Introduction
Multiple myeloma (MM) is a malignant disorder of
clonal plasma cells. It accounts for 1% of all cancers diagnosed
and >10% of hematological malignancies (1). Advances in therapeutic strategies have
resulted in improvements in the median survival of MM patients over
the past decade from 3–4 years to 7–8 years (2). However, MM remains incurable due to
its inevitable recurrence and progression, in which drug resistance
remains a major problem (3,4).
In recent years, scholars and the general public
have been paying increasing attention to resveratrol. The latter
can ‘sensitize’ resistant cells to chemotherapeutic agents by
overcoming one or more mechanisms of chemoresistance (5). Various drug-resistant tumors,
including lung carcinoma, pancreatic cancer, acute myeloid
leukemia, promyelocytic leukemia, and MM, can be sensitized by
resveratrol (6–9). Pterostilbene
(3,5-dimethoxy-40-hydroxystilbene; Pter) is found mainly in
blueberries, grapes and tree wood (10). Pter is a natural dimethylated analog
of resveratrol but is superior to the latter with regard to
liposolubility and bioavailability, which potentially makes it a
more potent anticarcinogenic compound than resveratrol (10,11).
Pter has been demonstrated to execute apoptosis in tumors of the
bladder, breast, colon, lung, pancreas, and stomach, as well as
against leukemia cells (11). In
bladder cancers, Pter has been found to inhibit the growth of
sensitive and chemoresistant cancer cells by inducing cell cycle
arrest, autophagy and apoptosis (12). Hence, Pter could be a new and
promising agent for the treatment of chemoresistant cancer cells in
the bladder. Pter and 3′-hydroxypterostilbene have been proved to
be effective apoptosis-inducing agents in multiple-drug resistant
(MDR) and BCR-ABL-expressing leukemia cells, suggesting its
important role in the treatment of resistant hematologic
malignancies (13). However, the
effects of Pter and its exact pharmacologic mechanisms on other
drug-resistant hematologic malignancies (especially in
chemoresistant MM) are not known.
Apoptosis is a programmed form of cell death with
typical morphologic features including cell shrinkage, chromatin
condensation, DNA hydrolysis, nuclear fragmentation, and formation
of apoptotic bodies (14,15). The process is triggered by two
principle mechanisms: the death receptor-mediated (extrinsic)
pathway when death receptors are actived by bonding to
corresponding death ligands or the mitochondrial (intrinsic)
pathway initiated through release of mitochondrial intermembrane
space proteins (15). Both pathways
converge to activate a series of cysteine-dependent
aspartate-specific proteases called caspases which are generally
divided into two groups: the initiator caspases (caspase-2, −8, −9,
and −10) and the effector caspases (caspase-3, −6, and −7)
(16,17). Caspase-8, a key initiator caspase,
could be actived by death ligands and receptors (Fas/FasL) via the
extrinsic pathway, which is the first step of caspase cascade
(16,17). Caspase-3, one of the most important
effector caspases, is a protease system that directly leads to the
disintegration of apoptotic cells and is the center in the
regulation of apoptosis (16,17).
Cleavage of caspase-3 triggered the inactivation of poly(ADP)ribose
polymerase (PARP) and DNA fragmentation which are the hallmarks of
apoptosis (16,17). In general, the initiator caspases
are usually autoactivated by particular pro-apoptotic stimuli while
the effector caspases could be activated after proteolytic cleavage
by the initiator caspases and then cause the cleavage or
degradation of various specific substrates, leading ultimately to
cell apoptosis (15–17).
Herein, we investigated the antiproliferative and
pro-apoptotic effects of Pter on bortezomib-resistant H929R cells
and explored the related mechanism of action. We found that Pter
inhibited proliferation and induced caspase-dependent apoptosis as
well as S-phase arrest of H929R cells. Loss of mitochondrial
membrane potential (MMP) in Pter-treated cells was examined by flow
cytometry. Moreover, downregulation of expression of phosphorylated
Akt and upregulation of expression of phosphorylated p38 MAPK were
observed by western blotting. Significantly, synergism between Pter
and the histone deacetylase inhibitors (HDACIs) panobinostat or
vorinostat resulted in toxicity to H929R cells. Thus, our work in
the present study supported that Pter might be a promising natural
compound for relapsed/refractory myeloma therapy, especially
against myeloma resistant to bortezomib chemotherapy.
Materials and methods
Cells and cell culture
The human MM line H929 was purchased from American
Type Culture Collection (Manassas, VA, USA). The human
bortezomib-resistant MM line H929R was a kind gift from Professor
Jian Hou (Department of Hematology, Changzheng Hospital, The Second
Military Medical University, Shanghai, China). Human peripheral
blood mononuclear cells (PBMCs) were separated by Ficoll-Hypaque
density gradient centrifugation. Primary CD138+ MM cells
were obtained from the bone marrow of MM patients relapsed on
bortezomib chemotherapy using magnetic bead selection (Miltenyi
Biotech, Auburn, CA, USA). Peripheral blood samples and bone marrow
samples were obtained from patients or healthy donors following
acquisition of the study participants' written informed consent.
The study was approved by the Shanghai Tenth People's Hospital
Institutional Review Board. Bortezomib-resistant cell line H929R
was cultured in the presence of 50 nM bortezomib. All the cells
mentioned above were grown in suspension in RPMI-1640 medium
(Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10%
fetal bovine serum and 1% penicillin streptomycin-glutamine, and
incubated at 37°C in an atmosphere of 5% carbon dioxide.
Reagents and antibodies
Pter (purity, 98%) was purchased from J&K
Chemicals (Shanghai, China). A stock solution of Pter (100 mM) was
dissolved in dimethyl-sulfoxide (DMSO; Sigma-Aldrich, St. Louis,
MO, USA) and stored in the dark at −20°C. Antibodies against p38
MAPK (#9212), Phospho-p38 MAPK (Thr180/Tyr182, #9211), P13 Kinase
p85 (19H8) (#4257), Akt (#9272), Phospho-Akt (Thr308, #9275),
Phosphate-Akt (Ser473, #9271), SAPK/JNK (#9252), phosphate-SAPK/JNK
(Thr183/Tyr185, #9251), Phospho-Histone H2A.X (Ser139, #9718),
Phospho-Chk1 (Ser296, #2349), cdc25A (#3652), CDK2 (#2546), cleaved
caspase-3 (Asp175, #9661), cleaved caspase-8 (#9496), PARP (#9532)
and β-actin (#3700) were purchased from Cell Signaling Technology
(Beverly, MA, USA). Antibodies against CyclinA2 (#1547) were
purchased from Epitomic (Burlingame, CA, USA).
Fluorescence-conjugated secondary antibodies were purchased from
Cell Signaling Technology. Cell Counting Kit-8 (CCK-8) was
purchased from Dojindo (Mashikimachi, Japan). A MMP assay kit with
JC-1 dye was purchased from Beyotime Institute of Biotechnology
(Haimen, China).
Assays to measure cell proliferation
and cytotoxicity
CCK-8 assays were undertaken to assess the viability
of cancer cells with increasing concentrations of Pter or
bortezomib. Cells (4×105 cells/ml) were seeded in
96-well plates with different concentrations of drugs for 24, 48,
or 72 h. Then, CCK-8 solution (10 µl/well) was added to each well
and inculcated for a further 2 h at 37°C in an atmosphere of 5%
CO2. Finally, absorbance was measured at 450 nm using a
microplate reader.
Apoptosis assays
Cells (4×105 cells/ml) were seeded in
6-well plates with different concentrations of Pter for 48 h or
with 30 µM Pter for different time points. Then, cells were stained
with Annexin V/propidium iodide (PI) (BD Pharmingen, Franklin
Lakes, NJ, USA) and analyzed by flow cytometry following
manufacturer instructions. Early (Annexin V+,
PI−) and late (Annexin V+, PI+)
apoptotic cells were counted.
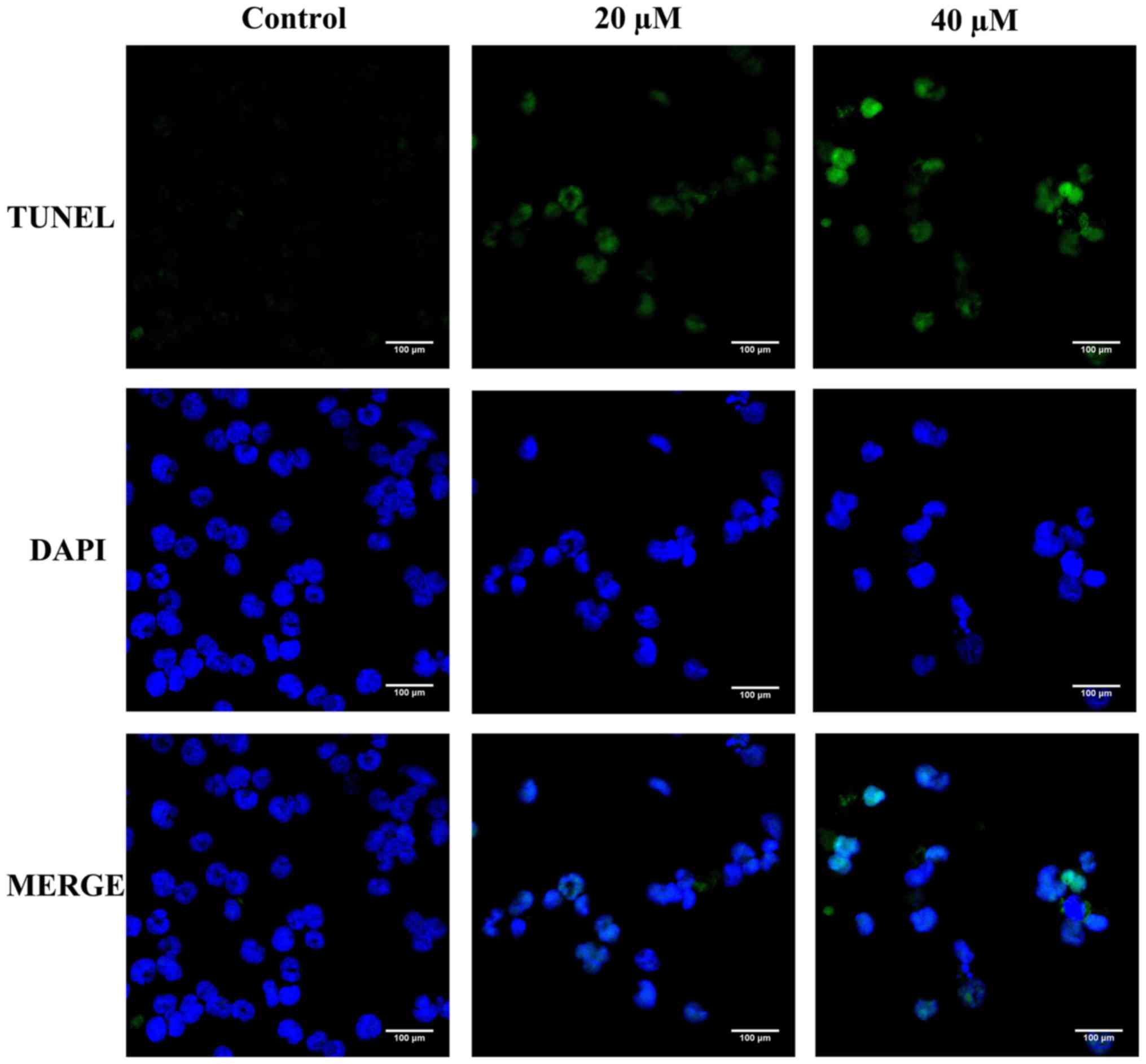
TUNEL (terminal deoxynucleotide
transferase dUTP nick-end labeling)/DAPI
(4,6-diamidino-2-phenylindole) double-staining assay
TUNEL/DAPI double-staining was used to detect the
apoptotic effect of Pter on the morphology of H929R cells. Cells
(4×105 cells/ml) were seeded in 6-well plates with
different concentrations of Pter for 24 h. Then, the suspension was
removed, and cells were fixed with 4% paraformaldehyde
(Sigma-Aldrich) for 25 min. Samples were examined by TUNEL assay
according to the manufacturer's protocol (Promega Corp., Madison,
WI, USA). Finally, DAPI (dilution 1:50,000; Sigma) was used to
stain the nuclei. After treatment, cell apoptosis were analyzed
using a fluorescence microscope (Zeiss Axiovert 25; Carl Zeiss,
Jena, Germany).
Mitochondrial transmembrane potential
assay
To determine whether the apoptosis induced by Pter
was associated with activation of the mitochondrial apoptotic
pathway, changes of mitochondrial transmembrane potential (MMP) in
the apoptotic process were examined by flow cytometry using an MMP
assay kit with JC-1 dye. Cells (4×105 cells/ml) were
cultured with different concentrations of Pter for 24 h. Then,
cells were washed with ice-cold phosphate-buffered saline (PBS) and
incubated with 0.5 ml JC-1 stain in a 37°C incubator for 20 min.
Finally, flow cytometry was carried out according to manufacturer's
instructions.
Western blotting
Cells were treated with different concentrations of
Pter and then lysed using lysis buffer (100 mM Tris-HCl, pH 6.8, 4%
sodium dodecyl sulfate, 20% glycerol). Equal amounts of proteins
(30 µg per lane) were separated on 10% or 12.5% sodium dodecyl
sulfate-polyacrylamide gels, transferred to nitrocellulose
membranes, blocked with 5% skimmed milk or 5% bovine serum albumin
for 1 h, and incubated with primary antibodies (1:1,000) at 4°C
overnight. Finally, membranes were treated with
fluorescence-conjugated secondary antibodies (1:1,000) at room
temperature for 60 min and detected by a two-color infrared laser
imaging system (Odyssey; Li-Cor, Lincoln, NE, USA).
Statistical analyses
Data are the mean ± standard deviation (SD).
Statistical significance was determined by the Student's t-test or
one-way ANOVA for multiple comparisons with SPSS v22.0 (IBM,
Armonk, NY, USA). p<0.05 was considered significant. All
experiments were carried out at least thrice.
Results
Pter inhibits proliferation of H929R
cells in a dose-dependent manner
H929R cells were obtained by increasing
extracellular concentrations of bortezomib stepwise over 8 months
(18). To verify the resistance in
H929R cells, H929 and H929R cells were exposed to different
concentrations of bortezomib for 48 h and the half-maximal
inhibitory concentration (IC50) was confirmed by CCK8
assays. Bortezomib inhibited proliferation of H929 cells
effectively (IC50=13.5±2.38 nM), whereas H929R cells
were more resistant to bortezomib (IC50=143.2±4.34
nM).
We first tested the antiproliferative activity of
Pter in bortezomib-resistant H929R and drug-naive H929 cells. H929R
and H929 cells were treated with 10, 20, 30, 40 and 50 µM Pter for
48 h and the toxic effect of Pter in both cell lines was examined
via CCK8 assays. We found that Pter significantly inhibited
proliferation of H929R and H929 cells in a dose-dependent manner
(Fig. 1A). To compare the
anti-proliferative effect of Pter on both cell lines, the
IC50 was then calculated by CalcuSyn. The
IC50 after 48 h of Pter treatment is 34.8±1.42 and
22.83±1.13 µM in H929R and H929 cells, respectively. Compared with
the growth-inhibitory effect of Pter on drug-naive H929 cells, the
agent is also largely capable of inhibiting the proliferation of
bortezomib-resistant cells, suggesting that Pter might be a
promising natural compound for relapsed/refractory myeloma therapy,
especially against myeloma resistant to bortezomib
chemotherapy.
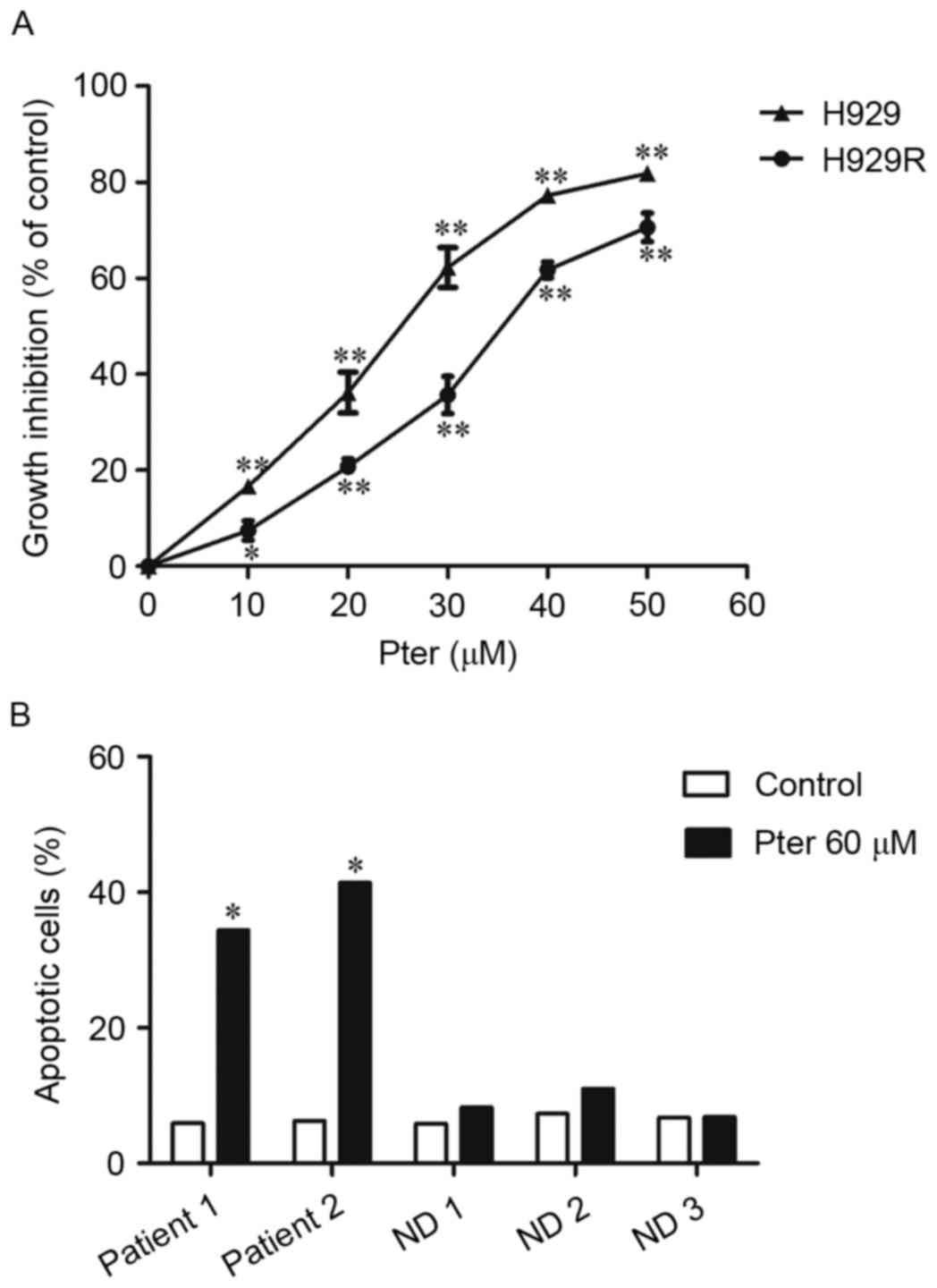 | Figure 1.Pter inhibits proliferation of H929R
cells in a dose-dependent manner. H929R and H929 cells were treated
with 10, 20, 30, 40 and 50 µM Pter for 48 h and the
growth-inhibitory effect of Pter in both cell lines was examined
via CCK8 assays (A). Primary CD 138+ MM cells from two
MM patients relapsed on bortezomib chemotherapy and PBMCs from
three health donors were treated with medium or Pter (60 µM, 24 h),
stained with Annexin V/PI and analyzed via flow cytometry (B).
*p<0.05, **p<0.01, compared to the vehicle control group;
Pter, pterostilbene; ND, normal donor; PBMCs, peripheral blood
mononuclear cells. |
Herein, the effects and the related mechanisms of
Pter in bortezomib-resistant myeloma were mainly explored. H929R
cells were then treated with Pter for different times. However,
time-dependent cytotoxicity was not observed within the given
concentration range (data not shown). Additionally, cytotoxicity
was also observed (by Annexin V/PI double-staining using flow
cytometric analyses) in primary CD 138+ cells isolated
from MM patients relapsed on bortezomib without any obvious effects
on PBMCs when Pter (60 µM, 24 h) was administered (Fig. 1B), suggesting that Pter might be a
safe agent for treatment of MM.
Pter induces apoptosis of H929R cells
in a dose- and time-dependent manner
Apoptosis is characterized by cell shrinkage,
chromatin condensation, DNA hydrolysis, nuclear fragmentation, and
formation of apoptotic bodies (14,15).
TUNEL/DAPI double-staining was first carried out to detect the
apoptotic effect of Pter on the morphology of H929R cells. Compared
with the control, Pter (20 or 40 µM, 24 h) induced a series of
morphological changes due to apoptosis (Fig. 2).
To investigate further the effects of Pter on H929R
cells, Annexin V/PI double-staining was assessed via flow
cytometry. When cells were treated with different concentrations of
Pter (0, 20, or 40 µM) for 24 h or with 30 µM Pter at different
time points (0, 12, 24 or 48 h), a significant apoptotic effect of
Pter on H929R cells was noted (Fig. 3A
and B). This phenomenon was associated with an increase in
expression of caspase-3, caspase-8 and poly(ADP)ribose polymerase
(PARP) cleavage proteins (Fig.
3C).
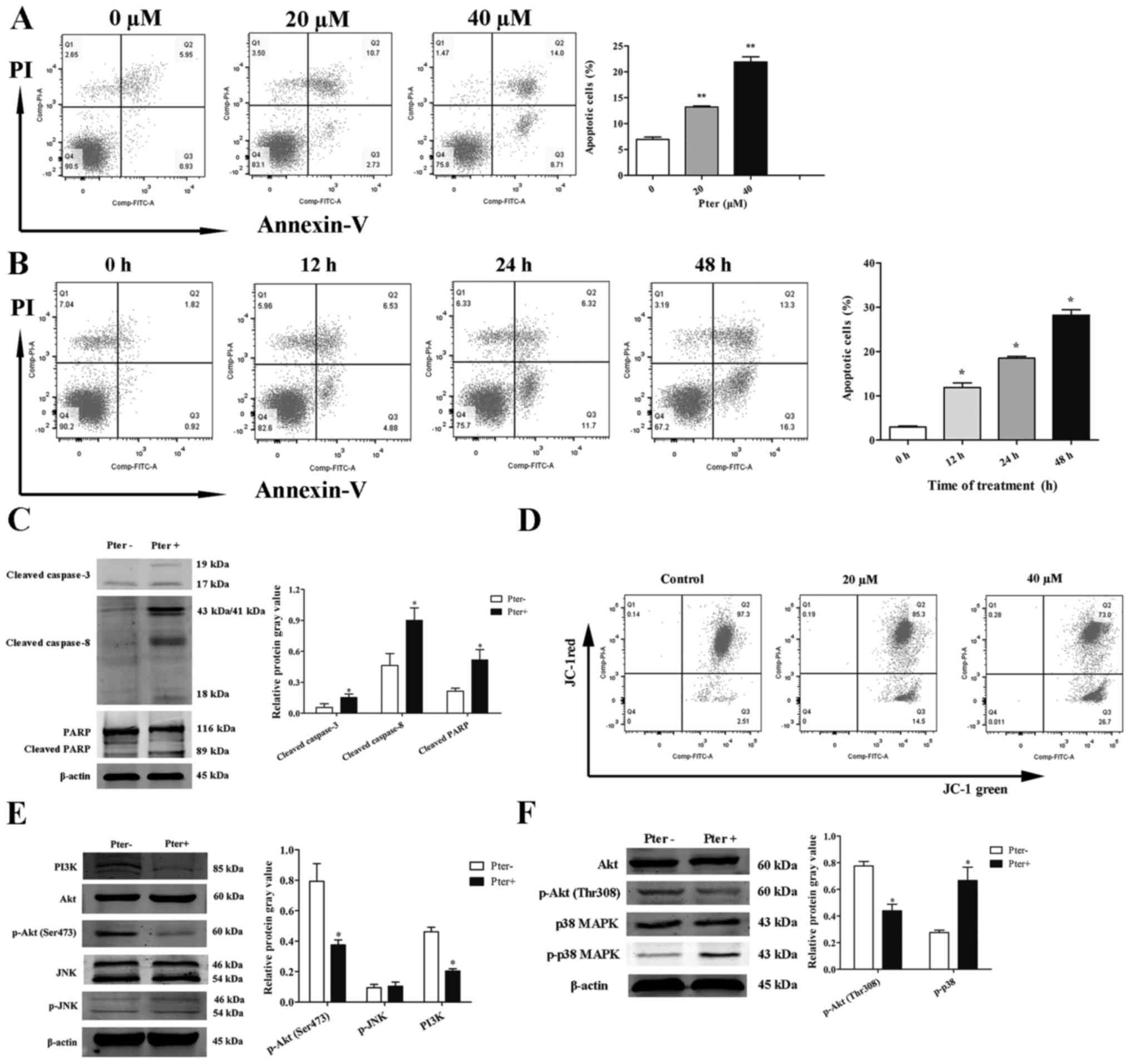 | Figure 3.Pter induces apoptosis of H929R cells
in a dose-and time-dependent manner. Quantitative analyses of
apoptosis were done via Annexin V/propidium iodide (PI)
double-staining using flow cytometry. H929R cells were treated with
different concentrations of Pter for 24 h (A) or with 30 µM Pter at
different time points (B). H929R cells were treated with medium or
Pter (30 µM, 48 h). Western blotting for caspase-3, caspase-8, PARP
cleaved proteins and quantification of relative cleaved protein
expression (cleaved proteins gray values/β-actin gray values) (C).
Cells were treated with different concentrations of Pter for 24 h
and changes in MMP were detected through JC-1 staining (D). Cells
were treated with medium or Pter (30 µM, 48 h). Western blotting
for PI3 kinase p85 (PI3K), phospho-Akt (Ser473), phosphate-SAPK/JNK
(p-JNK), phospho-Akt (Thr308), phospho-p38 MAPK (p-p38 MAPK) and
quantification of relative proteins expression (phosphorylated
proteins gray values/corresponding total proteins gray values, PI3K
gray values/β-actin gray values) (E). Data are the mean ± SD.
*p<0.05, **p<0.01 compared with the control group. Pter,
pterostilbene. |
Loss of mitochondrial transmembrane potential (MMP)
is one of the key events in apoptosis focus on mitochondria
(19,20). Activation of the mitochondrial
apoptotic pathway could be indirectly by determining whether the
MMP collapses (19,20). Next, changes in MMP in the apoptotic
process were examined by flow cytometry using an MMP assay kit with
JC-1 dye. A shift of red fluorescence in the control group to green
fluorescence in drug-treated group was noted, showing that MMP
levels in H929R cells treated with Pter (20 or 40 µM, 24 h) were
much lower than those in the control group (Fig. 3D).
Studies have shown that the MAPK signaling pathway
(especially c-Jun N-terminal kinase (JNK) and p38 MAPK) is
essential for Pter-mediated activation of caspases (21). Another study has reported that a
potent pan-PI3K/Akt inhibitor enhances the apoptotic effects of
bortezomib in bortezomib-resistant cells significantly, suggesting
the roles of these signaling pathways in Pter-induced apoptosis
(22). Therefore, we ascertained
(by western blotting) if JNK, p38MAPK or PI3K/Akt signaling
pathways were activated in Pter-treated H929R cells. H929R cells
were cultured with medium alone or Pter (30 µM) for 48 h, and then
levels of PI3 kinase p85 (PI3K), phospho-Akt (Thr308), phospho-Akt
(Ser473), phosphorylation of p38 MAPK and JNK were measured.
Downregulation of expression of PI3K, phosphorylated Akt and
upregulation of expression of phosphorylated p38 MAPK were
observed, but there were no changes in expression of phosphorylated
JNK (Fig. 3E).
These findings suggested that Pter induced apoptosis
of H929R cells in a dose-and time-dependent manner, and that this
effect was associated with a caspase-dependent cell death pathway
and loss of MMP. Also, Akt and p38 MAPK signaling pathways might be
involved in Pter-treated H929R cells.
Pter triggers marked recruitment of
H929R cells in the S phase of the cell cycle
To explore further the toxic effects of Pter on
H929R cells, we next undertook cell cycle analyses on cells with
medium alone or Pter using flow cytometry. After treatment of H929R
cells with Pter (30 µM; 0, 6, 12, and 24 h), a remarkable
accumulation of cells in the S phase was observed compared with
that in the control (>1.5-fold) (Fig. 4A and B). To investigate the
molecular mechanisms, a series of proteins related to DNA damage
and S-phase arrest was evaluated (using western blotting) in H929R
cells treated with medium or Pter (30 µM, 24 h). Levels of
phospho-histone H2A.X, a sensitive marker for DNA double-strand
breaks (DSBs) contributing to genomic instability and cancer
treatment (23) and
phospho-checkpoint kinase 1 (p-Chk1) proteins were upregulated
significantly and proteins of cell division cycle 25 homologue A
(cdc25A), cyclinA2 and cyclin-dependent kinase 2 (CDK2) were
decreased notably in Pter-treated groups (Fig. 4C). In conclusion, these findings
suggested that DNA damage and S-phase arrest might be involved in
Pter-related toxicity in H929R.
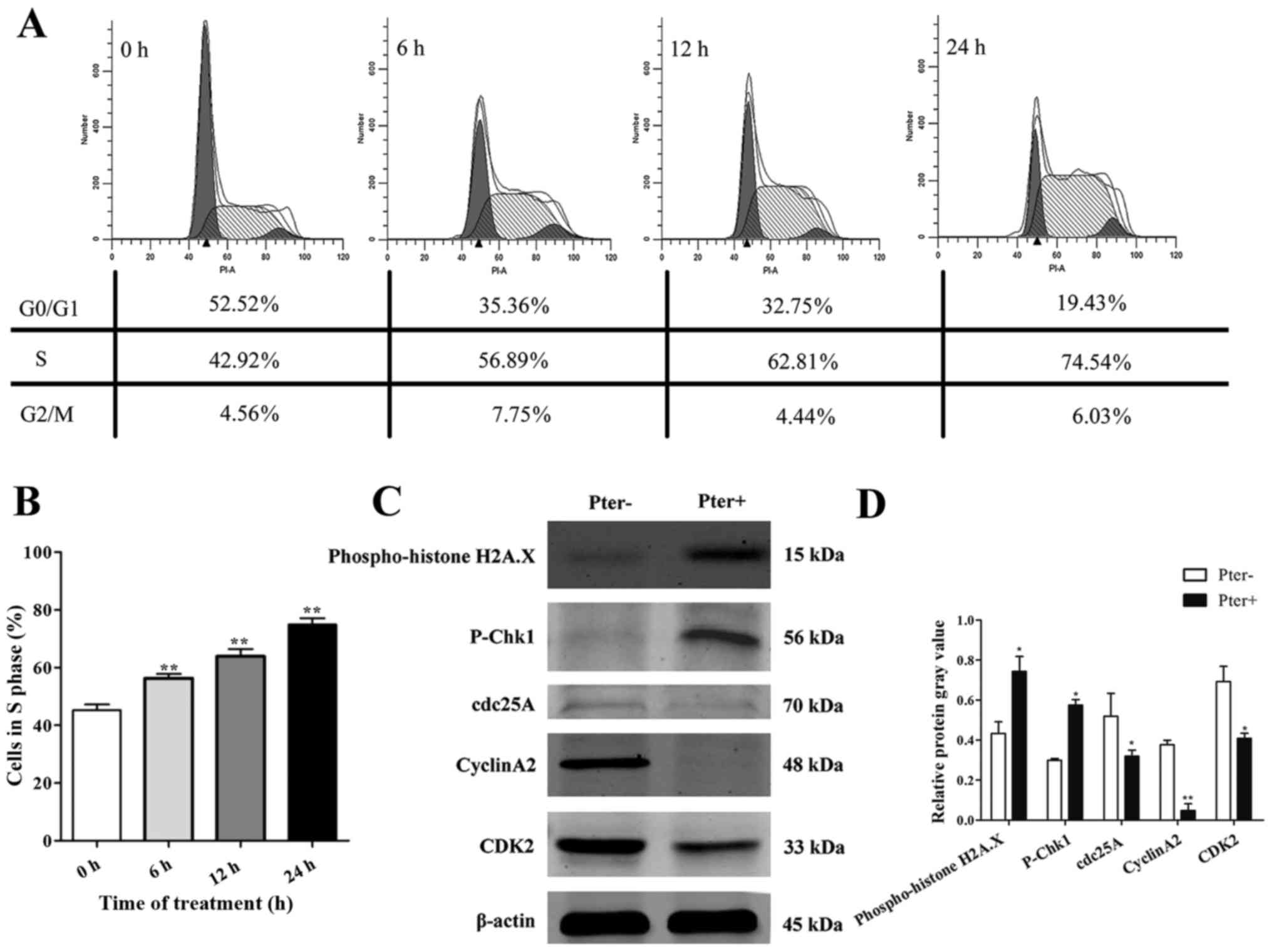 | Figure 4.Pter triggers marked recruitment of
H929R cells in the S phase of the cell cycle. Cell cycle analyses
of H929R cells treated with medium alone or Pter (30 µM) using flow
cytometry. These data represented one of three experiments (A). The
percentage of the S phase at 0, 6, 12, and 24 h was 45.19±2.05%,
56.27±1.51%, 63.88±2.48%, and 74.9±2.16%, respectively (B). Cells
were treated with medium or Pter (30 µM, 24 h). Western blotting
for a series of proteins related to S-phase arrest and DNA damage.
Quantification of relative protein expression (specific protein
gray values/β-actin gray values) (C). Data are the mean ± SD.
*p<0.05, **p<0.01 compared with the control group. Pter,
pterostilbene. |
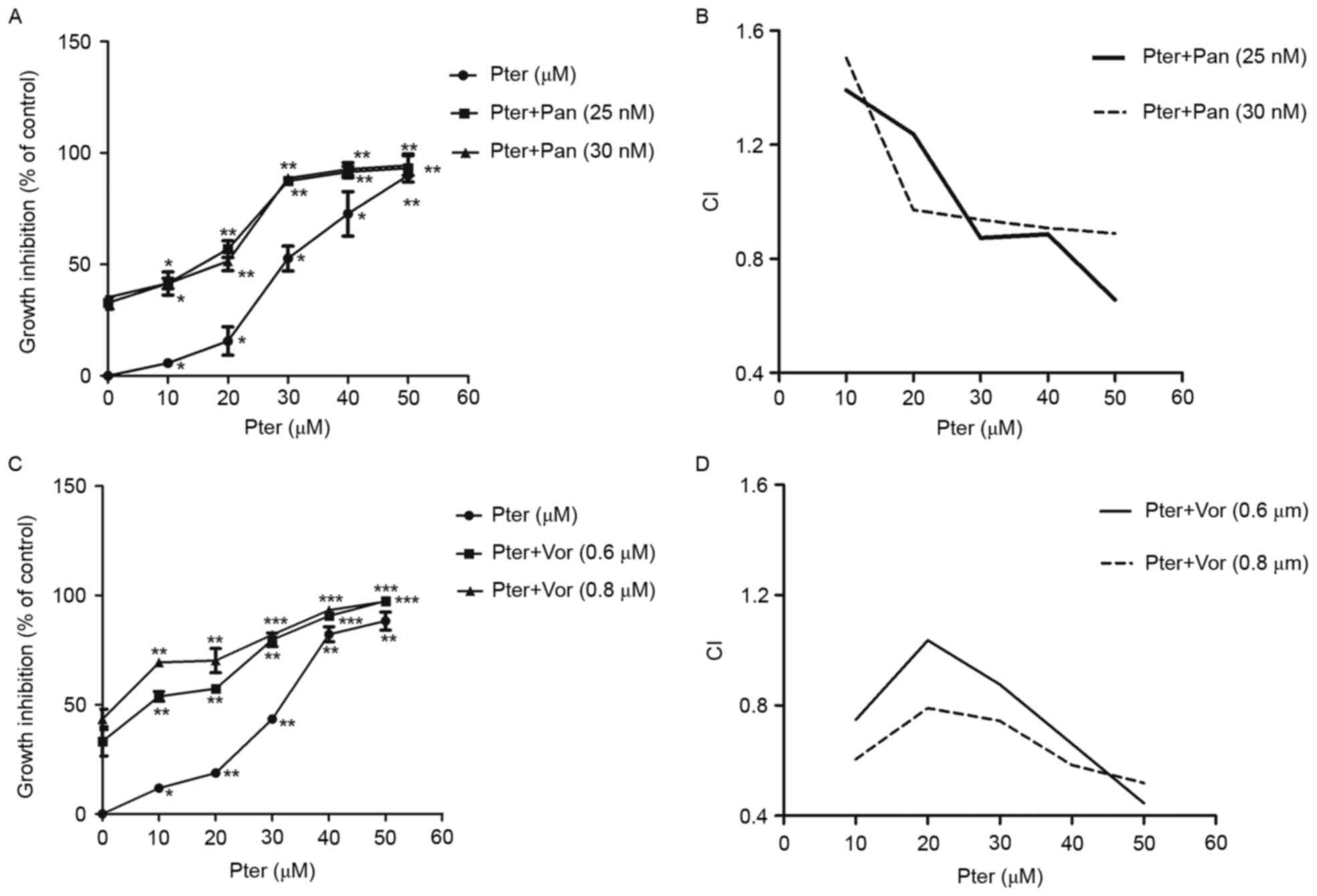
Pter in combination with HDACIs results in cytoxic
effect on H929R cells in a synergistic manner. Combination
chemotherapy is one of the most effective strategies for patients
with relapsed/refractory MM. To explore the potential of Pter for
MM, we assessed the cytotoxic effects of Pter combined with HDACIs
panobinostat or vorinostat. First, H929R cells were treated with
different concentrations of panobinostat or vorinostat for 48 h,
and the IC50 values of the two drugs in H929R cells were
32.5±0.91 nM and 0.8±0.35 µM, respectively. Then, panobinostat (25
or 30 nM) or vorinostat (0.6 or 0.8 µM) was added to H929R cells
treated with increasing concentrations of Pter (0, 10, 20, 30, 40,
and 50 µM, 48 h) and the toxic effects on H929R cells were
evaluated by CCK8 assays. The combination treatment with
panobinostat (25 or 30 nM) or vorinostat (0.6 or 0.8 µM) and
increasing concentrations of Pter sharply increased the growth
inhibition of H929R cells (Fig. 5A and
C). A synergistic effect was confirmed by combination index
(CI) values using the Chou-Talalay method. As indicated in Fig. 5B and D, median dose effect analyses
showed the combination of Pter with HDACis, especially vorinostat
induced synergistic cytotoxicity, with a CI <1.0 in H929R
cells.
Discussion
As a first-generation proteasome inhibitor (PI),
bortezomib is approved by the US Food and Drug Administration (FDA)
for treatment of relapsed/refractory or newly diagnosed multiple
myeloma (MM) (24). Bortezomib
alone or in combination with other drugs (e.g. doxorubicin,
melphalan, dexamethasone, immunomodulators) has considerable
therapeutic clinical efficacy, but resistance and relapse are
inevitable (24,25). Until now, the mechanisms of
bortezomib resistance have been understood incompletely (3,25).
Worse still, once MM patients have become refractory to bortezomib,
poor outcome is inevitable, and median, overall survival, and
event-free survival will be <10 months (4). Hence, the need to find strategies to
overcome bortezomib resistance is urgent.
Resveratrol can sensitize cells resistant to
chemotherapeutic agents by overcoming one or more mechanisms of
chemoresistance (5). In MM,
resveratrol has been shown to enhance the apoptotic and
anti-proliferative potential of bortezomib and thalidomide through
downregulation of nuclear factor-κB (NF-κB), signal transducer and
activator of transcription 3 (STAT-3) pathways (6). In another study, resveratrol was
demonstrated to enhance the apoptotic potential of perifosine and
bortezomib in drug-refractory MM and T-cell leukemia cells by
enhancing recruitment of Fas/CD95 death receptors in the extrinsic
pathway of apoptosis (26).
Compared with resveratrol, Pter has similar pharmacologic benefits
but exhibits much greater bioavailability (95% vs. 20%) and much
longer half-life (105 vs. 14 min), which makes it more potent for
clinical use (10). Pter was found
to inhibit growth of chemoresistant human bladder-cancer cells by
inducing cell cycle arrest, autophagy and apoptosis (12). In docetaxel-induced MDR human lung
cancer cell lines, Pter has been shown to inhibit cellular growth,
cell cycle arrest, apoptosis and autophagy (27). In MDR leukemia cell lines, Pter was
discovered to induce apoptosis through a caspase-independent
mechanism, suggesting its utility in treatment of resistant
hematologic malignancies (13). In
addition, based on the anti-chemoresistant effects of Pter in other
tumor types, we investigated the toxic effects of Pter in the
bortezomib-resistant MM line H929R.
Our data showed that Pter inhibited proliferation of
H929R cells in a dose-dependent manner but time-dependent
cytotoxicity was not observed. Of note, in MDR leukemia, Pter has
been shown to induce dose-dependent inhibition of cell growth in
all cell lines tested, but the time-dependent cytotoxicity was not
mentioned (13). Studies have shown
that Pter can induce a concentration- and time-dependent
anti-proliferation effect in various tumor cell types, including
melanoma, breast adenocarcinoma, and lung cancer (27–29);
these findings are not entirely consistent with our findings. The
difference in results may be associated with the types of cancer
cells used in vitro studies. Therefore, further
investigations on different myeloma cell lines in vitro and
related animal models in vivo are needed.
In addition, our data suggested that Pter triggered
apoptosis of H929R cells in a dose- and time-dependent manner, a
result that is consistent with reports of apoptosis in melanoma
after treatment with Pter and inositol hexaphosphate (29). TUNEL/DAPI double-staining was first
done and we observed a series of morphologic changes due to
apoptosis. Furthermore, the mechanisms involved in Pter-treated
H929R cells were explored, and we found that both caspase-related
proteins as well as MMP, p38 MAPK and Akt signaling pathways were
associated with Pter-induced apoptosis. In human acute myeloid
leukemia cell lines, Pter has been shown to be capable of inducing
apoptosis through activation of caspase-8, −9 and −3, an
MMP-dependent pathway, ERK1/2 and JNK, results that are consistent
with our findings to a certain extent (13). Various studies have shown that
activation of the PI3K/Akt signaling pathway often results in
bortezomib resistance and MM recurrence (22,30).
In bortezomib-resistant cell lines (H929R, RPMI-8226R), a potent
pan-PI3K inhibitor has been reported to downregulate phospho-Akt
(p-Akt) expression markedly (22).
In a previous study exploring the role of PI3K/Akt in the
regulation of Azadirachtin-induced autophagy in SL-1 cells, Shao
et al demonstrated decreased PI3K and phospho-Akt proteins
in SL-1 cells following Azadirachtin treatment (31). Herein, we observed downregulation of
PI3K, phospho-Akt (Ser473) and phospho-Akt (Thr308) proteins after
exposure of H929R to Pter, suggesting that PI3K/Akt signaling
pathway might be involved in Pter-induced apoptosis.
S-phase arrest is mediated by DNA damage activating
Chk1/2, which inactivates cdc25A and downstream cyclinA2 and CDK2
(32). We found that Pter induced
significant arrest of the S phase in H929R cells. Meanwhile,
western blotting showed that expression of phospho-histone H2A.X, a
sensitive marker for DNA DSBs that contributes to genomic
instability and cancer treatment (23), and p-Chk1 proteins were upregulated
significantly and that expression of proteins of cdc25A, CDK2 and
cyclinA2 were decreased considerably. These findings suggest that
Pter could induce DNA damage and could result in S-phase arrest of
H929R cells.
Mutations in apoptotic pathways, DNA damage, DNA
repair or mitotic-checkpoint pathways can permit the survival or
continued growth of cells with genomic abnormalities, thereby
enhancing the likelihood of malignant transformation (33). Among them, the mitotic-checkpoint
pathways and their associated proteins have critical roles in cell
cycle arrest and cancer therapy (33–36).
In general, the checkpoint pathway could check DNA damage and then
forwards specific information to the protein cores of cell cycle
machinery or replication apparatus, resulting in cell cycle arrest
and DNA repair (34–36). However, if cellular damage cannot be
properly repaired, checkpoint signaling pathway would convey
information to apoptotic protein cores and upregulate
proapoptotic-related proteins, leading to apoptosis (35,36).
Therefore, the anti-myeloma effects of Pter in H929R cells might be
attributed to inhibition of MM cell proliferation and induction of
cell apoptosis via mitotic-checkpoint signaling pathway.
Studies have reported that Pter combined with
inositol-6-phosphate (IP6) showed synergistic growth inhibition in
melanoma (29). Herein, we report
for the first time that Pter combined with an HDACI (panobinostat
or vorinostat) could inhibit proliferation of H929R cells with CI
values <1. As the first FDA-approved HDACI for patients with
relapsed/refractory MM who have undergone treatments previously
(including bortezomib), panobinostat has been shown to be an
effective agent in bortezomib-resistant myeloma cells and
bortezomib-refractory MM patients (37,38).
Vorinostat (an oral non-selective class-I and class-II HDACI), has
also been shown to be a potent anti-myeloma agent in preclinical
and clinical studies (39,40). Therefore, the synergistic effects of
Pter and a HDACI (panobinostat or vorinostat) might be important
for relapsed/refractory MM. This synergistic effect suggests that
the serious side effects of HDACIs (panobinostat or vorinostat)
could be reduced via combination with Pter. However, further
validation and exploration of their cooperative effects and safety
profiles are needed.
In conclusion, our findings suggest that the
anti-myeloma activity of Pter in the bortezomib-resistant line
H929R involves inhibition of cell proliferation, apoptosis
induction, and S-phase arrest. Moreover, certain caspase-related
proteins, loss of MMP, and activation of p38 MAPK and Akt signaling
pathways are associated with Pter-induced apoptosis. Furthermore,
our data first showed the synergistic effects of Pter in
combination with HDACIs (panobinostat or vorinostat), which might
be important for clinical trials of such combinations and related
safety-profile studies.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (81372391, 81570190, 81529001,
81302699, 31271496, 81600174 and 81300443), and ‘Personalized
Medicines-Molecular Signature-based Drug Discovery and
Development’, Strategic Priority Research Program of the Chinese
Academy of Sciences, grant no. XDA12020309.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Anderson KC: The 39th David A. Karnofsky
Lecture: Bench-to-bedside translation of targeted therapies in
multiple myeloma. J Clin Oncol. 30:445–452. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang WC and Lin SF: Mechanisms of drug
resistance in relapse and refractory multiple myeloma. BioMed Res
Int. 2015:3414302015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kumar SK, Lee JH, Lahuerta JJ, Morgan G,
Richardson PG, Crowley J, Haessler J, Feather J, Hoering A, Moreau
P, et al: International Myeloma Working Group: Risk of progression
and survival in multiple myeloma relapsing after therapy with IMiDs
and bortezomib: A multicenter international myeloma working group
study. Leukemia. 26:149–157. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gupta SC, Kannappan R, Reuter S, Kim JH
and Aggarwal BB: Chemosensitization of tumors by resveratrol. Ann
NY Acad Sci. 1215:150–160. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bhardwaj A, Sethi G, Vadhan-Raj S,
Bueso-Ramos C, Takada Y, Gaur U, Nair AS, Shishodia S and Aggarwal
BB: Resveratrol inhibits proliferation, induces apoptosis, and
overcomes chemoresistance through down-regulation of STAT3 and
nuclear factor-kappaB-regulated antiapoptotic and cell survival
gene products in human multiple myeloma cells. Blood.
109:2293–2302. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu Y, He W, Gao X, Li B, Mei C, Xu R and
Chen H: Resveratrol overcomes gefitinib resistance by increasing
the intracellular gefitinib concentration and triggering apoptosis,
autophagy and senescence in PC9/G NSCLC cells. Sci Rep.
5:177302015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kato A, Naiki-Ito A, Nakazawa T, Hayashi
K, Naitoh I, Miyabe K, Shimizu S, Kondo H, Nishi Y, Yoshida M, et
al: Chemopreventive effect of resveratrol and apocynin on
pancreatic carcinogenesis via modulation of nuclear phosphorylated
GSK3β and ERK1/2. Oncotarget. 6:42963–42975. 2015.PubMed/NCBI
|
|
9
|
Yaseen A, Chen S, Hock S, Rosato R, Dent
P, Dai Y and Grant S: Resveratrol sensitizes acute myelogenous
leukemia cells to histone deacetylase inhibitors through reactive
oxygen species-mediated activation of the extrinsic apoptotic
pathway. Mol Pharmacol. 82:1030–1041. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Estrela JM, Ortega A, Mena S, Rodriguez ML
and Asensi M: Pterostilbene: Biomedical applications. Crit Rev Clin
Lab Sci. 50:65–78. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McCormack D and McFadden D: Pterostilbene
and cancer: Current review. J Surg Res. 173:e53–e61. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen RJ, Ho CT and Wang YJ: Pterostilbene
induces autophagy and apoptosis in sensitive and chemoresistant
human bladder cancer cells. Mol Nutr Food Res. 54:1819–1832. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tolomeo M, Grimaudo S, Di Cristina A,
Roberti M, Pizzirani D, Meli M, Dusonchet L, Gebbia N, Abbadessa V,
Crosta L, et al: Pterostilbene and 3′-hydroxypterostilbene are
effective apoptosis-inducing agents in MDR and BCR-ABL-expressing
leukemia cells. Int J Biochem Cell Biol. 37:1709–1726. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hollville E and Martin SJ: Measuring
apoptosis by microscopy and flow cytometry. Curr Protoc Immunol.
112:1–24. 2016.
|
|
15
|
Koff JL, Ramachandiran S and
Bernal-Mizrachi L: A time to kill: Targeting apoptosis in cancer.
Int J Mol Sci. 16:2942–2955. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shi Y: Mechanisms of caspase activation
and inhibition during apoptosis. Mol Cell. 9:459–470. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fan T-J, Han L-H, Cong RS and Liang J:
Caspase family proteases and apoptosis. Acta Biochim Biophys Sin
(Shanghai). 37:719–727. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhu R, Xi H, Li YH, Jiang H, Zou JF and
Hou J: Establishment of a bortezomib-resistant myeloma cell line
and differential proteins analysis by MALDI-OF-MS. Zhejiang Da Xue
Xue Bao Yi Xue Ban. 38:445–452. 2009.(In Chinese). PubMed/NCBI
|
|
19
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan
JA, Tammareddi A, Moore VG, Deng J, Anderson KC, Richardson P, Tai
YT, et al: Pretreatment mitochondrial priming correlates with
clinical response to cytotoxic chemotherapy. Science.
334:1129–1133. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hsiao PC, Chou YE, Tan P, Lee WJ, Yang SF,
Chow JM, Chen HY, Lin CH, Lee LM and Chien MH: Pterostilbene
simultaneously induced G0/G1-phase arrest and MAPK-mediated
mitochondrial-derived apoptosis in human acute myeloid leukemia
cell lines. PLoS One. 9:e1053422014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu W, Chen Y, Xiang R, Xu W, Wang Y, Tong
J, Zhang N, Wu Y and Yan H: Novel phosphatidylinositol 3-kinase
inhibitor BKM120 enhances the sensitivity of multiple myeloma to
bortezomib and overcomes resistance. Leuk Lymphoma. 58:428–437.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bonner WM, Redon CE, Dickey JS, Nakamura
AJ, Sedelnikova OA, Solier S and Pommier Y: GammaH2AX and cancer.
Nat Rev Cancer. 8:957–967. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen D, Frezza M, Schmitt S, Kanwar J and
Dou QP: Bortezomib as the first proteasome inhibitor anticancer
drug: Current status and future perspectives. Curr Cancer Drug
Targets. 11:239–253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Murray MY, Auger MJ and Bowles KM:
Overcoming bortezomib resistance in multiple myeloma. Biochem Soc
Trans. 42:804–808. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Reis-Sobreiro M, Gajate C and Mollinedo F:
Involvement of mitochondria and recruitment of Fas/CD95 signaling
in lipid rafts in resveratrol-mediated antimyeloma and antileukemia
actions. Oncogene. 28:3221–3234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hsieh MJ, Lin CW, Yang SF, Sheu GT, Yu YY,
Chen MK and Chiou HL: A combination of pterostilbene with autophagy
inhibitors exerts efficient apoptotic characteristics in both
chemosensitive and chemoresistant lung cancer cells. Toxicol Sci.
137:65–75. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang Y, Yan X, Duan W, Yan J, Yi W, Liang
Z, Wang N, Li Y, Chen W, Yu S, et al: Pterostilbene exerts
antitumor activity via the Notch1 signaling pathway in human lung
adenocarcinoma cells. PLoS One. 8:e626522013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schneider JG, Alosi JA, McDonald DE and
McFadden DW: Effects of pterostilbene on melanoma alone and in
synergy with inositol hexaphosphate. Am J Surg. 198:679–684. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ikeda H, Hideshima T, Fulciniti M, Perrone
G, Miura N, Yasui H, Okawa Y, Kiziltepe T, Santo L, Vallet S, et
al: PI3K/p110{delta} is a novel therapeutic target in multiple
myeloma. Blood. 116:1460–1468. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shao X, Lai D, Zhang L and Xu H: Induction
of autophagy and apoptosis via PI3K/AKT/TOR pathways by
azadirachtin A in Spodoptera litura cells. Sci Rep. 6:354822016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wahl DR and Lawrence TS: Integrating
chemoradiation and molecularly targeted therapy. Adv Drug Deliv
Rev. 109:74–83. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kastan MB and Bartek J: Cell-cycle
checkpoints and cancer. Nature. 432:316–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Golubnitschaja O: Cell cycle checkpoints:
The role and evaluation for early diagnosis of senescence,
cardiovascular, cancer, and neurodegenerative diseases. Amino
Acids. 32:359–371. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Walworth NC: Cell-cycle checkpoint
kinases: Checking in on the cell cycle. Curr Opin Cell Biol.
12:697–704. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pietenpol JA and Stewart ZA: Cell cycle
checkpoint signaling: Cell cycle arrest versus apoptosis.
Toxicology 181–182. 475–481. 2002. View Article : Google Scholar
|
|
37
|
Raedler LA: Farydak (panobinostat): First
HDAC inhibitor approved for patients with relapsed multiple
myeloma. Am Health Drug Benefits. 9:84–87. 2016.PubMed/NCBI
|
|
38
|
Corrales-Medina FF, Manton CA, Orlowski RZ
and Chandra J: Efficacy of panobinostat and marizomib in acute
myeloid leukemia and bortezomib-resistant models. Leuk Res.
39:371–379. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Maiso P, Carvajal-Vergara X, Ocio EM,
López-Pérez R, Mateo G, Gutiérrez N, Atadja P, Pandiella A and San
Miguel JF: The histone deacetylase inhibitor LBH589 is a potent
antimyeloma agent that overcomes drug resistance. Cancer Res.
66:5781–5789. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Afifi S, Michael A, Azimi M, Rodriguez M,
Lendvai N and Landgren O: Role of histone deacetylase inhibitors in
relapsed refractory multiple myeloma: A focus on vorinostat and
panobinostat. Pharmacotherapy. 35:1173–1188. 2015. View Article : Google Scholar : PubMed/NCBI
|