Introduction
Decoy receptor 3 (DcR3), a soluble molecule
belonging to the tumor necrosis factor receptor superfamily
(TNFRSF), was first identified as a decoy receptor of Fas ligand
(FasL) and inhibitor of FasL-induced apoptosis (1). DcR3 also neutralizes the biological
effects of two other TNFSF members, namely, LIGHT (TNFSF14) and
TNF-like molecule 1A (TL1A/TNFSF15) (2–4). DcR3
can be defined as an immunomodulator on the basis of its
neutralizing effects on FasL, LIGHT and TL1A (4–7). DcR3
is upregulated in tumor cells and inflammatory diseases (8–11).
DcR3 in serum can be used as a biomarker to predict cancer invasion
and progression of inflammation (12–15).
DcR3 also acts as an effector molecule to modulate cell function
through non-decoy activities, including the effects on cell
adhesion and differentiation (16–18).
Liver cancer is a common malignancy worldwide.
Cancerous liver tissues yield high DcR3 expression, and this
expression is correlated with tumor differentiation, serosal
invasion and liver metastases (13,19,20).
Nevertheless, the precise mechanisms of DcR3 in liver cancer
progression and metastasis remain unclear.
E-cadherin, a classical member of the cadherin
superfamily, is a calcium-dependent cell-cell adhesion glycoprotein
(21). The E-cadherin-catenin
complex plays a key role in cellular adhesion (22,23).
The loss of E-cadherin function or expression has been implicated
in cancer progression and metastasis (24,25).
In the present study, DcR3 treatment caused HepG2 cell cytoskeleton
remodeling, inhibited E-cadherin expression, and promoted cell
migration. Immunohistochemical analysis revealed that E-cadherin
and DcR3 exhibited an opposite expression trend in liver carcinoma
tissues. The present study also demonstrated the functional
mechanism of DcR3 in cancer cell migration and provides a
theoretical basis for the use of a DcR3 antagonist to treat liver
cancer.
Materials and methods
Clinical samples
Tissue samples from three patients with hepatic
carcinoma and biliary tract disease were collected during surgical
resection performed at the Shenzhen Second People's Hospital
(Shenzhen, China). Liver tumor and non-tumor liver tissues were
fixed and immediately used to prepare tissue slices. All of the
samples were obtained with patient consent and approval of the
Institutional Animal Care and Use Committee, Shenzhen Institutes of
Advanced Technology.
Cell culture and transfection
The human hepatocarcinoma cell line HepG2 or the
normal liver cell line L02 were obtained from the Shanghai
Institute of Cell Biology (Shanghai, China). Both cell lines were
maintained in Dulbecco's modified Eagle's medium (DMEM),
supplemented with 10% fetal bovine serum (FBS) (Gibco, Carlsbad,
CA, USA), and 100 µg/ml each of penicillin-streptomycin (HyClone,
Logan, UT, USA) in 5% CO2 at 37°C.
In 6-well plates, 2×105 cells/well were
cultured overnight and transfected with 2 µg PLVX–IRES-ZsGreen-DcR3
plasmid with Lipofectamine® 2000 (Invitrogen, Carlsbad,
CA, USA). The cells transfected with an empty vector were used as a
blank control.
Western blot analysis
HepG2 cells were treated with 3 µg/ml DcR3-Fc or
IgG1 as control, or transfected with the indicated plasmids. At 48
h after transfection or treatment, cells were harvested and
determined by the antibodies indicated in the figures. Cell pellets
were lysed in RIPA (Thermo Fisher Scientific Inc., Waltham, MA,
USA), 1 mM phenylmethylsulfonyl fluoride (PMSF) (Beyotime,
Shanghai, China) and 1% protease inhibitor cocktail (Thermo Fisher
Scientific, Inc.). Lysates were normalized for total protein (25
µg) and loaded on 8–12% sodium dodecyl sulfate-polyacrylamide gel,
electrophoresed, and transferred to a polyvinylidene fluoride
(PVDF) membrane (Millipore, Kenilworth, NJ, USA), followed by
blocking with 5% skimmed milk at room temperature for 1 h. The
membrane was incubated with primary antibodies overnight at 4°C,
and rinsed with Tris-buffered saline with Tween-20. The blots were
then incubated with horseradish peroxidase (HRP)-conjugated
secondary antibody (KPL) for 1 h at room temperature. Detection was
performed using EMD Millipore Luminata™ Western HRP
Chemiluminescence Substrates (WBLUR0500). Nuclear and cytoplasmic
extracts were isolated with NE-PER™ Nuclear and Cytoplasmic
Extraction Reagents (78833) purchased from Thermo Fisher
Scientific. The defined sections of the film were scanned for image
capture and quantification using Adobe Photoshop software (CS4;
(Adobe Systems, Inc., San Jose, CA, USA) and ImageJ software
(National Institutes of Health, Bethesda, MD, USA).
RNA isolation and real-time
quantitative PCR
Total RNA was isolated using TRIzol reagent
(Invitrogen) according to the manufacturer's instructions. RNA
samples were reverse transcribed with oligo (dT) and M-MLV Reverse
Transcriptase (Takara, Tokyo, Japan). A mixture of 1 µg RNA, 4 µl
5X RT mix, 1 µl primer mix, and nuclease-free water were made up to
a 20-µl volume. The reverse transcription step was as follow: 37°C
for 15 min; 85°C for 5 sec, and then stored at −20°C. Real-time
quantitative PCR analysis was performed with specific primers for
human E-cadherin (forward, 5′-TGGAGGAATTCTTGCTTTGC-3′ and reverse,
5′-CGTACATGTCAGCCAGCTTC-3′) in a CFX96 Touch™ Real-Time PCR
Detection System (Bio-Rad, Hercules, CA, USA) with SYBR qPCR mix
(Takara). Relative levels of gene expression were determined using
GAPDH as the control (forward, 5′-ATCTGGCACCACACCTTCTAC-3′ and
reverse, 5′-CAGCCAGGTCCAGACGCAGG-3′). SYBR-Green PCR Master Mix 2
µl, forward and reverse primers 200 nM, cDNA template 100 ng, and
ddH2O up to 10 µl volume was mixed together. PCR
conditions consisted of the following: 95°C for 3 min for
denaturation; 95°C for 5 sec for annealing; and 60°C for 40 sec for
extension, for 40 cycles. The threshold cycle for each sample was
selected from the linear range and converted to a starting quantity
by interpolation from a standard curve generated on the same plate
for each set of primers. The E-cadherin mRNA levels were normalized
for each well to the GAPDH mRNA levels using the 2−ΔΔCt
method.
Immunofluorescent assay
L02 or HepG2 cells were washed with
phosphate-buffered saline (PBS) and fixed at room temperature with
4% polyformaldehyde for 10 min, permeated with 0.1% Triton X-100
for 7 min, blocked for 30 min with 1% BSA, and incubated
sequentially with the indicated primary and secondary antibodies.
4,6-Diamidino-2-phenylindole (DAPI) (Beyotime) was used to label
the nuclei. Phalloidin-Rhodamine (Thermo Fisher Scientific) was
used for F-actin staining.
Flow cytometry
L02 or HepG2 cells were collected and washed with
PBS, fixed and permeabilized with Fix/Perm solution (BioLegend, San
Diego, CA, USA) before intracellular staining. After 15 min, the
cells were washed twice with Perm/Wash buffer, and incubated with
the DcR3 antibody at 4°C for 1 h. Cells were washed with PBS and
incubated with the FITC-goat anti-rabbit antibody at 4°C for 30
min. Cells were washed with PBS twice. The intracellular
fluorescence of FITC was detected by FCM after excitation at 488
nm. Fluorescence emissions at 530 nm from 10,000 cells were
collected, amplified and scaled to generate a single-parameter
histogram.
Immunochemistry
The sample sections were deparaffinized and
rehydrated. After boiling in a microwave oven, the antigen was
retrieved with a 0.01 M sodium citrate buffer (pH 6.0) at a
sub-boiling temperature for 20 min. The following steps were
performed with the SP kit (9001; ZSGB-BIO, Beijing, China).
Shortly, the sections were incubated with 3% hydrogen peroxide for
10 min to block endogenous peroxidase. After 15 min of
pre-incubation in 5% normal goat serum to prevent non-specific
staining, the samples were incubated with the antibody to DcR3
(Abcam, Cambridge, UK) at 4°C overnight. Secondary antibody was
added and incubated for 30 min. The sections were incubated in
horseradish enzyme-labeled chain avidin solution for 30 min at room
temperature. Color was developed with a diaminobenzidine (DAB)
substrate kit. Counterstaining was performed with hematoxylin.
Wound healing assay
Confluent cell cultures were grown on 6-well plates.
Wounds were made with the tip of a micropipette. DcR3-Fc was added
to the culture medium at a final concentration of 3 µg/ml. IgG1 was
added as a control. Wound closure speed was analyzed as indicated
in the legend.
Transwell assay
HepG2 cells were treated with 3 µg/ml DcR3-Fc or IgG
for 24 h, and then were trypsinized and resuspended in DMEM without
FBS before plating on the upper layer of the Transwell with an 8-µm
pore-size membrane at a cell density of 1×104. DMEM
containing 5% FBS was added to the lower layer. After 15 h, the
cells remaining on the top surface were scratched off. The cells on
the lower surface were fixed in methanol, stained with 0.5% crystal
violet (Beyotime), and images were captured under a microscope. The
intact Transwell was dissolved in 33% acetic acid, and the
supernatant was detected for absorption values with a
spectrophotometer at 590 nm.
Materials
DcR3-Fc and human IgG1 proteins were purchased from
Sino Biological, Inc. (Beijing, China). Anti-DcR3, anti-E-cadherin
and anti-IκBα antibodies were purchased from Abcam. Anti-p65 was
purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Anti-tubulin was purchased from Abmart (Shanghai, China).
Statistical analysis
All experiments were repeated at least three times
or noted otherwise. Data are expressed as mean ± SD. The t-test was
performed for inter-group comparisons. Values with p<0.05 were
considered to show significant differences.
Results
DcR3 regulates colony scattering of
HepG2 cells and decreases E-cadherin expression
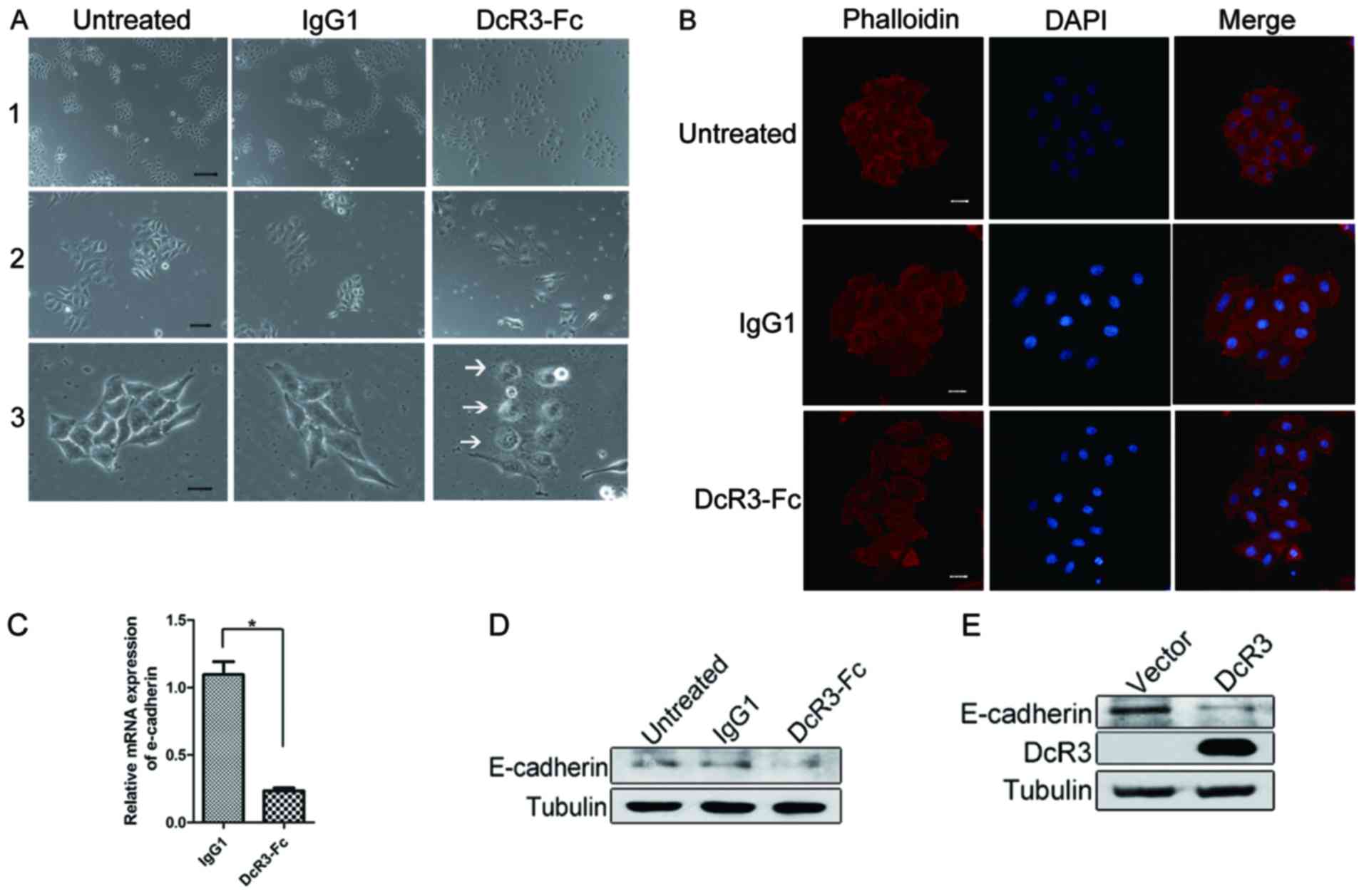
HepG2 cells were examined using a colony scattering
assay to analyze the function of DcR3 in regulating cell migration.
The colony scattering assay, mimicking certain aspects of tumor
invasion, reveals the ability of epithelial tumor cells to detach
from colonies in culture. The cells were plated at very low
density, and the morphological characteristics of the colonies were
evaluated 5 days after plating. The colonies were compact in the
control or IgG1 groups, and >90% of the cells in a colony
contained cell-cell junctions. By contrast, the cells were
scattered in the DcR3 treatment group, and <20% of the cells
formed junctions (Fig. 1A). In the
cells detached from the scattered colonies, numerous protrusions
were formed on the membrane edge of these cells (Fig. 1A, lane 3, arrows). Philloidin
staining revealed that DcR3 promoted actin remodeling and revealed
a scattered phenotype (Fig. 1B).
This finding indicated that DcR3 triggered changes in cell
morphology and enhanced the ability of cells to detach from the
colonies.
DcR3 treatment disrupts colony scattering and causes
cytoskeleton remodeling in HepG2 cells. To investigate the role of
DcR3 in the regulation of cell-cell adhesion, we detected whether
E-cadherin, a key molecule in the regulation of intercellular
adhesion, was regulated by DcR3. The mRNA of E-cadherin was
significantly downregulated by DcR3 treatment (Fig. 1C). The same effect was observed at
the protein level (Fig. 1D). DcR3
expression also inhibited E-cadherin expression (Fig. 1E). Thus, DcR3 is a negative
regulator of E-cadherin.
DcR3 and E-cadherin expression levels
are inversely correlated in hepatocarcinoma cell lines and
tissues
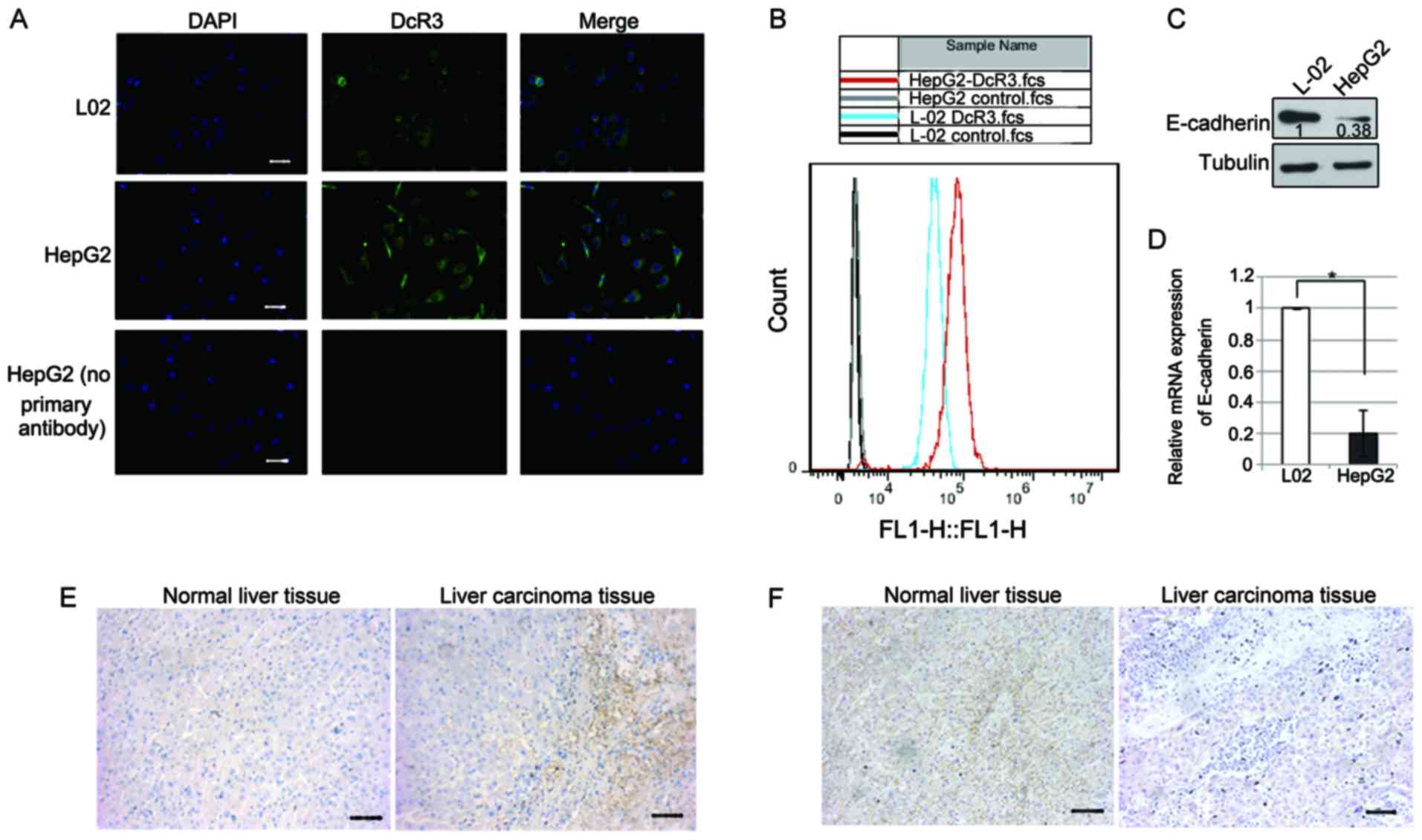
To understand the significance of the role of DcR3
in hepatocarcinoma, we detected DcR3 and E-cadherin expression
levels in the normal liver cell line L02 and the hepatoma cell line
HepG2 by performing immunofluorescent assays. The DcR3 expression
level was higher in the HepG2 cells than that noted in the L02
cells (Fig. 2A). To confirm this
observation, we detected the DcR3 expression using flow cytometry.
The DcR3 expression level in HepG2 cells was higher than that in
the L-02 cells (Fig. 2B). Protein
and mRNA quantification showed that E-cadherin was downregulated in
the HepG2 cells as compared with that in the L-02 cells (Fig. 2C and D). Importantly,
immunohistochemical staining showed that DcR3 was almost
undetectable in the non-tumor liver tissues (from patients with
biliary tract disease) but was upregulated in the liver cancer
tissues (Fig. 2E). Inversely,
E-cadherin was located in the cell junction in non-tumor tissue,
but was almost undetectable in liver cancer tissue (Fig. 2F). Therefore, hepatocarcinomas
exhibited low E-cadherin expression but high DcR3 levels.
DcR3 promotes cancer cell
migration
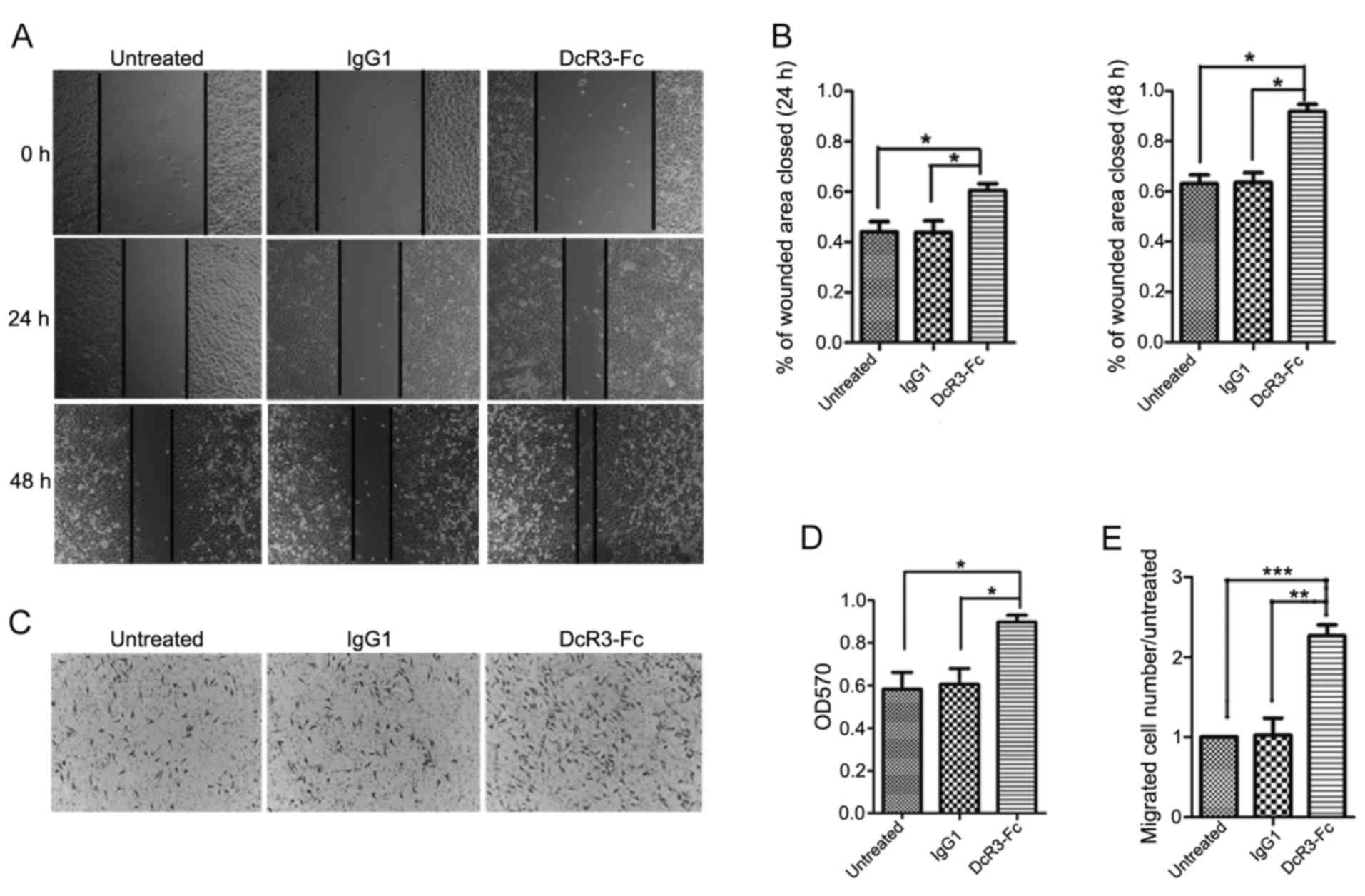
Considering that E-cadherin plays a crucial role in
cell adhesion and migration, we analyzed the effect of DcR3 on the
migratory ability of HepG2 cells. The wound healing assay
demonstrated that the addition of the DcR3-containing supernatant
caused a strong increase in cell migration at 24 and 48 h (Fig. 3A and B). To confirm this trend, we
detected the cell migratory ability using a Transwell assay. We
observed that DcR3 greatly enhanced HepG2 cell migration (Fig. 3C-E). These results also indicated
that DcR3 plays a positive role in cancer cell migration.
DcR3 induces p65 cytoplasm-nuclear
translocation
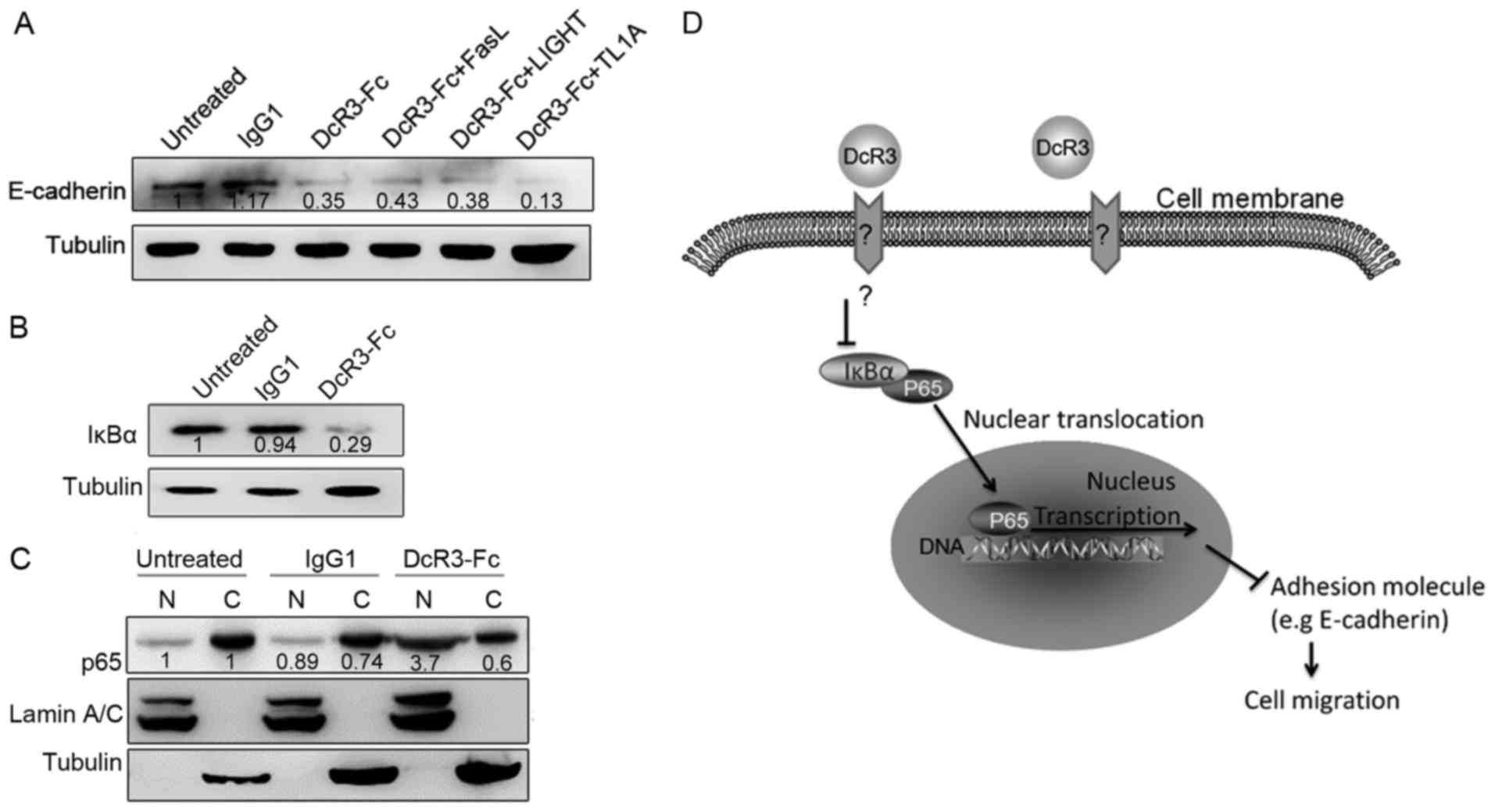
To investigate whether DcR3 inhibits E-cadherin
expression via its decoy function, we treated HepG2 cells by
simultaneously adding DcR3 and its ligand FasL, LIGHT or TL1A, to
the culture medium. The addition of these ligands did not affect
the function of DcR3 in the regulation of E-cadherin expression
(Fig. 4A). Thus, DcR3 inhibited
E-cadherin expression via its non-decoy function.
To investigate the mechanism of the function of DcR3
in E-cadherin regulation, we explored the involved signaling
pathway. DcR3 regulates the NFκB signaling pathway in monocytes.
E-cadherin is also a target of p65 (26,27).
Consequently, we investigated whether DcR3 activates NFκB signaling
in HepG2 cells. DcR3 significantly downregulated IkBα expression
(Fig. 4B). Furthermore, DcR3
treatment of HepG2 cells markedly increased the nuclear
translocation of the NF-κB subunit p65 (Fig. 4C). These data suggest that the NFκB
signaling cascade is an essential component in the involvement of
E-cadherin expression for the DcR3-mediated migration response.
Discussion
DcR3 can be defined as a novel immunosuppressant on
the basis of its neutralizing effects on FasL, LIGHT and TL1A. DcR3
is expressed by tumor cells from various lineages, including
adenocarcinomas of the colon, rectum (28,29),
lung (30) and gastric cancer
(31), hepatocellular carcinoma
(13,20) and in chronic liver diseases
(19), which frequently lead to
cancer formation. Increased DcR3 levels in serum or tissues were
found to be correlated with poor prognosis and resistance to
treatment in some cancer patients (13). In addition to its neutralizing
effect, DcR3 also acts as an effector molecule to modulate cell
function via non-decoy activities, including the regulation of DC
and macrophage differentiation that leads to Th2 polarization
(32,33), M2 macrophage differentiation
(34), and cytoskeleton remodeling
(16,17).
DcR3 can induce actin reorganization in human
monocytes, and this protein triggers multiple signaling molecules,
such as PKC and phosphatidylinositol 3-kinase (PI3K) (35). Furthermore, DcR3 induces
NFκB-mediated expression of ICAM-1, VCAM-1 and IL-8 by monocytes;
consequently, their binding to endothelial cells is enhanced
(17). DcR3-Fc was found to act on
THP-1 monocytes and differentiated macrophages to increase the
expression level of integrin α4. Thus, cell aggregation and
proliferation are promoted and apoptosis is reduced (18). DcR3 is upregulated in cancer cells;
thus, these observations suggest its important roles in modulating
the migration and trafficking of monocytes/macrophages in the tumor
microenvironment.
E-cadherin plays an important role in cell adhesion
by forming adherent junctions to bind cells within tissues. The
loss of E-cadherin expression has been defined as a hallmark of
EMT. EMT is a process by which epithelial cells lose their cell
polarity and cell-cell adhesion, and they gain migratory and
invasive properties to become mesenchymal stem cells. DcR3 was
found to induce IκB kinase activation, IκB degradation, and p65
nuclear translocation in human microvascular endothelial cells
(17). E-cadherin is a target of
p65, which represses E-cadherin expression and enhances the
epithelial-to-mesenchymal transition of mammary epithelial cells
via ZEB-1 and ZEB-2 (36). In
addition, TGF-β is one of the most critical factors involved in EMT
regulation. TGF-β promotes EMT through both Smad-dependent and
Smad-independent manner (37). The
relationship between DcR3 and the TGF-β pathway will be the
research of interest in a future study. A model of the function of
DcR3 in cell migration regulation is shown in Fig. 4D. DcR3 controls the expression of
E-cadherin and the p65 translocation of HepG2 cells. Thus, it
elicits double effects in tumor metastasis regulation and immune
modulation. The blocking of DcR3 may be applied as an effective
therapeutic strategy to prevent tumor metastasis.
In conclusion, the present study demonstrated that
DcR3 is overexpressed in hepatic carcinoma tissues and cell lines.
DcR3-Fc treatment inhibited E-cadherin expression, enhanced tumor
cell migration in vitro, and promoted p65 nuclear
translocation. These findings revealed the mechanism underlying the
ability of DcR3 to regulate cell migration. Therefore, DcR3 may be
a potential target for the gene therapy of hepatic carcinoma.
Acknowledgements
The present study was supported by the Shenzhen
Basic Research Program (JCYJ20150630114942293,
JCYJ20140416180228582 and JCYJ20160229201353324), the Nature
Science Foundation of China for Young Scholar grant (81501356), the
Shenzhen Peacock Next-generation Monoclonal Antibody Drug Research
and Development Program (1110140040347265), the Fourth Group of
Talents in Guangdong Province (2014–1), the Shenzhen Engineering Laboratory
(2014–1677), and the Shenzhen Technology Study program
(JSGG20160229202150023).
References
|
1
|
Pitti RM, Marsters SA, Lawrence DA, Roy M,
Kischkel FC, Dowd P, Huang A, Donahue CJ, Sherwood SW, Baldwin DT,
et al: Genomic amplification of a decoy receptor for Fas ligand in
lung and colon cancer. Nature. 396:699–703. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yu KY, Kwon B, Ni J, Zhai Y, Ebner R and
Kwon BS: A newly identified member of tumor necrosis factor
receptor superfamily (TR6) suppresses LIGHT-mediated apoptosis. J
Biol Chem. 274:13733–13736. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Migone TS, Zhang J, Luo X, Zhuang L, Chen
C, Hu B, Hong JS, Perry JW, Chen SF, Zhou JX, et al: TL1A is a
TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell
costimulator. Immunity. 16:479–492. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gill RM and Hunt JS: Soluble receptor
(DcR3) and cellular inhibitor of apoptosis-2 (cIAP-2) protect human
cytotrophoblast cells against LIGHT-mediated apoptosis. Am J
Pathol. 165:309–317. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin WW and Hsieh SL: Decoy receptor 3: A
pleiotropic immunomodulator and biomarker for inflammatory
diseases, autoimmune diseases and cancer. Biochem Pharmacol.
81:838–847. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wan X, Shi G, Semenuk M, Zhang J and Wu J:
DcR3/TR6 modulates immune cell interactions. J Cell Biochem.
89:603–612. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Siakavellas SI, Sfikakis PP and Bamias G:
The TL1A/DR3/DcR3 pathway in autoimmune rheumatic diseases. Semin
Arthritis Rheum. 45:1–8. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu Y, Han B, Sheng H, Lin M, Moore PA,
Zhang J and Wu J: Clinical significance of detecting elevated serum
DcR3/TR6/M68 in malignant tumor patients. Int J Cancer.
105:724–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Otsuki T, Tomokuni A, Sakaguchi H, Aikoh
T, Matsuki T, Isozaki Y, Hyodoh F, Ueki H, Kusaka M, Kita S, et al:
Over-expression of the decoy receptor 3 (DcR3) gene in peripheral
blood mononuclear cells (PBMC) derived from silicosis patients.
Clin Exp Immunol. 119:323–327. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bai C, Connolly B, Metzker ML, Hilliard
CA, Liu X, Sandig V, Soderman A, Galloway SM, Liu Q, Austin CP, et
al: Overexpression of M68/DcR3 in human gastrointestinal tract
tumors independent of gene amplification and its location in a
four-gene cluster. Proc Natl Acad Sci USA. 97:1230–1235. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ge Z, Sanders AJ, Ye L, Wang Y and Jiang
WG: Expression of death decoy receptor-3 (DcR3) in human breast
cancer and its functional effects on breast cancer cells in vitro.
J Exp Ther Oncol. 9:109–118. 2011.PubMed/NCBI
|
|
12
|
Tu HF, Liu CJ, Liu SY, Chen YP, Yu EH, Lin
SC and Chang KW: Serum decoy receptor 3 level: A predictive marker
for nodal metastasis and survival among oral cavity cancer
patients. Head Neck. 33:396–402. 2011.PubMed/NCBI
|
|
13
|
Bamias G, Gizis M, Delladetsima I, Laoudi
E, Siakavellas SI, Koutsounas I, Kaltsa G, Vlachogiannakos J,
Vafiadis-Zouboulis I, Daikos GL, et al: Elevated serum levels of
the antiapoptotic protein decoy-receptor 3 are associated with
advanced liver disease. Can J Gastroenterol Hepatol.
2016:26370102016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bamias G, Kaltsa G, Siakavellas SI,
Papaxoinis K, Zampeli E, Michopoulos S, Zouboulis-Vafiadis I and
Ladas SD: High intestinal and systemic levels of decoy receptor 3
(DcR3) and its ligand TL1A in active ulcerative colitis. Clin
Immunol. 137:242–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ao R, Du YQ, Wang Y, Chen YS and Wang BY:
MMP-2 and DcR3 expression in esophageal cancer tissue and
correlation with patient survival. Int J Clin Exp Med. 6:700–705.
2013.PubMed/NCBI
|
|
16
|
Hsu MJ, Lin WW, Tsao WC, Chang YC, Hsu TL,
Chiu AW, Chio CC and Hsieh SL: Enhanced adhesion of monocytes via
reverse signaling triggered by decoy receptor 3. Exp Cell Res.
292:241–251. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang CR, Hsieh SL, Ho FM and Lin WW: Decoy
receptor 3 increases monocyte adhesion to endothelial cells via
NF-kappa B-dependent up-regulation of intercellular adhesion
molecule-1, VCAM-1, and IL-8 expression. J Immunol. 174:1647–1656.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tateishi K, Miura Y, Hayashi S, Takahashi
M and Kurosaka M: DcR3 protects THP-1 macrophages from apoptosis by
increasing integrin alpha4. Biochem Biophys Res Commun.
389:593–598. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim S, Kotoula V, Hytiroglou P, Zardavas D
and Zhang L: Significance of increased expression of decoy receptor
3 in chronic liver disease. Dig Liver Dis. 41:591–598. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen G and Luo D: Expression of decoy
receptor 3 in liver tissue microarrays. Natl Med J India.
21:275–278. 2008.PubMed/NCBI
|
|
21
|
Nagar B, Overduin M, Ikura M and Rini JM:
Structural basis of calcium-induced E-cadherin rigidification and
dimerization. Nature. 380:360–364. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Benham-Pyle BW, Pruitt BL and Nelson WJ:
Cell adhesion. Mechanical strain induces E-cadherin-dependent Yap1
and β-catenin activation to drive cell cycle entry. Science.
348:1024–1027. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ishiyama N, Lee SH, Liu S, Li GY, Smith
MJ, Reichardt LF and Ikura M: Dynamic and static interactions
between p120 catenin and E-cadherin regulate the stability of
cell-cell adhesion. Cell. 141:117–128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Perl AK, Wilgenbus P, Dahl U, Semb H and
Christofori G: A causal role for E-cadherin in the transition from
adenoma to carcinoma. Nature. 392:190–193. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nagafuchi A, Shirayoshi Y, Okazaki K,
Yasuda K and Takeichi M: Transformation of cell adhesion properties
by exogenously introduced E-cadherin cDNA. Nature. 329:341–343.
1987. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Paul A, Danley M, Saha B, Tawfik O and
Paul S: PKCζ promotes breast cancer invasion by regulating
expression of E-cadherin and Zonula Occludens-1 (ZO-1) via
NFκB-p65. Sci Rep. 5:125202015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yu W, Xu YC, Tao Y, He P, Li Y, Wu T, Zhu
YP, Li J, Wu JX and Dai J: DcR3 regulates the growth and metastatic
potential of SW480 colon cancer cells. Oncol Rep. 30:2741–2748.
2013.PubMed/NCBI
|
|
29
|
Mild G, Bachmann F, Boulay JL, Glatz K,
Laffer U, Lowy A, Metzger U, Reuter J, Terracciano L, Herrmann R,
et al: DCR3 locus is a predictive marker for 5-fluorouracil-based
adjuvant chemotherapy in colorectal cancer. Int J Cancer.
102:254–257. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang Y, Luo J, He R, Huang W, Li Z, Li P,
Dang Y, Chen G and Li S: Expression and clinicopathological
implication of DcR3 in lung cancer tissues: A tissue microarray
study with 365 cases. Onco Targets Ther. 9:4959–4968. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zheng XF, Miao LY, Li S and Wang YB:
Promotive effects of Dcr3 gene on the occurrence and progression of
gastric cancer and its mechanism. Hepatogastroenterology.
61:880–884. 2014.PubMed/NCBI
|
|
32
|
Hsu TL, Chang YC, Chen SJ, Liu YJ, Chiu
AW, Chio CC, Chen L and Hsieh SL: Modulation of dendritic cell
differentiation and maturation by decoy receptor 3. J Immunol.
168:4846–4853. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu SF, Liu TM, Lin YC, Sytwu HK, Juan HF,
Chen ST, Shen KL, Hsi SC and Hsieh SL: Immunomodulatory effect of
decoy receptor 3 on the differentiation and function of bone
marrow-derived dendritic cells in nonobese diabetic mice: From
regulatory mechanism to clinical implication. J Leukoc Biol.
75:293–306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chang YC, Hsu TL, Lin HH, Chio CC, Chiu
AW, Chen NJ, Lin CH and Hsieh SL: Modulation of macrophage
differentiation and activation by decoy receptor 3. J Leukoc Biol.
75:486–494. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Weissinger D, Tagscherer KE,
Macher-Göppinger S, Haferkamp A, Wagener N and Roth W: The soluble
Decoy Receptor 3 is regulated by a PI3K-dependent mechanism and
promotes migration and invasion in renal cell carcinoma. Mol
Cancer. 12:1202013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chua HL, Bhat-Nakshatri P, Clare SE,
Morimiya A, Badve S and Nakshatri H: NF-kappaB represses E-cadherin
expression and enhances epithelial to mesenchymal transition of
mammary epithelial cells: Potential involvement of ZEB-1 and ZEB-2.
Oncogene. 26:711–724. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|