Introduction
NRAS mutations occur in ~3–5% of metastatic
colorectal carcinoma (mCRC) patients and have been associated with
lower disease control and response rates to the epidermal growth
factor receptor (EGFR)-targeted monoclonal antibody cetuximab
(1–8). Because patients with wild-type
KRAS mCRC that harbor NRAS activating mutations do
not derive benefit from the administration of cetuximab, all major
international clinical guidelines recommend restricting its use to
mCRC patients with wild-type RAS tumors (6,9,10).
Although previous data indicated that NRAS might provide
similar or identical oncogenic signals to those of KRAS, as
they are not typically found in the same tumor (11,12),
accumulating evidence suggests very distinct clinical consequences
for the mutually exclusive KRAS- and NRAS-mutant mCRC
subsets (13,14). Dissimilar biological consequences
for mutations of KRAS and NRAS, which appear to be
selected under distinct tumorigenic contexts, underlie their
clinical distinction in mCRC patients. Accordingly, NRAS
mutations, which appear to arise specifically under settings of
continuous exposure to apoptotic stimuli in the context of chronic
inflammation, provide a MAPK-related distinct, prosurvival
signaling environment that mutational activation of KRAS
does not (14).
An important unresolved question arising from the
above observation is whether the apparently unique phenotype of
mutant NRAS can be exploited as a therapeutic strategy to
circumvent the refractoriness to cetuximab. Whereas most studies
have focused on investigating the downstream effectors of KRAS
signaling for bypassing the response of KRAS-mutant mCRC
cells to anti-EGFR therapy, almost nothing is known about the
specific pathways employed by NRAS-mutant mCRC cells that
render them unresponsive to cetuximab. Here we used isogenic mCRC
cell lines to explore signaling pathways specifically engaged by
the most common oncogenic NRAS Q61K variant upon challenge
of mCRC cells with cetuximab. We provide evidence for an unexpected
deregulation of the erythropoietin-producing hepatocellular (Eph)
receptor tyrosine kinase (RTK)/ephrin ligand cell communication
system (EphA2/ephrin-A1), which negatively influences cetuximab
efficacy in NRAS-mutant mCRC cells.
Materials and methods
Cell lines
The X-MAN™ isogenic cell lines SW48 NRAS-WT
(NRAS+/+) and SW48 NRASQ61K/+
(cat no. HD 103-017), were purchased from Horizon Discovery Ltd.
(Cambridge, UK) and maintained following the manufacturer's
instructions in RPMI-1640 medium with 2 mmol/l L-glutamine, 25
mmol/l sodium bicarbonate and 10% fetal bovine serum.
Drugs and materials
Cetuximab was provided by the Hospital Universitari
de Girona Dr Josep Trueta Pharmacy. Bioactive recombinant human
EphrinA1/Fc (EA1-Fc; cat no. 6417-A1) was purchased from R&D
Systems (Minneapolis, MN, USA) and dissolved in PBS.
Cell proliferation
Cells were plated in 24-well plates at 5,000
cells/well and incubated for 18 h in a humidified atmosphere
containing 5% carbon dioxide at 37°C to allow for attachment, after
which a zero-time point was determined. Cells were grown in regular
medium with or without 100 µg/ml cetuximab and counted with a
Coulter Counter (Coulter Electronics Inc., Hialeah, FL, USA). All
assays were performed at least twice in triplicate.
Phospho-proteome profiling
Phospho-receptor screening was performed using
Proteome Profiler Human Phospho-RTK array (R&D Systems)
according to the manufacturer's instructions. Densitometry analyses
of the scanned phospho-arrays were carried out using Carestream
Molecular Imaging Software (Carestream Health, Rochester, NY,
USA).
RNA isolation and reverse
transcription
Total RNA was extracted from cells using Nucleospin
RNA plus kit (Macherey-Nagel GmbH & Co. KG) according to the
manufacturer's instructions. Two micrograms of total RNA was
reverse-transcribed into cDNA using High Capacity cDNA Reverse
Transcription kit (Thermo Fisher Scientific, Carlsbad, CA, USA)
according to the manufacturer's instructions. RNA concentration and
quality were determined in an ND-1000 spectrophotometer (NanoDrop™
ND-1000, NanoDrop Technologies, USA).
Gene expression
cDNA (50 ng) were assayed in triplicate according to
established protocols using a QuantStudio™ 7 Flex Real-Time PCR
system (Thermo Fisher Scientific) with an automated baseline and
threshold cycle detection. GAPDH and ACTB were used as reference
genes. Primers and fluorescent probes for EPHA1, EPHA2, EFNA1,
EFNA2, GAPDH, and ACTB were obtained from Thermo Fisher Scientific
(TaqMan Gene Expression assays: assay ID Hs00358886-m1,
Hs01023290_m1, Hs00178313_m1, Hs00171656-m, Hs99999902_m1, and
Hs99999903_m1, respectively). Data were analyzed using the Thermo
Fisher Cloud software (Thermo Fisher Scientific).
Real-time cell growth rates
Proliferation was monitored in real time using the
xCELLigence RTCA DP Instrument (ACEA Biosciences, San Diego, CA,
USA). Cellular growth rate was determined by the slope of the
growth curve using the RTCA Software Package 1.2. We conducted the
normalization at one-time point before the treatment.
PathScan sandwich immunoassay
The PathScan® Intracellular Signaling
array kit (cat no. 7323; Cell Signaling Technology, Danvers, MA,
USA) was used as per the manufacturer's instructions.
Statistical analysis
Data are presented as mean ± SD from at least three
independent experiments. Two-group comparisons were performed using
Student's t-test. Comparisons of means of ≥3 groups were performed
by ANOVA, and the existence of individual differences tested by
Scheffé's multiple contrasts. P-values <0.01 were considered to
be statistically significant. All statistical tests were
two-sided.
Results
Heterozygous knock-in of the NRAS
activating mutation Q61K is sufficient to allow escape from
cetuximab-induced cell growth inhibition
We used an in vitro mCRC model of isogenic
pairs of SW48 colon cancer cell lines in which one allele of the
endogenous NRAS gene contained a heterozygous knock-in of
the c.181C>A activating mutation resulting in an amino acid
substitution from glutamine (Q) to lysine (K) at position 61
(NRASQ61K/+). We previously reported that,
whereas a strong reduction of cell viability was noted for parental
NRAS+/+ cells cultured in the presence of 100
µg/ml cetuximab, NRASQ61K/+ cells were fully
refractory to cetuximab-induced cell viability (15).
NRASQ61K/+ cells fail to
activate EphA1 receptor tyrosine kinase in response to
cetuximab
We first examined the changes in the
phospho-proteome of isogenic NRAS+/+ and
NRASQ61K/+ cells using the commercially available
Proteome Profiler Human Phospho-RTK array kit. Phospho-RTK
profiling revealed that the SW48-based model of mCRC mostly depends
on EGFR signaling to proliferate since EGFR (HER1) was the tyrosine
kinase receptor more significantly active among the 42 different
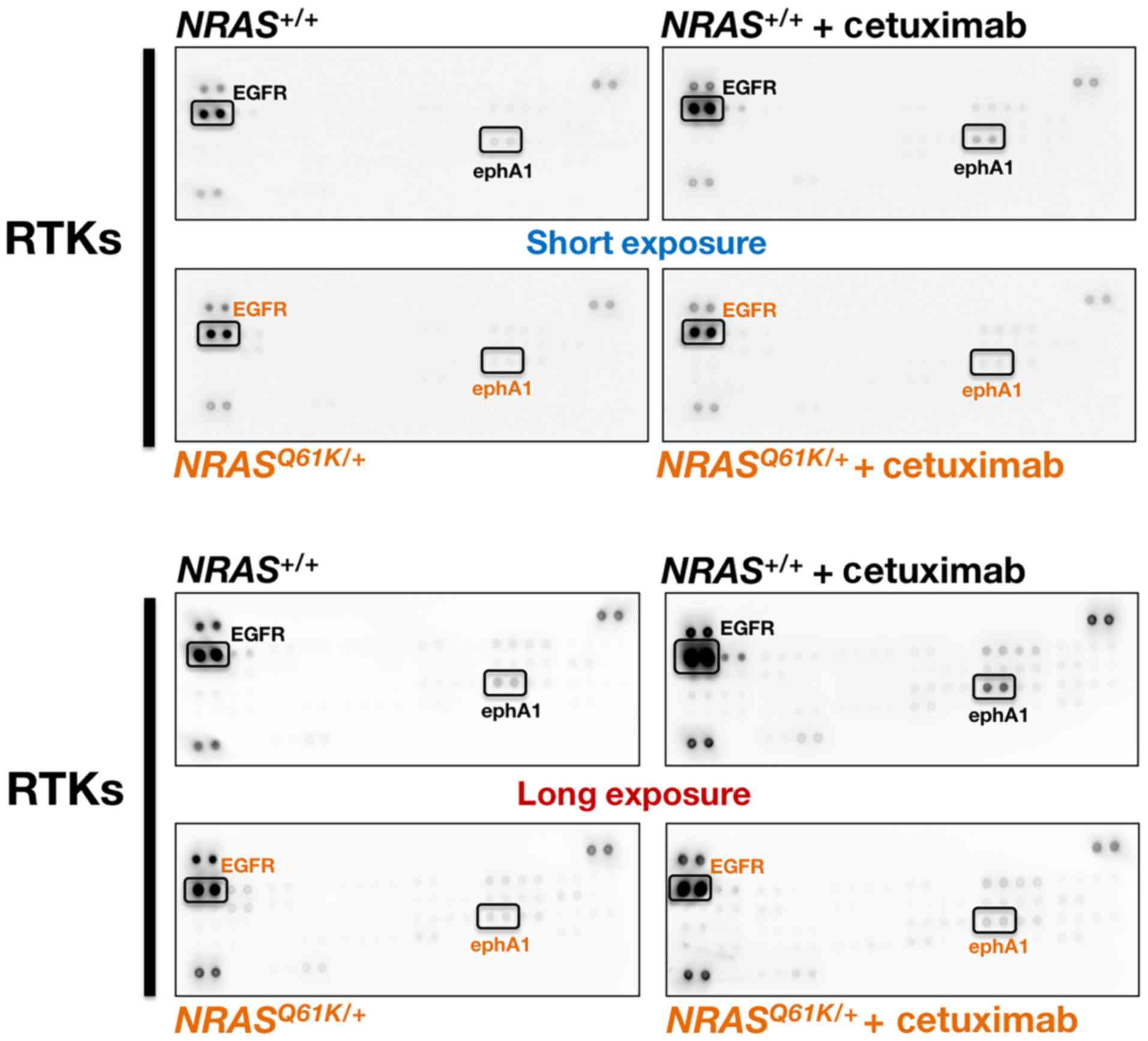
phospho-receptor tyrosine kinases included in the array (Fig. 1). Treatment of
NRAS+/+ and NRASQ61K/+ with
cetuximab was found to further enhance the
phosphorylation/activation status of EGFR (Fig. 1), a phenomenon that was likely due
to cetuximab-induced EGFR homodimerization and autophosphorylation
as previously reported in non-small-cell lung cancer cells, head
and neck squamous carcinoma cells, and triple-negative breast
cancer cells (16–18).
Closer inspection of the relative levels of tyrosine
phosphorylation detected by the array indicated that
NRAS+/+ cells, albeit modestly, activated the
endogenous ephrin receptor ephA1 in response to cetuximab, and this
was more obvious with a longer exposure of the membrane. By
contrast, ephA1 was not activated in cetuximab-refractory
NRASQ61K/+ cells in response to cetuximab
(Fig. 1).
Cetuximab fails to downregulate EphA2
in NRAS-mutant mCRC cells
The EPH gene family is the largest subfamily of
RTKs, including at least 16 receptors and 9 ligands for Eph
kinases, termed ephrins (19–22).
We performed quantitative real-time RT-PCR in SW48 cells to detect
the expression of the transcripts encoding EPHA1 and EPHA2
receptors and EFNA1 (ephrin-A1) and EFNA2 (ephrin-A2) ligands. When
NRAS+/+ cells were compared with
NRASQ61K/+ cells, a trend towards lower
expression of the EPHA1 transcript was detected in
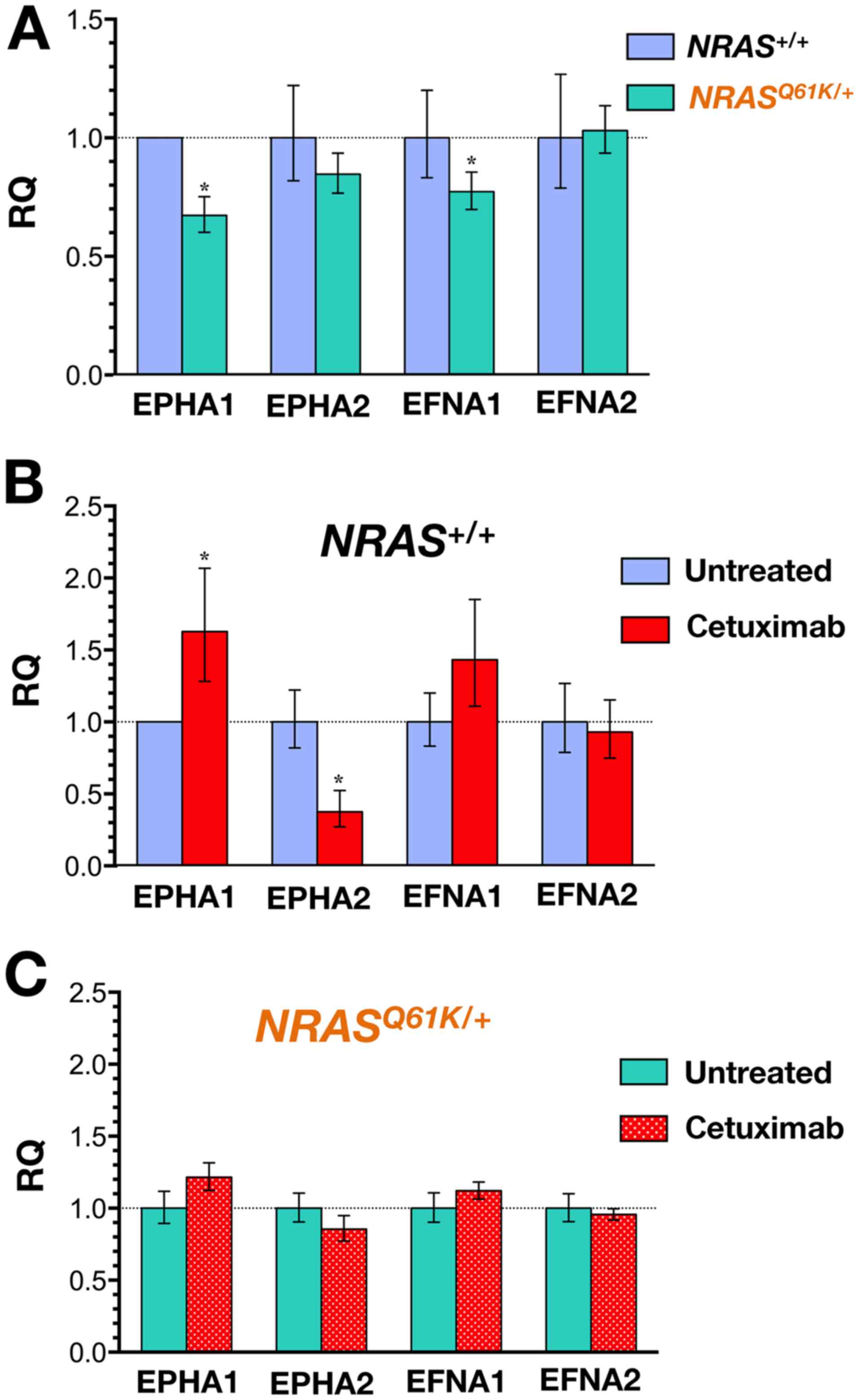
NRASQ61K/+ cells (Fig. 2A).
A completely different picture emerged when the
expression of EPHA1, EPHA2, EFNA1, and EFNA2 transcripts was
evaluated following 48-h exposure to cetuximab (Fig. 2B). Whereas the baseline expression
of EPHA1 remained unaltered in cetuximab-resistant
NRASQ61K/+ cells, a significant 1.5-fold
upregulation of the EPHA1 transcript occurred in
cetuximab-responsive NRAS+/+ cells. Furthermore,
cetuximab treatment resulted in a robust and significant >3-fold
downregulation of EPHA2 in NRAS+/+ cells whereas
the expression of EPHA2 remained unaltered in
NRASQ61K/+ cells. A trend towards higher
expression of EFNA1 ligand accompanied the downregulation of EPHA2
in cetuximab-responsive NRAS+/+ cells. By
contrast, cetuximab treatment failed to change the expression of
EFNA1 and EFNA2 ligands in cetuximab-resistant
NRASQ61K/+ cells (Fig. 2C).
Stimulation of ephA2 with a soluble
recombinant version of the ephrin-A1 ligand restores cetuximab
responsiveness in NRAS-mutant cells
Because ligand-independent cross-talk between ephA2
and other oncogenic pathways (e.g., PI3K/AKT and RAS/ERK) results
in tumor promotion (19–22), whereas ligand-induced ephA2
signaling triggers intrinsic tumor suppressive signaling involving
the blockade of PI3K/AKT and RAS/ERK pathways (21–24),
we hypothesized that the ephA2/ephrin-A1 axis might operate as a
molecular switch determining the responsiveness/unresponsiveness of
cetuximab in NRAS wild-type and NRAS-mutant mCRC
cells. To question whether the loss of ligand-dependent signaling
changed the function of ephA2 to a ‘protector’ against cetuximab in
NRAS-mutant mCRC cells, we took advantage of the
well-documented observation that stimulation of tumor cells with
ephrin-A1-Fc (eA1-Fc), a soluble recombinant ephrin-A1 ligand fused
to the Fc portion of human immunoglobulin G (IgG), leads to
tyrosine phosphorylation of ephA2 and its downregulation (25–28).
Cell proliferation rates of
NRAS+/+ and NRASQ61K/+ cells cultured with or
without cetuximab, eA1-Fc, or cetuximab plus eA1-Fc were
dynamically calculated using impedance technology (Fig. 3A)
The cell proliferation rate for
NRASQ61K/+ cells treated with cetuximab was
significantly higher than that for cetuximab-treated
NRAS+/+ cells, confirming the refractoriness of
NRASQ61K/+ cells to the anti-proliferative
effects of cetuximab. A small reduction in cell proliferation
occurred in NRASQ61K/+ cells treated with eA1-Fc
but not in NRAS+/+ cells. The addition of eA1-Fc
failed to alter the ability of cetuximab to significantly reduce
the proliferation rate of NRAS+/+ cells.
Interestingly, co-treatment with eA1-Fc and cetuximab fully
restored the capacity for cetuximab to inhibit the growth of
NRASQ61K/+ cells.
Stimulation of ephA2 with eA1-Fc
suppresses cetuximab-unresponsive hyperactivation of MAPK and AKT
in NRAS-mutant cells
To confirm that engagement of the ligand-dependent
tumor-suppressive branch of ephA2 signaling synergistically
sensitized NRAS-mutant cells to cetuximab via suppression of
the pro-oncogenic ligand-independent branch of ephA2 signaling, we
used commercially available slide-based antibody arrays to
simultaneously assess multiple well-characterized intracellular
signaling molecules (Fig. 3B). We
confirmed that pharmacological mimicking of ligand-dependent
stimulation of ephA2 with eA1-Fc resulted in a significant
pan-tyrosination of ephA2. Of note, eA1-Fc-induced activation of
ephA2 was stronger in cetuximab-refractory
NRASQ61K/+ cells than in cetuximab-responsive
NRAS+/+ cells. Co-treatment with eA1-Fc decreased
the cetuximab-unresponsive hyperactivation of MAPK in
NRAS-mutant cells and decreased also the
cetuximab-unresponsive hyperactivation of AKT in NRAS-mutant
cells. Thus, co-treatment with eA1-Fc and cetuximab generates a
phospho-signaling signature in NRAS-mutant cells reminiscent
to that observed in cetuximab-treated NRAS+/+
cells. Indeed, the strong activation of ephA2 with eA1-Fc that
occurred in NRAS-mutant cells decreased AKT to levels lower
than those observed in NRAS+/+ cells, where no
further changes in the deactivation of MAPK and AKT induced by
cetuximab occurred when NRAS+/+ cells were
co-exposed to eA1-Fc (Fig. 3B).
Discussion
In recent years, the ephrin RTKs and ephrin ligands
have been established as integral drivers of cancer formation and
progression (19–22). Here we provide evidence that
dysregulation of the ephrin-A1/ephA2 signaling axis plays an
unexpected role in determining the refractoriness of
NRAS-mutant mCRC cells to the EGFR-targeted monoclonal
antibody cetuximab.
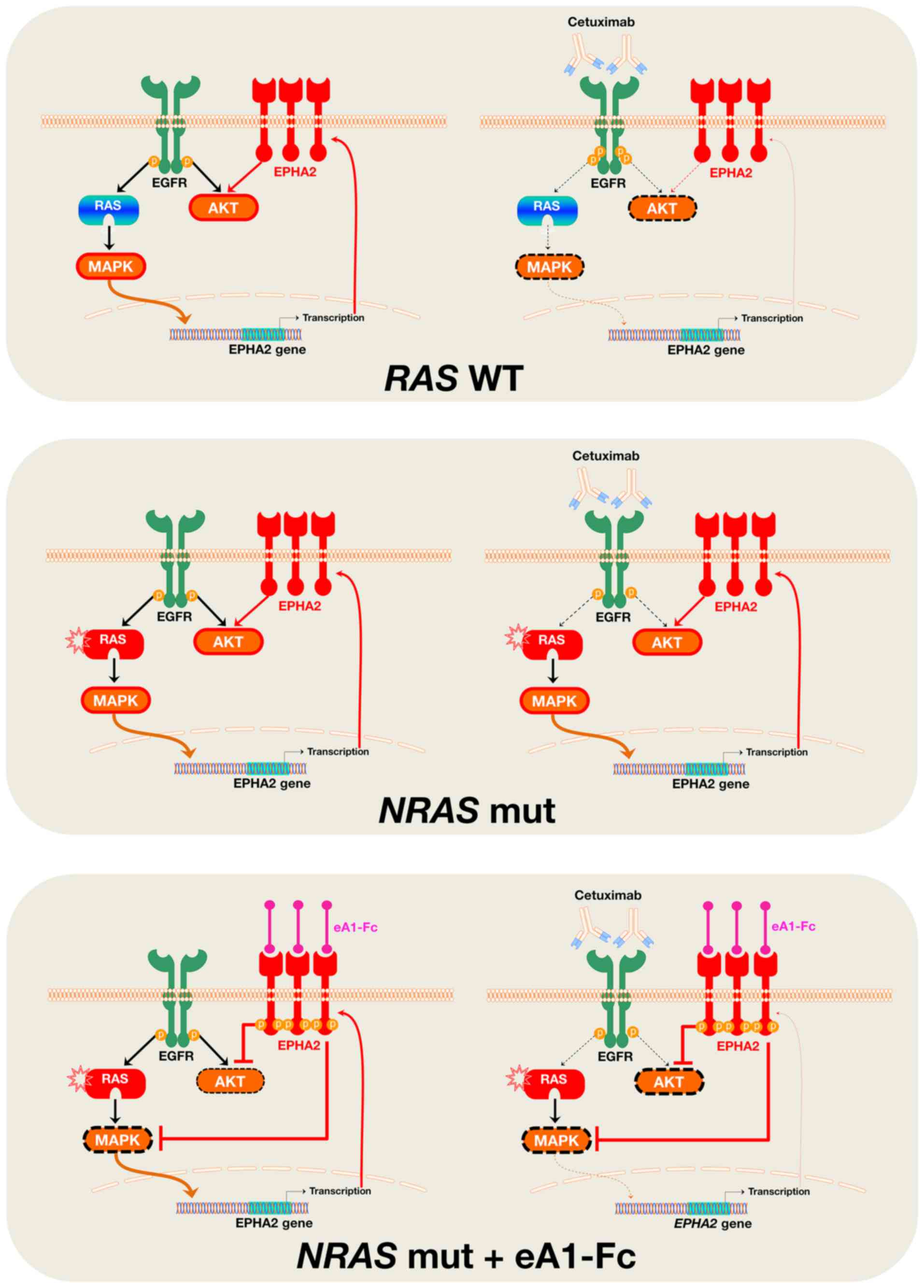
Our findings suggest that suppression of the
ligand-independent tumor-promoting signaling of ephA2 might be part
of the complex molecular mechanism through which cetuximab exerts
its growth inhibitory effects against EGFR-dependent RAS
wild-type mCRC cells. Because the ephA2 protein can directly
interact with EGFR (29–32), and EPHA2 is a direct transcriptional
target of the Ras-Raf-MAPK pathway (33–35),
cetuximab-induced blockade of EGFR signaling and subsequent
downregulation of MAPK activity leads to a reduction in EPHA2
expression in wild-type RAS mCRC cells. In NRAS-mutant mCRC
cells, however, the incapacity of cetuximab to block MAPK activity
impedes the establishment of the feedback loop that negatively
regulates EPHA2 expression (Fig.
4), which ultimately translates into the unresponsiveness of
‘NRAS-protected’ EPHA2 to the downregulatory effects of
cetuximab.
Our claim that cetuximab-directed reduction of EPHA2
expression might be part of the mechanism of action of cetuximab is
supported by the finding that function-based targeting of ephA2
signaling was sufficient to fully restore the function of cetuximab
in NRAS-mutant mCRC cells. Treatment with eA1-Fc, which
generates phenotypes similar to those generated by siRNA-mediated
or antisense oligonucleotide-mediated genetic knockdown of ephA2
(25–28,36–38),
efficiently converted NRAS-mutant cells into RAS
wild-type cells in terms of cetuximab functioning and efficacy.
Upon restoration of the ligand-dependent tumor-suppressive
signaling of ephA2 via stimulation with recombinant ephrin-A1, the
constitutively active MAPK signaling of NRAS-mutant mCRC
cells was inhibited in the presence of cetuximab. Moreover,
supporting and expanding earlier studies attributing
ligand-dependent ephA2 activation to suppression of the AKT-mTOR
pathway in cancer cells (23,39),
treatment with eA1-Fc synergistically interacted with cetuximab to
suppress AKT activation in NRAS-mutant cells. Indeed, we
observed a significantly stronger phosphorylation of the tyrosine
residues of the ephA2 receptor following stimulation with eA1-Fc in
NRAS-mutant cells (and a more significant decrease in
phosphorylation of AKT) than in RAS wild-type cells,
strongly suggesting that NRAS-mutant mCRC cells
constitutively exhibit an accelerated
phosphorylation/dephosphorylation cross-talk between ephA2 and
AKT.
Our findings are in line and expand on recent
studies demonstrating the involvement of ephA2 in the resistance to
small molecule EGFR tyrosine kinase inhibitors, such as erlotinib
and gefitinib, in lung cancer (32,40),
vemurafenib in melanoma (41), and
the anti-HER2 monoclonal antibody trastuzumab in breast cancer
(42–44). It might be argued that the strength
of evidence provided by the sole isogenic cell line pair
(NRAS+/+ vs NRASQ61K/+) used in
our current approach precludes any general extrapolation to mCRC
patients. However, it should be noted that, while elucidating new
molecular processes contributing to CRC pathogenesis using
ephA2high-sorted cell subpopulations with stem-like
features purified from a chemically-induced model of sporadic colon
carcinogenesis, De Robertis et al recently reported that
dysregulated expression of the ephA2 receptor accompanied by
downregulation of the ligand EFNA1 might operate as a novel
mechanism of resistance to cetuximab that can be considered an
alternative to KRAS mutations (45). We conclude that, even in the absence
of constitutive overexpression of ephA2 in NRAS-mutant
cells, dysregulated signaling of the ephrin-A1/ephA2 axis suffices
to overcome the inhibition of EGFR signaling imposed by cetuximab.
Future studies should examine whether the altered functioning of
the ephrin-A1/ephA2 axis might confer stem-like properties to
NRAS-mutant mCRC cells, thus explaining the shortened
survival and lack of response to anti-EGFR treatment of
NRAS-mutant mCRC patients (1–9).
Because both ephA2 and EFNA1 are recognized as novel
biomarkers of benefit from cetuximab-based therapy in mCRC
independently of the KRAS mutation status (45–47),
our current findings might help to delineate the ephrin-A1/ephA2
signaling axis as a common mechanism of cetuximab resistance
involving all mCRC patients. Moreover, the fact that cetuximab
functioning apparently involves also the upregulation of ephA1,
whose reduced expression correlates with poor differentiation,
invasion, metastasis and poor overall survival in CRC (48), further underscores the unappreciated
relevance of ephrin receptors and ephrin ligands in the
clinico-molecular management of mCRC.
In conclusion, our results reveal that: a) the
clinical benefit of cetuximab in mCRC might involve the suppression
of the ligandless oncogenic state of the ephA2 receptor; b)
imparting ligand-dependent tumor suppressing signaling through
ephA2 restores the responsiveness of NRAS-mutant mCRC cells
to cetuximab. The fact that NRAS-mutant mCRC cells
molecularly behave like RAS wild-type cells upon ephrin-A1
signaling to ephA2, in terms of cetuximab efficacy, might open new
therapeutic opportunities to clinically widen the usage of
cetuximab in mCRC patients.
Acknowledgements
This study was supported by grants from the
Ministerio de Ciencia e Innovación (Grant SAF2016-80639-P), Plan
Nacional de I+D+I, Spain and the Agència de Gestió d'Ajuts
Universitaris i de Recerca (AGAUR) (grant 2014 SGR229), Departament
d'Economia I Coneixement, Catalonia, Spain, to Javier A. Menendez.
Bernardo Queralt and Javier A. Menendez thank a charity collection
organized by Fundació Roses Contra el Càncer (Roses, Girona,
Catalonia) that allowed this line of research to be initiated in
2014. The Metabolism and Cancer Laboratory is supported by an
unrestricted grant from the Joan Armangué family (Girona,
Catalonia). The authors would like to thank Dr Kenneth McCreath for
editorial support.
References
|
1
|
De Roock W, Claes B, Bernasconi D, De
Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V,
Papamichael D, Laurent-Puig P, et al: Effects of KRAS, BRAF, NRAS,
and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy
in chemotherapy-refractory metastatic colorectal cancer: A
retrospective consortium analysis. Lancet Oncol. 11:753–762. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Peeters M, Oliner KS, Parker A, Siena S,
Van Cutsem E, Huang J, Humblet Y, Van Laethem JL, André T, Wiezorek
J, et al: Massively parallel tumor multigene sequencing to evaluate
response to panitumumab in a randomized phase III study of
metastatic colorectal cancer. Clin Cancer Res. 19:1902–1912. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Misale S, Di Nicolantonio F,
Sartore-Bianchi A, Siena S and Bardelli A: Resistance to anti-EGFR
therapy in colorectal cancer: From heterogeneity to convergent
evolution. Cancer Discov. 4:1269–1280. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Meriggi F, Vermi W, Bertocchi P and
Zaniboni A: The emerging role of NRAS mutations in colorectal
cancer patients selected for anti-EGFR therapies. Rev Recent Clin
Trials. 9:8–12. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bronte G, Silvestris N, Castiglia M,
Galvano A, Passiglia F, Sortino G, Cicero G, Rolfo C, Peeters M,
Bazan V, et al: New findings on primary and acquired resistance to
anti-EGFR therapy in metastatic colorectal cancer: Do all roads
lead to RAS? Oncotarget. 6:24780–24796. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hecht JR, Douillard JY, Schwartzberg L,
Grothey A, Kopetz S, Rong A, Oliner KS and Sidhu R: Extended RAS
analysis for anti-epidermal growth factor therapy in patients with
metastatic colorectal cancer. Cancer Treat Rev. 41:653–659. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schirripa M, Cremolini C, Loupakis F,
Morvillo M, Bergamo F, Zoratto F, Salvatore L, Antoniotti C,
Marmorino F, Sensi E, et al: Role of NRAS mutations as prognostic
and predictive markers in metastatic colorectal cancer. Int J
Cancer. 136:83–90. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hsu HC, Thiam TK, Lu YJ, Yeh CY, Tsai WS,
You JF, Hung HY, Tsai CN, Hsu A, Chen HC, et al: Mutations of
KRAS/NRAS/BRAF predict cetuximab resistance in metastatic
colorectal cancer patients. Oncotarget. 7:22257–22270.
2016.PubMed/NCBI
|
|
9
|
Taniguchi H, Yamazaki K, Yoshino T, Muro
K, Yatabe Y, Watanabe T, Ebi H, Ochiai A and Baba E; Tsuchihara K;
Japanese Society of Medical Oncology: Japanese Society of Medical
Oncology Clinical Guidelines: RAS (KRAS/NRAS) mutation testing in
colorectal cancer patients. Cancer Sci. 106:324–327. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Allegra CJ, Rumble RB, Hamilton SR, Mangu
PB, Roach N, Hantel A and Schilsky RL: Extended RAS gene mutation
testing in metastatic colorectal carcinoma to predict response to
anti-epidermal growth factor receptor monoclonal antibody therapy:
American Society of Clinical Oncology Provisional Clinical Opinion
Update 2015. J Clin Oncol. 34:179–185. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Irahara N, Baba Y, Nosho K, Shima K, Yan
L, Dias-Santagata D, Iafrate AJ, Fuchs CS, Haigis KM and Ogino S:
NRAS mutations are rare in colorectal cancer. Diagn Mol Pathol.
19:157–163. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Janakiraman M, Vakiani E, Zeng Z, Pratilas
CA, Taylor BS, Chitale D, Halilovic E, Wilson M, Huberman K, Filho
JC Ricarte, et al: Genomic and biological characterization of exon
4 KRAS mutations in human cancer. Cancer Res. 70:5901–5911. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Haigis KM, Kendall KR, Wang Y, Cheung A,
Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A,
Sebolt-Leopold J, Shannon KM, et al: Differential effects of
oncogenic K-Ras and N-Ras on proliferation, differentiation and
tumor progression in the colon. Nat Genet. 40:600–608. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang Y, Velho S, Vakiani E, Peng S, Bass
AJ, Chu GC, Gierut J, Bugni JM, Der CJ, Philips M, et al: Mutant
N-RAS protects colorectal cancer cells from stress-induced
apoptosis and contributes to cancer development and progression.
Cancer Discov. 3:294–307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Queralt B, Cuyàs E, Bosch-Barrera J,
Massaguer A, De Llorens R, Martin-Castillo B, Brunet J, Salazar R
and Menendez JA: Synthetic lethal interaction of cetuximab with
MEK1/2 inhibition in NRAS-mutant metastatic colorectal cancer.
Oncotarget. 7:82185–82199. 2016.PubMed/NCBI
|
|
16
|
Mandic R, Rodgarkia-Dara CJ, Zhu L, Folz
BJ, Bette M, Weihe E, Neubauer A and Werner JA: Treatment of HNSCC
cell lines with the EGFR-specific inhibitor cetuximab (Erbitux)
results in paradox phosphorylation of tyrosine 1173 in the
receptor. FEBS Lett. 580:4793–4800. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yoshida T, Okamoto I, Okabe T, Iwasa T,
Satoh T, Nishio K, Fukuoka M and Nakagawa K: Matuzumab and
cetuximab activate the epidermal growth factor receptor but fail to
trigger downstream signaling by Akt or Erk. Int J Cancer.
122:1530–1538. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oliveras-Ferraros C, Vazquez-Martin A,
López-Bonet E, Martín-Castillo B, Del Barco S, Brunet J and
Menendez JA: Growth and molecular interactions of the anti-EGFR
antibody cetuximab and the DNA cross-linking agent cisplatin in
gefitinib-resistant MDA-MB-468 cells: New prospects in the
treatment of triple-negative/basal-like breast cancer. Int J Oncol.
33:1165–1176. 2008.PubMed/NCBI
|
|
19
|
Pasquale EB: Eph-ephrin bidirectional
signaling in physiology and disease. Cell. 133:38–52. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pasquale EB: Eph receptors and ephrins in
cancer: Bidirectional signalling and beyond. Nat Rev Cancer.
10:165–180. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wykosky J and Debinski W: The EphA2
receptor and ephrinA1 ligand in solid tumors: Function and
therapeutic targeting. Mol Cancer Res. 6:1795–1806. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Beauchamp A and Debinski W: Ephs and
ephrins in cancer: ephrin-A1 signalling. Semin Cell Dev Biol.
23:109–115. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Miao H, Li DQ, Mukherjee A, Guo H, Petty
A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, et al: EphA2
mediates ligand-dependent inhibition and ligand-independent
promotion of cell migration and invasion via a reciprocal
regulatory loop with Akt. Cancer Cell. 16:9–20. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Petty A, Myshkin E, Qin H, Guo H, Miao H,
Tochtrop GP, Hsieh JT, Page P, Liu L, Lindner DJ, et al: A small
molecule agonist of EphA2 receptor tyrosine kinase inhibits tumor
cell migration in vitro and prostate cancer metastasis in vivo.
PLoS One. 7:e421202012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Carter N, Nakamoto T, Hirai H and Hunter
T: EphrinA1-induced cytoskeletal re-organization requires FAK and
p130(cas). Nat Cell Biol. 4:565–573. 2002.PubMed/NCBI
|
|
26
|
Duxbury MS, Ito H, Zinner MJ, Ashley SW
and Whang EE: Ligation of EphA2 by Ephrin A1-Fc inhibits pancreatic
adenocarcinoma cellular invasiveness. Biochem Biophys Res Commun.
320:1096–1102. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wykosky J, Gibo DM, Stanton C and Debinski
W: EphA2 as a novel molecular marker and target in glioblastoma
multiforme. Mol Cancer Res. 3:541–551. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Walker-Daniels J, Riese DJ II and Kinch
MS: c-Cbl-dependent EphA2 protein degradation is induced by ligand
binding. Mol Cancer Res. 1:79–87. 2002.PubMed/NCBI
|
|
29
|
Ramnarain DB, Park S, Lee DY, Hatanpaa KJ,
Scoggin SO, Otu H, Libermann TA, Raisanen JM, Ashfaq R, Wong ET, et
al: Differential gene expression analysis reveals generation of an
autocrine loop by a mutant epidermal growth factor receptor in
glioma cells. Cancer Res. 66:867–874. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Larsen AB, Pedersen MW, Stockhausen MT,
Grandal MV, van Deurs B and Poulsen HS: Activation of the EGFR gene
target EphA2 inhibits epidermal growth factor-induced cancer cell
motility. Mol Cancer Res. 5:283–293. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Larsen AB, Stockhausen MT and Poulsen HS:
Cell adhesion and EGFR activation regulate EphA2 expression in
cancer. Cell Signal. 22:636–644. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Koch H, Busto ME, Kramer K, Médard G and
Kuster B: Chemical proteomics uncovers EPHA2 as a mechanism of
acquired resistance to small molecule EGFR kinase inhibition. J
Proteome Res. 14:2617–2625. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Miao H, Wei BR, Peehl DM, Li Q, Alexandrou
T, Schelling JR, Rhim JS, Sedor JR, Burnett E and Wang B:
Activation of EphA receptor tyrosine kinase inhibits the Ras/MAPK
pathway. Nat Cell Biol. 3:527–530. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Macrae M, Neve RM, Rodriguez-Viciana P,
Haqq C, Yeh J, Chen C, Gray JW and McCormick F: A conditional
feedback loop regulates Ras activity through EphA2. Cancer Cell.
8:111–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pratt RL and Kinch MS: Ligand binding
up-regulates EphA2 messenger RNA through the mitogen-activated
protein/extracellular signal-regulated kinase pathway. Mol Cancer
Res. 1:1070–1076. 2003.PubMed/NCBI
|
|
36
|
Liu F, Park PJ, Lai W, Maher E,
Chakravarti A, Durso L, Jiang X, Yu Y, Brosius A, Thomas M, et al:
A genome-wide screen reveals functional gene clusters in the cancer
genome and identifies EphA2 as a mitogen in glioblastoma. Cancer
Res. 66:10815–10823. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Carles-Kinch K, Kilpatrick KE, Stewart JC
and Kinch MS: Antibody targeting of the EphA2 tyrosine kinase
inhibits malignant cell behavior. Cancer Res. 62:2840–2847.
2002.PubMed/NCBI
|
|
38
|
Shao H, Pandey A, O'Shea KS, Seldin M and
Dixit VM: Characterization of B61, the ligand for the Eck receptor
protein-tyrosine kinase. J Biol Chem. 270:5636–5641. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Miao H, Gale NW, Guo H, Qian J, Petty A,
Kaspar J, Murphy AJ, Valenzuela DM, Yancopoulos G, Hambardzumyan D,
et al: EphA2 promotes infiltrative invasion of glioma stem cells in
vivo through cross-talk with Akt and regulates stem cell
properties. Oncogene. 34:558–567. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Amato KR, Wang S, Tan L, Hastings AK, Song
W, Lovly CM, Meador CB, Ye F, Lu P, Balko JM, et al: EPHA2 blockade
overcomes acquired resistance to EGFR kinase inhibitors in lung
cancer. Cancer Res. 76:305–318. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Miao B, Ji Z, Tan L, Taylor M, Zhang J,
Choi HG, Frederick DT, Kumar R, Wargo JA, Flaherty KT, et al: EPHA2
is a mediator of vemurafenib resistance and a novel therapeutic
target in melanoma. Cancer Discov. 5:274–287. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhuang G, Brantley-Sieders DM, Vaught D,
Yu J, Xie L, Wells S, Jackson D, Muraoka-Cook R, Arteaga C and Chen
J: Elevation of receptor tyrosine kinase EphA2 mediates resistance
to trastuzumab therapy. Cancer Res. 70:299–308. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Menyhárt O, Santarpia L and Győrffy B: A
comprehensive outline of trastuzumab resistance biomarkers in HER2
overexpressing breast cancer. Curr Cancer Drug Targets. 15:665–683.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Youngblood VM, Kim LC, Edwards DN, Hwang
Y, Santapuram PR, Stirdivant SM, Lu P, Ye F, Brantley-Sieders DM
and Chen J: The ephrin-A1/EPHA2 signaling axis regulates glutamine
metabolism in HER2-positive breast cancer. Cancer Res.
76:1825–1836. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
De Robertis M, Loiacono L, Fusilli C,
Poeta ML, Mazza T, Sanchez M, Marchionni L, Signori E, Lamorte G,
Vescovi AL, et al: Dysregulation of EGFR pathway in EphA2 cell
subpopulation significantly associates with poor prognosis in
colorectal cancer. Clin Cancer Res. 23:159–170. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pentheroudakis G, Kotoula V, De Roock W,
Kouvatseas G, Papakostas P, Makatsoris T, Papamichael D, Xanthakis
I, Sgouros J, Televantou D, et al: Biomarkers of benefit from
cetuximab-based therapy in metastatic colorectal cancer:
Interaction of EGFR ligand expression with RAS/RAF, PIK3CA
genotypes. BMC Cancer. 13:492013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Strimpakos A, Pentheroudakis G, Kotoula V,
De Roock W, Kouvatseas G, Papakostas P, Makatsoris T, Papamichael
D, Andreadou A, Sgouros J, et al: The prognostic role of ephrin A2
and endothelial growth factor receptor pathway mediators in
patients with advanced colorectal cancer treated with cetuximab.
Clin Colorectal Cancer. 12:267–274.e2. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dong Y, Wang J, Sheng Z, Li G, Ma H, Wang
X, Zhang R, Lu G, Hu Q, Sugimura H, et al: Downregulation of EphA1
in colorectal carcinomas correlates with invasion and metastasis.
Mod Pathol. 22:151–160. 2009. View Article : Google Scholar : PubMed/NCBI
|