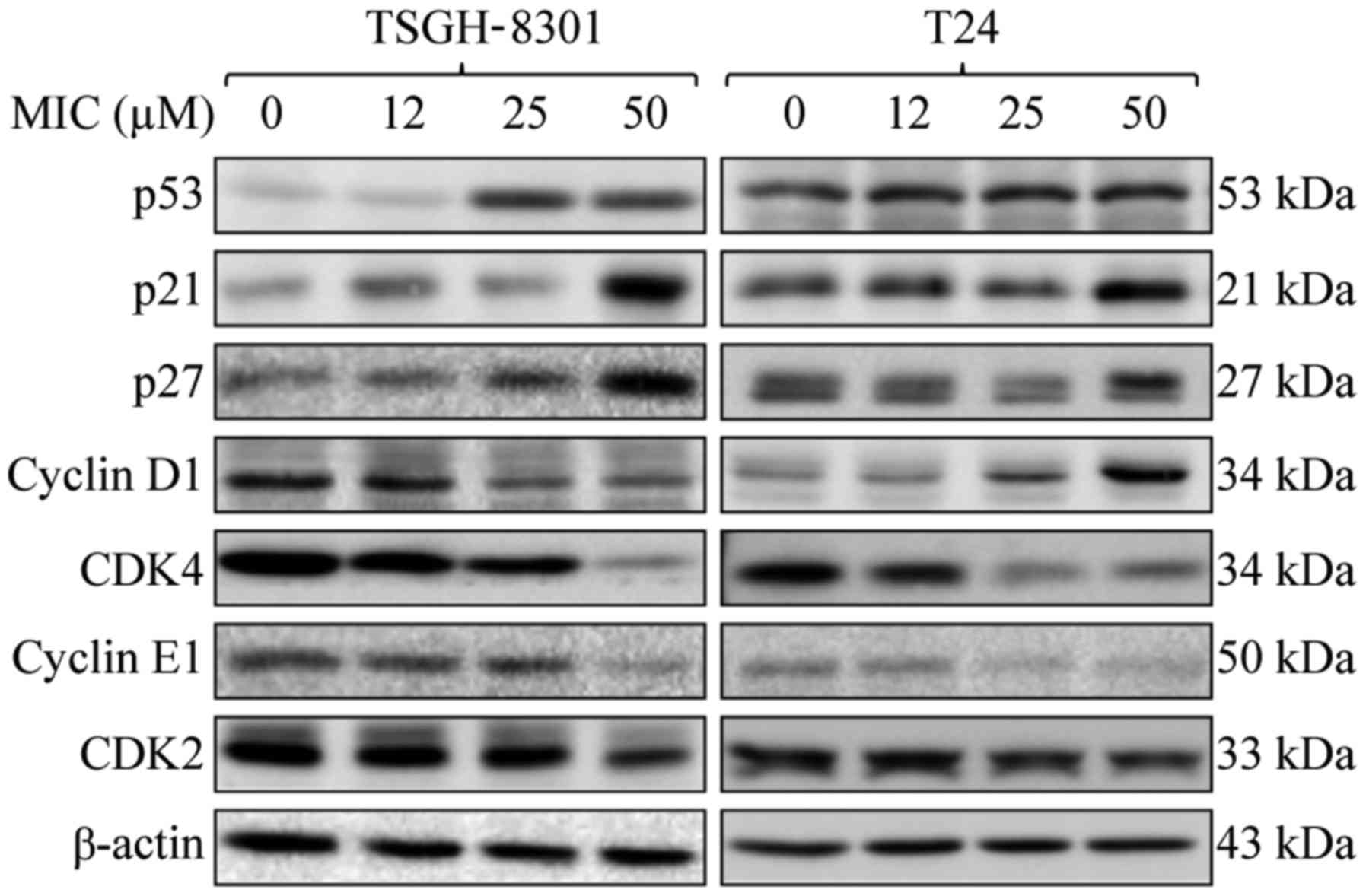
MIC increases p21 and p27 levels, and
inhibits cyclin E1, CDK2 and CDK4 expression in T24 and TSGH-8301
cells
We then investigated the roles of cell cycle arrest
and p53 in MIC-induced growth inhibition of bladder cancer cells,
using western blot analysis. The levels of p21 and p27 were
significantly increased, but those of cyclin E1, CDK2 and CDK4 were
decreased after treatment with 25 or 50 µM MIC in the TSGH-8301 and
T24 cells, as compared to these levels in the untreated cells
(Fig. 4). Notably, MIC, at 25 and
50 µM, stimulated p53 expression in the TSGH-8301 (wild-type)
cells, but not in the T24 (p53 mutant) cells as compared to the
untreated cells. However, 25 or 50 µM MIC inhibited expression of
cyclin D1, which is required for G1/S transition (20), in the TSGH-8301 cells, resulting in
growth arrest. However, MIC at these concentrations stimulated
cyclin D1 expression in the T24 cells. Thus, the TSGH-8301 cells
were more sensitive to MIC-induced G0/G1 cell cycle arrest than the
T24 cells, suggesting that cyclin D1 and possibly p53 are involved
in cell cycle arrest. We hypothesized that, in p53-mutant T-24
cells, MIC-stimulated expression of cyclin D1 decreased the
susceptibility of these cells to MIC-induced G0/G1 cell cycle
arrest as compared with the TSGH-8301 cells.
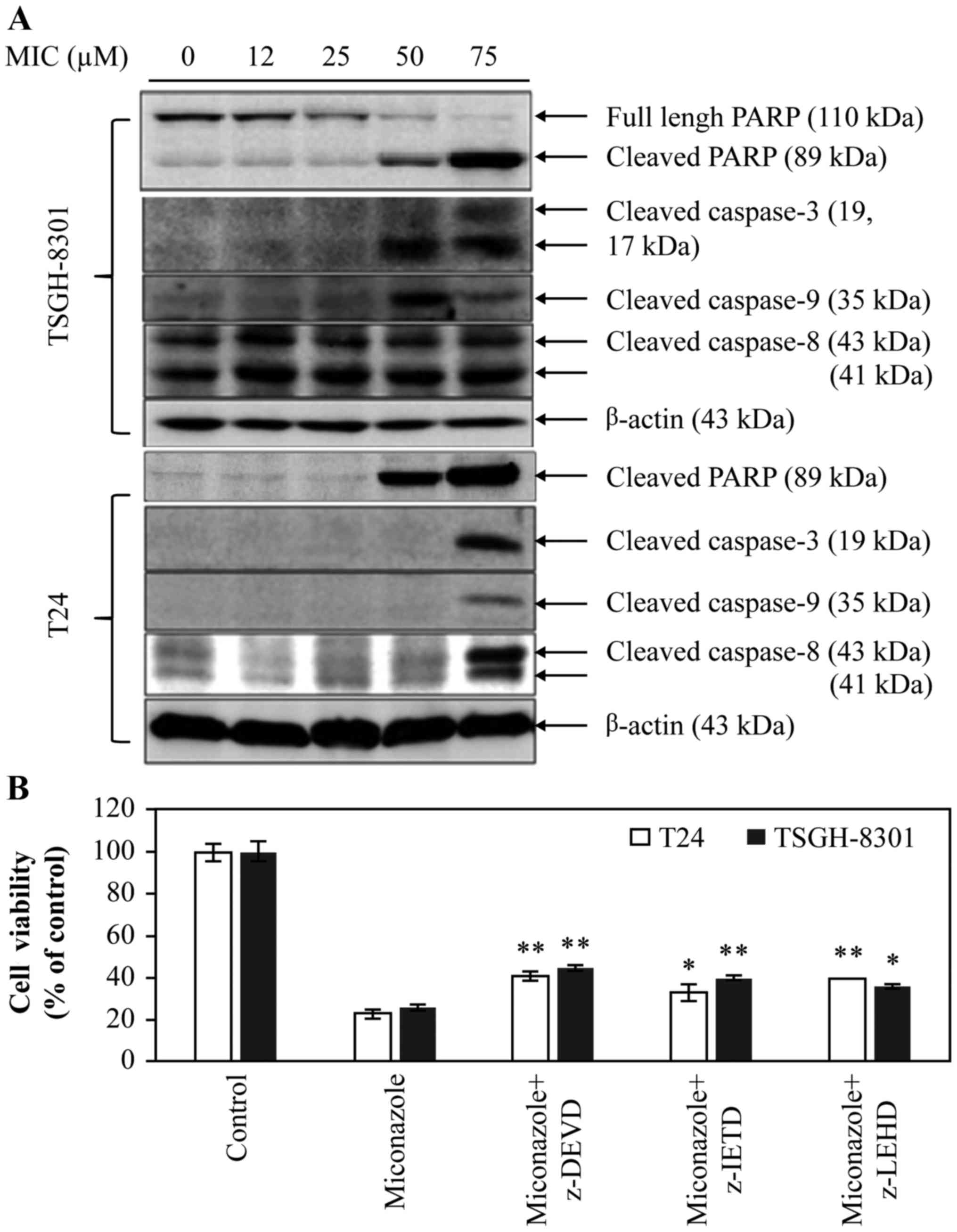
MIC induces caspase-dependent
apoptosis in T24 and TSGH-8301 cells
Apoptotic pathways include the mitochondrial
‘intrinsic pathway’ and a DR-related ‘extrinsic pathway’, which
involves activation of caspase-9, −8, and −3/-7, and cleavage of
PARP (21). Thus, to clarify which
apoptotic pathway is induced by MIC, levels of the
cleaved/activated forms of caspase-8, −9 and −3, and PARP were
determined by western blotting. Cleavage of PARP was induced in the
TSGH-8301 and T24 cells treated with 50 and 75 µM MIC for 24 h
(Fig. 5A). Therefore, both the
mitochondrial and DR pathways were found to be involved in
MIC-induced apoptosis in these bladder cancer cells. Moreover, MIC
induced a higher level of apoptosis in the TSGH-8301 cells when
compared to the T24 cells. Importantly, the level of the cleaved
form of caspase-8 (~41 kDa) was significantly higher in the
TSGH-8301 cells than that in the T24 cells after treatment with ≥12
µM MIC. To determine the roles of capases in MIC-induced apoptosis,
the cells were treated with 50 µM MIC in the presence of vehicle
only (control), or 10 µM caspase-3, −8 or −9 inhibitors (z-DEVD,
z-IETD and z-LEHD, respectively) for 24 h. As shown in Fig. 5B, these caspase inhibitors partially
reversed MIC-induced apoptosis in both the T24 and TSGH-8301 cell
lines (*p<0.05; **p<0.01). Notably, in the T24 cells, there
was still a moderate difference between cells treated with MIC plus
caspase-8 inhibitor and those treated with MIC alone
(*p<0.05).
MIC induces DR-mediated apoptosis
independent of p53 in T24 and TSGH-8301 cells
To confirm that MIC induces extrinsic apoptosis, we
determined the effect of MIC on the expression of proteins in the
DR pathway in the T24 and TSGH-8301 cells. Treatment with 50 µM MIC
increased expression of DR5 (Fig.
6A), but moderately decreased DR4 levels in both cell lines.
However, treatment of these cells with 75 µM MIC almost completely
abolished the expression of DR4, DR5 and β-actin (an internal
control) (Fig. 6A). This suggests
the non-specific cytotoxicity of MIC at 75 µM. To confirm that DR5
is involved in MIC-induced apoptosis in both cell types, we
performed MTT assay. Pre-treatment of cells with anti-DR5 blocking
antibody significantly attenuated MIC-induced apoptosis in both
types of bladder cancer cell lines (Fig. 6B), indicating that the DR5-mediated
apoptosis pathway is involved in MIC-induced apoptosis in bladder
cancer cells. To define the role of p53 in MIC-induced cell death,
T24 and TSGH-8301 cells were treated with 50 µM MIC without or with
1 and 10 µM PFT-α (a p53 inhibitor) for 24 h. The cell survival was
then determined using MTS assay. As shown in Fig. 6C, PFT-α at 1 µM, significantly
reversed MIC-induced cytotoxicity in both cell types, particularly
TSGH-8301 cells which express wild-type p53. PFT-α treatment
attenuated MIC-increased expression of cleaved PARP in the T24 and
TSGH-8301 cells and cyclin D1 only in the T24 cells (Fig. 6D). PFT-α treatment attenuated
MIC-increased expression of cleaved PARP in the T24 and TSGH-8301
cells and cyclin D1 in the TSGH-8301 cells (Fig. 6D). These results suggest that MIC
induced apoptosis in T24 cells in a p53-independent, but cyclin
D1-dependent manner.
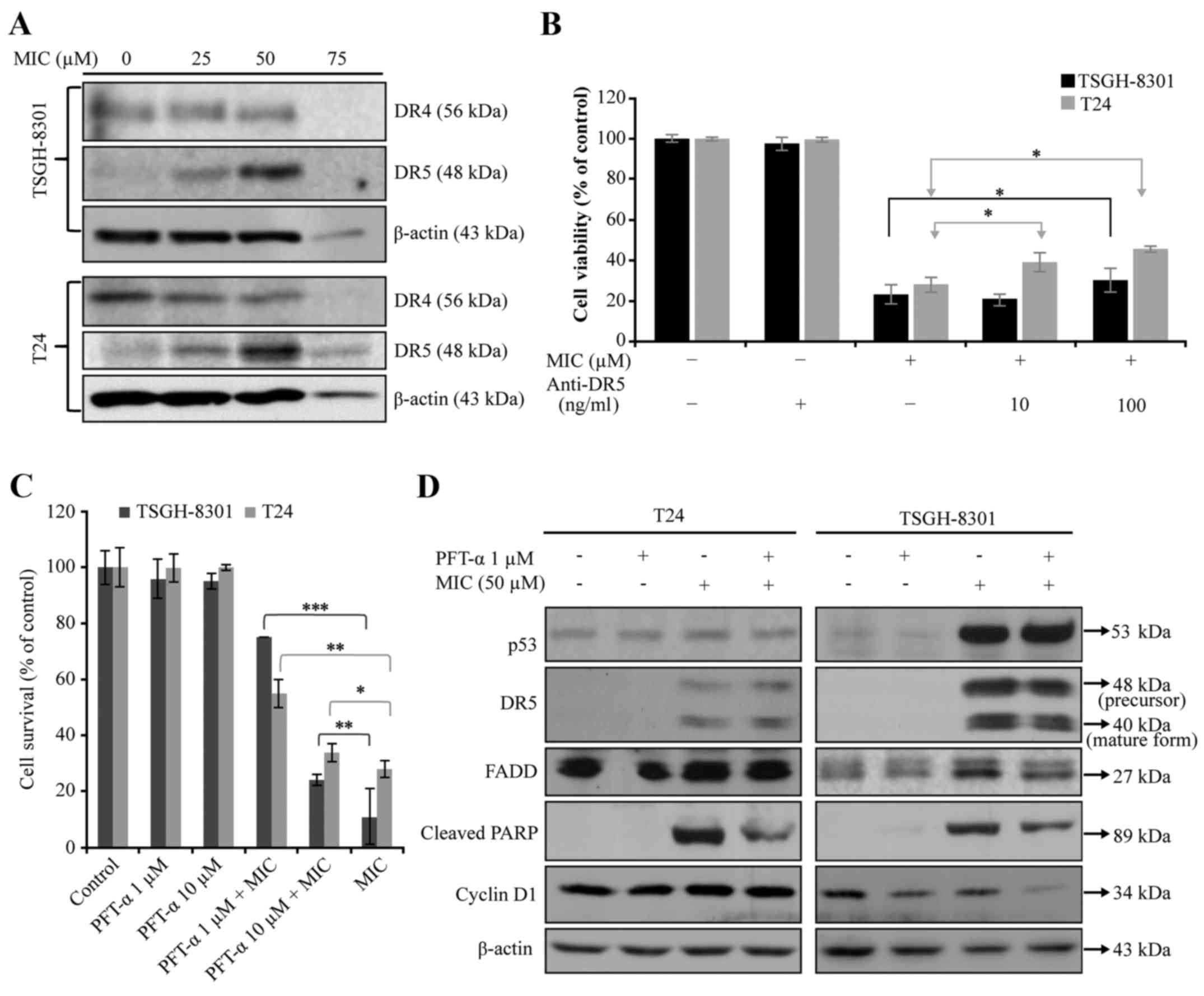 | Figure 6.Effects of MIC without or with PFT-α
(a putative p53 inhibitor) or anti-DR5 mAb on the expression of
proteins involved in (A and D) death receptor (DR)-mediated
apoptosis and (B and C) cell viability in T24 and TSGH-8301 cells.
(A) T24 and TSGH-8301 cells were treated with 0, 25, 50 or 75 µM
MIC for 24 h. Western blotting of cell lysates was performed with
antibodies against DR4, DR5 and β-actin. (B) T24 and TSGH-8301
cells were incubated with the blocking anti-DR5 mAb, at 10 or 100
ng/ml for 2 h at 37°C before being co-cultured with MIC at 50 µM.
After incubation of cells with MIC (50 µM) ± 10 or 100 ng/ml of
anti-DR5 antibody for 24 h at 37°C, cell viability was assayed with
MTT assay. (B and C) Data are mean ± SD of three independent
experiments; *p<0.05, **p<0.01, ***p<0.001. (C) T24 and
TSGH-8301 cells were treated with 50 µM MIC ± 1 and 10 µM PFT-α
(p53 inhibitor) for 24 h. The cell survival was then determined
using MTS assay. (D) T24 and TSGH-8301 cells were treated with
vehicle only or 50 µM MIC ± 1 µM PFT-α for 24 h. Western blotting
of cell lysates was performed using antibodies against p53, DR5,
FADD, cleaved PARP, cyclin D1 and β-actin. |
MIC induces apoptosis through the
ROS-mediated mitochondrial pathway in T24 and TSGH-8301 cells
To confirm that the intrinsic, mitochondrial pathway
is involved in MIC-induced bladder cancer cell apoptosis, we
measured Δψm in MIC-treated T24 and TSGH-8301 cells using the
fluorescent cationic dye JC-1; loss of Δψm is an indicator of
mitochondrial damage during apoptosis (22). JC-1 aggregates in normally polarized
mitochondria, emitting red fluorescence, but in apoptotic
depolarized cells, where Δψm is reduced, JC-1 is diffused as
monomers throughout the cells and emits green fluorescence. A
concentration-dependent decrease in red fluorescence was observed
and the ratios of green/red fluorescence were estimated in the 75
µM MIC-treated T24 and TSGH-8301 cells (98.1/1.6 and 97.8/1.9%,
respectively; Fig. 7A and B). This
indicated that MIC treatment reduced Δψm in both cancer cell
lines.
We then examined the expression of the proapoptotic
protein Bax (23), which triggers
cytochrome c release, and of Bcl-2, an anti-apoptotic
protein, which inhibits cytochrome c release (24). Treatment of T24 and TSGH-8301 cells
with >50 µM MIC for 24 h resulted in increased expression of Bax
and/or cleaved Bax protein, and decreased expression of Bcl-2
protein in the T24 cells (Fig. 7C).
Since loss of Δψm promotes the release of cytochrome c and
Smac/DIABLO into the cytosol (21),
we determined the levels of cytochrome c and Smac/DIABLO in
the cytosolic fractions of T24 and TSGH-8301 cells that were
treated with higher concentrations of MIC (50 and 75 µM). As shown
in Fig. 7D, MIC increased
cytochrome c and Smac/DIABLO levels in the cytosol in a
concentration-dependent manner in both T24 and TSGH-8301 cells.
Taken together, these results indicated that the mitochondrial
pathway was also involved in the MIC-induced apoptosis. Since ROS
can alter the cellular redox state and Δψm (25,26),
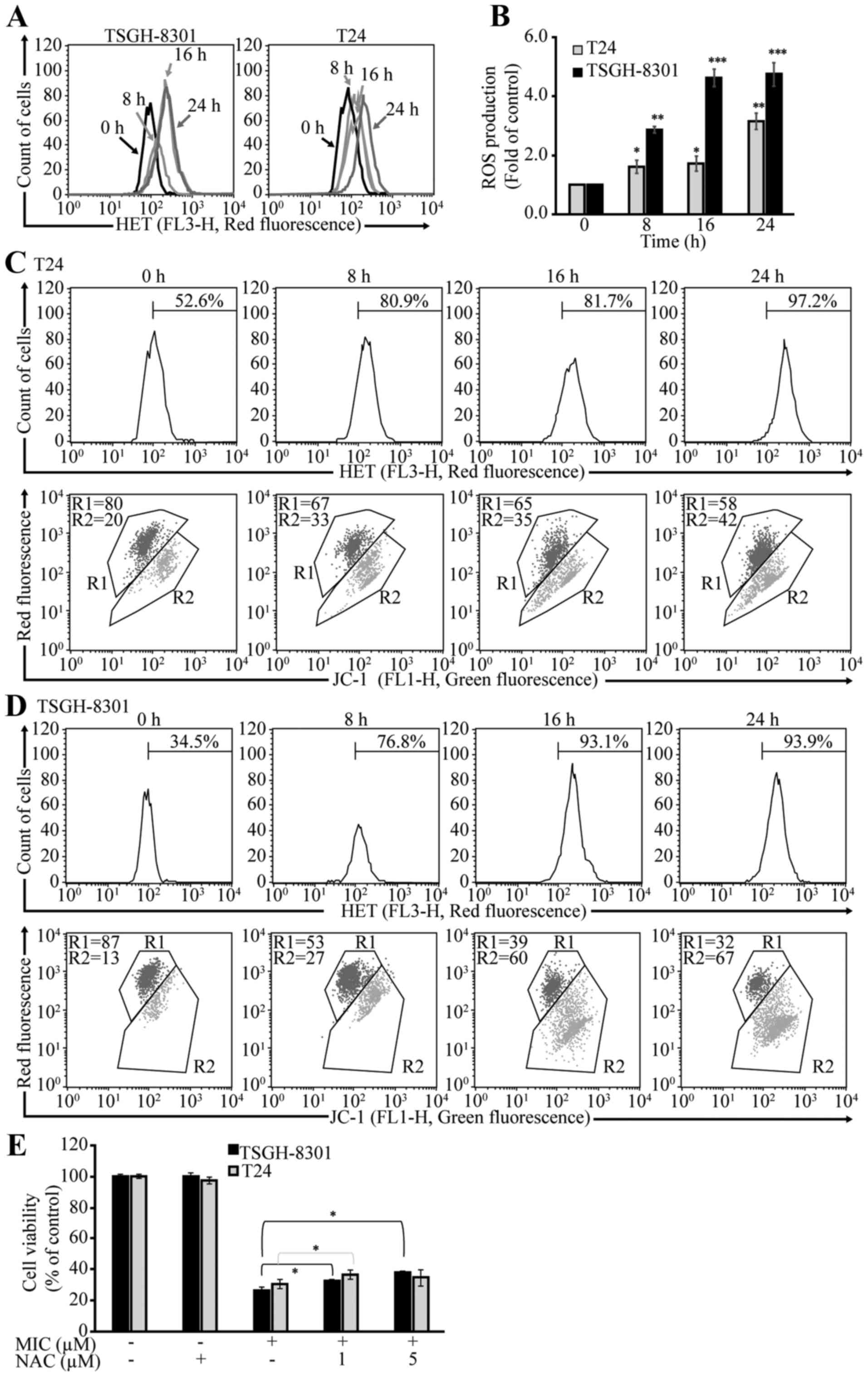
we investigated whether MIC-treated T24 and TSGH-8301 cells produce
ROS. DHE staining yielded increased red fluorescence intensity
(Fig. 8A) and ROS production
(Fig. 8B) in both MIC-treated cell
lines over time (8–24 h). Furthermore, JC-1 staining yielded
increasing green fluorescence intensity in both cell lines during
the same period (Fig. 8C and D),
indicating a reduction in Δψm. Moreover, pre-treatment of these
cells with the antioxidant N-acetyl-L-cysteine (NAC)
increased cell survival rates (~10%) in both types of cells
(Fig. 8E). Collectively, these
results suggest that MIC-induced mitochondrial-mediated apoptosis
is associated with ROS production in bladder cancer cells.
Discussion
In the present study, we demonstrated that MIC
inhibits cell growth in human bladder cancer cells in vitro
by inducing both mitochondrial- and DR5-mediated apoptosis.
The cytotoxic effects of MIC in both bladder cancer
cell lines were similar to that previously reported for colon
cancer cells (COLO205; IC50 ~50 µM) (12). MIC caused cell cycle arrest at G0/G1
transition in both the T24 and TSGH-8301 cells. The
tumor-suppressor p53 plays a pivotal role in cell growth arrest and
apoptosis (27,28). Western blotting revealed the
elevation of p21 and p27 kinase inhibitors in both MIC-treated
bladder cancer cells and a decrease in cyclin E1, CDK2 and CDK4
kinase. Given the differential expression of tumor-suppressor p53
and cyclin D1 kinase in the two bladder cancer cell lines used, we
hypothesized that the effect of MIC on cyclin D1 expression in
these cells is dependent on the p53 status. MIC induced
downregulation of cyclin D1 in the TSGH-8301 cells (expressing
wild-type p53), but not in the T24 cells (expressing p53 mutant).
This was evidenced by the additive effect of MIC and PFT-α on the
downregulation of cyclin D1 in the TSGH-8301 cells (Fig. 6D). The downstream protein of p53,
p21/WAF1 (also known as cyclin-dependent kinase inhibitor), which
binds to and inhibits the activity of cyclin-CDK2 and CDK4/6
complexes, functions as a regulator of cell cycle progression at
the G1 and S phases (29). The
expression of the corresponding gene is tightly controlled by p53,
through which p21 mediates p53-dependent cell cycle G1
phase arrest in response to a variety of stress stimuli (30).
Additionally, p27 belongs to the Cip/Kip
family of CDK inhibitors, which prevents activation of cyclin
E-CDK2 or cyclin D-CDK4 complexes, thus controlling cell cycle
progression at G1 (31). Therefore,
MIC may cause cell death in both bladder cancer cell lines by
arresting the G0/G1 phase of the cell cycle. The magnitude of
MIC-induced apoptosis in the two bladder cancer cell lines was
found to be associated with the p53 status. These results also
supported the hypothesis that MIC-induced cell-growth inhibition in
both bladder cancer cell lines involves the p53 pathway.
Furthermore, higher MIC doses (50 and 75 µM) induced apoptosis in
both bladder cancer cell lines, as demonstrated by DNA
fragmentation and increased caspase-9, −8 and −3, and PARP levels.
Caspases play pivotal roles in the initiation and execution of the
DR- and mitochondrial-mediated apoptotic pathways. Our data also
revealed that MIC-induced apoptosis in both bladder cancer cell
lines was associated with the p53 status, and that TSGH-8301 cells
were more susceptible than T24 cells to MIC-induced cell apoptosis.
Notably, a putative p53 inhibitor, PFT-α, at 1 and 10 µM,
effectively protected both TSGH-8301 and T24 cell types from MIC
(50 µM)-induced apoptosis or cell death as compared to cells
treated with MIC alone, regardless of the presence or absence of
p53 mutation in these cells (Fig.
6C). We reason that PFT-α protects against MIC-induced
apoptosis or cell death in a p53-independent, but in a cyclin
D1-dependent manner in T24 cells. MIC (50 µM) increased the
expression of cyclin D1 in the T24 cells, but attenuated cyclin D1
expression in the TSGH-8301 cells as compared with the control
cells treated without MIC. PFT-α is known to protect cancer cells
(HCT116) from DNA damage-induced apoptosis by a p53-independent
mechanism involving cyclin D1 (32). Notably, PFT-α did not affect the
expression of p53 and DR5, but protected against apoptosis in both
the T24 and TSGH-8301 cells treated with MIC as evidenced by
decreased levels of cleaved PARP in these cells (Fig. 6D). The reason that PFT-α did not
significantly affect the expression of p53, or other
transcriptional factors, such as NF-κB (33,34),
C/EBP homologous protein (35,36),
Elk 1 (2) and YY1 (37) is not clear. It may be under the
influence of DR5 expressed in these cells treated with MIC.
Activation of caspase-8 is associated with the DR signaling cascade
and the extrinsic pathway (38).
Our results revealed that both bladder cancer cell lines treated
with ≥50 µM MIC for 24 h exhibited upregulation of caspase-8 and
DR5. However, apoptosis was partially blocked with an anti-DR5
blocking antibody. Thus, we speculate that MIC induces apoptosis
through the DR pathway by activation of caspase-8 and the
downstream caspase-3 in both bladder cancer cell lines.
ROS cause Δψm dysfunction and release of cytochrome
c, which activates caspase-3, leading to
mitochondrial-mediated apoptosis (39). MIC has been shown to induce actin
cytoskeleton stabilization and ROS accumulation upon killing of
yeast cells (7). We demonstrated
that MIC induced mitochondrial-mediated apoptosis in the T24 and
TSGH-8301 bladder cancer cells, as determined by Δψm decrease, the
release of cytochrome c and Smac/DIABLO, activation of
caspase-9 and −3, and a change in the Bax/Bcl-2 ratio (23). Treatment with higher doses of MIC
enhanced ROS levels in both bladder cancer cell lines, which
paralleled the decrease of Δψm over the same period, but this
effect was blocked by the antioxidant NAC. Thus, MIC-induced
apoptosis in the T24 and TSGH-8301 bladder cancer cells involved
the mitochondrial pathway and was modulated by ROS generation.
In conclusion, we demonstrated that MIC caused G0/G1
cell cycle arrest in human bladder cancer cells, and thus, induced
apoptosis in these cells through both the extrinsic DR5-dependent
and intrinsic mitochondrial-mediated pathways. MIC appeared to
cause cell cycle arrest through both p53-independent and
ROS-dependent mechanisms, as evidenced by the inability of NAC to
influence the protective effect of PFT-α on MIC-induced cell cycle
arrest (data not shown). Since MIC has been shown to interact with
lipid rafts/caveolae in target cells (7), we hypothesized that MIC causes cell
cycle arrest and activates both apoptosis pathways, at least in
part, by interaction with lipid rafts/caveolae in the plasma
membrane, where DR5 transduces signaling (40). MIC has been widely used as an
antifungal agent without severe side effects, even with prolonged
use (5,41,42).
Thus, the anticancer effect of MIC may be of potential clinical
significance for the treatment of bladder cancer in humans.
Acknowledgements
The present study was supported by the Taichung
Veterans General Hospital, and the Hung Kung University (grant no.
TCVGH-HK1028007).
Glossary
Abbreviations
Abbreviations:
|
MIC
|
miconazole
|
|
PARP
|
poly(ADP-ribose) poly-merase
|
|
NAC
|
N-acetyl-L-cysteine
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
MTS
|
3-(4,5-dimethyl-thiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium,
inner salt
|
References
|
1
|
Garcia-Cuesta C, Sarrion-Pérez MG and
Bagán JV: Current treatment of oral candidiasis: A literature
review. J Clin Exp Dent. 6:e576–e582. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Oh YT, Liu X, Yue P, Kang S, Chen J,
Taunton J, Khuri FR and Sun SY: ERK/ribosomal S6 kinase (RSK)
signaling positively regulates death receptor 5 expression through
co-activation of CHOP and Elk1. J Biol Chem. 285:41310–41319. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Diehl KB: Topical antifungal agents: An
update. Am Fam Physician. 54:1687–1692. 1996.PubMed/NCBI
|
|
4
|
Nawrot U, Nowicka J, Juszczak K and Gusin
B: Susceptibility to antifungal agents of Candida species isolated
from paediatric and adult patients with haematological diseases.
Mycoses. 48:385–390. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Piérard GE, Hermanns-Lê T, Delvenne P and
Piérard-Franchimont C: Miconazole, a pharmacological barrier to
skin fungal infections. Expert Opin Pharmacother. 13:1187–1194.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Colin P, Koenig P, Ouzzane A, Berthon N,
Villers A, Biserte J and Rouprêt M: Environmental factors involved
in carcinogenesis of urothelial cell carcinomas of the upper
urinary tract. BJU Int. 104:1436–1440. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
François IE, Bink A, Vandercappellen J,
Ayscough KR, Toulmay A, Schneiter R, van Gyseghem E, Van den Mooter
G, Borgers M, Vandenbosch D, et al: Membrane rafts are involved in
intracellular miconazole accumulation in yeast cells. J Biol Chem.
284:32680–32685. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Alvarez J, Montero M and Garcia-Sancho J:
High affinity inhibition of Ca2+-dependent K+
channels by cytochrome P-450 inhibitors. J Biol Chem.
267:11789–11793. 1992.PubMed/NCBI
|
|
9
|
Kaur R, Dwivedi AR, Kumar B and Kumar V:
Recent developments on 1,2,4-triazole nucleus in anticancer
compounds: A review. Anticancer Agents Med Chem. 16:465–489. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bruserud O: Effects of azoles on human
acute myelogenous leukemia blasts and T lymphocytes derived from
acute leukemia patients with chemotherapy-induced cytopenia. Int
Immunopharmacol. 1:2183–2195. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shahbazfar AA, Zare P, Ranjbaran M,
Tayefi-Nasrabadi H, Fakhri O, Farshi Y, Shadi S and Khoshkerdar A:
A survey on anticancer effects of artemisinin, iron, miconazole,
and butyric acid on 5637 (bladder cancer) and 4T1 (breast cancer)
cell lines. J Cancer Res Ther. 10:1057–1062. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu CH, Jeng JH, Wang YJ, Tseng CJ, Liang
YC, Chen CH, Lee HM, Lin JK, Lin CH, Lin SY, et al: Antitumor
effects of miconazole on human colon carcinoma xenografts in nude
mice through induction of apoptosis and G0/G1 cell cycle arrest.
Toxicol Appl Pharmacol. 180:22–35. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mun YJ, Lee SW, Jeong HW, Lee KG, Kim JH
and Woo WH: Inhibitory effect of miconazole on melanogenesis. Biol
Pharm Bull. 27:806–809. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chang HT, Chen WC, Chen JS, Lu YC, Hsu SS,
Wang JL, Cheng HH, Cheng JS, Jiann BP, Chiang AJ, et al: Effect of
miconazole on intracellular Ca2+ levels and
proliferation in human osteosarcoma cells. Life Sci. 76:2091–2101.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yuan SY, Hsu SL, Tsai KJ and Yang CR:
Involvement of mitochondrial pathway in Taxol-induced apoptosis of
human T24 bladder cancer cells. Urol Res. 30:282–288. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yuan SY, Cheng CL, Ho HC, Wang SS, Chiu
KY, Su CK, Ou YC and Lin CC: Nortriptyline induces mitochondria and
death receptor-mediated apoptosis in bladder cancer cells and
inhibits bladder tumor growth in vivo. Eur J Pharmacol.
761:309–320. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xia Z, Lundgren B, Bergstrand A, DePierre
JW and Nässberger L: Changes in the generation of reactive oxygen
species and in mitochondrial membrane potential during apoptosis
induced by the antidepressants imipramine, clomipramine, and
citalopram and the effects on these changes by Bcl-2 and
Bcl-XL. Biochem Pharmacol. 57:1199–1208. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bindokas VP, Jordán J, Lee CC and Miller
RJ: Superoxide production in rat hippocampal neurons: Selective
imaging with hydroethidine. J Neurosci. 16:1324–1336.
1996.PubMed/NCBI
|
|
19
|
Satoh T, Numakawa T, Abiru Y, Yamagata T,
Ishikawa Y, Enokido Y and Hatanaka H: Production of reactive oxygen
species and release of L-glutamate during superoxide
anion-induced cell death of cerebellar granule neurons. J
Neurochem. 70:316–324. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zuryń A, Litwiniec A, Gagat M, Drzewucka
J, Gackowska L and Grzanka A: The influence of arsenic trioxide on
the cell cycle, apoptosis and expression of cyclin D1 in the Jurkat
cell line. Acta Histochem. 116:1350–1358. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wyllie AH, Kerr JF and Currie AR: Cell
death: The significance of apoptosis. Int Rev Cytol. 68:251–306.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu CS, Chen YJ, Chen JJ, Shieh JJ, Huang
CH, Lin PS, Chang GC, Chang JT and Lin CC: Terpinen-4-ol induces
apoptosis in human nonsmall cell lung cancer in vitro and in vivo.
Evid Based Complement Alternat Med. 2012:8182612012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brunelle JK and Letai A: Control of
mitochondrial apoptosis by the Bcl-2 family. J Cell Sci.
122:437–441. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shimizu S, Narita M and Tsujimoto Y: Bcl-2
family proteins regulate the release of apoptogenic cytochrome c by
the mitochondrial channel VDAC. Nature. 399:483–487. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cheng SB, Wu LC, Hsieh YC, Wu CH, Chan YJ,
Chang LH, Chang CM, Hsu SL, Teng CL and Wu CC: Supercritical carbon
dioxide extraction of aromatic turmerone from Curcuma longa Linn.
induces apoptosis through reactive oxygen species-triggered
intrinsic and extrinsic pathways in human hepatocellular carcinoma
HepG2 cells. J Agric Food Chem. 60:9620–9630. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
el-Deiry WS, Harper JW, O'Connor PM,
Velculescu VE, Canman CE, Jackman J, Pietenpol JA, Burrell M, Hill
DE, Wang Y, et al: WAF1/CIP1 is induced in p53-mediated
G1 arrest and apoptosis. Cancer Res. 54:1169–1174.
1994.PubMed/NCBI
|
|
28
|
Wang H, Yang Z, Liu C, Huang S, Wang H,
Chen Y and Chen G: RBP-J-interacting and tubulin-associated protein
induces apoptosis and cell cycle arrest in human hepatocellular
carcinoma by activating the p53-Fbxw7 pathway. Biochem Biophys Res
Commun. 454:71–77. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gartel AL and Radhakrishnan SK: Lost in
transcription: p21 repression, mechanisms, and consequences. Cancer
Res. 65:3980–3985. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li H, Qian W, Weng X, Wu Z, Li H, Zhuang
Q, Feng B and Bian Y: Glucocorticoid receptor and sequential P53
activation by dexamethasone mediates apoptosis and cell cycle
arrest of osteoblastic MC3T3-E1 cells. PLoS One. 7:e370302012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
van de Mark K, Chen JS, Steliou K, Perrine
SP and Faller DV: Alpha-lipoic acid induces
p27Kip-dependent cell cycle arrest in non-transformed
cell lines and apoptosis in tumor cell lines. J Cell Physiol.
194:325–340. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sohn D, Graupner V, Neise D, Essmann F,
Schulze-Osthoff K and Jänicke RU: Pifithrin-alpha protects against
DNA damage-induced apoptosis downstream of mitochondria independent
of p53. Cell Death Differ. 16:869–878. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ravi R, Bedi GC, Engstrom LW, Zeng Q,
Mookerjee B, Gélinas C, Fuchs EJ and Bedi A: Regulation of death
receptor expression and TRAIL/Apo2L-induced apoptosis by NF-kappaB.
Nat Cell Biol. 3:409–416. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shetty S, Graham BA, Brown JG, Hu X,
Vegh-Yarema N, Harding G, Paul JT and Gibson SB: Transcription
factor NF-kappaB differentially regulates death receptor 5
expression involving histone deacetylase 1. Mol Cell Biol.
25:5404–5416. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yamaguchi H and Wang HG: CHOP is involved
in endoplasmic reticulum stress-induced apoptosis by enhancing DR5
expression in human carcinoma cells. J Biol Chem. 279:45495–45502.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yoshida T, Shiraishi T, Nakata S, Horinaka
M, Wakada M, Mizutani Y, Miki T and Sakai T: Proteasome inhibitor
MG132 induces death receptor 5 through CCAAT/enhancer-binding
protein homologous protein. Cancer Res. 65:5662–5667. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Baritaki S, Katsman A, Chatterjee D, Yeung
KC, Spandidos DA and Bonavida B: Regulation of tumor cell
sensitivity to TRAIL-induced apoptosis by the metastatic suppressor
Raf kinase inhibitor protein via Yin Yang 1 inhibition and death
receptor 5 up-regulation. J Immunol. 179:5441–5453. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang HT, Wu J, Wen M, Su LJ and Luo H:
Galangin induces apoptosis in hepatocellular carcinoma cells
through the caspase 8/t-Bid mitochondrial pathway. J Asian Nat Prod
Res. 14:626–633. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang F, Chen WD, Deng R, Zhang H, Tang J,
Wu KW, Li DD, Feng GK, Lan WJ, Li HJ, et al: Hirsutanol A, a novel
sesquiterpene compound from fungus Chondrostereum sp., induces
apoptosis and inhibits tumor growth through
mitochondrial-independent ROS production: Hirsutanol A inhibits
tumor growth through ROS production. J Transl Med. 11:322013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lim SC, Parajuli KR and Han SI: The
alkyllysophospholipid edelfosine enhances TRAIL-mediated apoptosis
in gastric cancer cells through death receptor 5 and the
mitochondrial pathway. Tumour Biol. 37:6205–6216. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Taguchi H, Miyaji M and Yoshida T:
Evaluation of miconazole activity contained in human serum to hypha
of Aspergillus fumigatus. Nippon Ishinkin Gakkai Zasshi. 41:41–44.
2000.(In Japanese). View Article : Google Scholar
|
|
42
|
Eichenfield LF and Bogen ML: Absorption
and efficacy of miconazole nitrate 0.25% ointment in infants with
diaper dermatitis. J Drugs Dermatol. 6:522–526. 2007.PubMed/NCBI
|