Introduction
Non-small cell lung cancer (NSCLC) is a leading
cause of cancer-related mortality worldwide. Overall 5-year
survival rate of NSCLC is lower than 5% (1,2). For
treatment of NSCLC, epidermal growth factor receptor-targeted
tyrosine kinase inhibitors (EGFR-TKIs) such as gefitinib, erlotinib
and afatinib have become first-line drugs for the NSCLC patients
harboring EGFR active mutations (3–5).
However, the insensitive subgroup of lung cancer cells can survive
under the EGFR-TKIs treatment. Thus, tumor recurrence and acquired
drug resistance is inevitable due to the repeated use of EGFR-TKIs
(6).
Recently, studies has demonstrated that cancer stem
cells (CSCs), which are a small subgroup of cells in tumor
possessing the ability of self-renewal, are capable of driving
tumorigenesis. Accumulating evidence indicated that lung cancer
stem cells are responsible for resistance to chemotherapeutics and
EGFR-TKIs (7–9). CSCs have become the target for
reversing the resistance of lung cancer to EGFR-TKIs.
MicroRNAs (miRNAs) are single-stranded RNAs with a
length of 21–25 nucleotides. They regulate cancer initiation and
progression by regulating the expression of cancer-related genes.
miRNAs usually act as negative regulators of gene expression by
binding to the mRNAs at the 3untranslated region (3′UTR) (10,11).
Accumulating evidence has indicated that aberrant miRNAs is
responsible to cancer survival, development and drug resistance
(12,13). In CSCs, it is reported that miRNAs
are usually dysregulated in various types of cancers. Correcting
the disorder of miRNAs in CSCs has become an important strategy for
cancer therapy (14–16). Previous research has identified
CD133 as a molecular marker of lung cancer stem cells (17). In the present study, we found that
the expression of miR-23a was significantly upregulated in CD133
positive PC9 CSCs. Inhibition of miR-23a increases the sensitivity
of lung cancer stem cells to erlotinib by upregulating the PTEN
expression.
Materials and methods
Cell culture and CSC isolation
The NSCLC cell line PC9 was purchased from the
American Type Culture Collection (ATCC; Manassas, VA, USA) and was
cultured in RPMI-1640 medium containing 10% fetal bovine serum
(FBS; Gibco, Waltham, MA, USA). For isolating CSCs of PC9 cell
line, FACSVantage (BD FACSCalibur; BD Biosciences, San Diego, CA,
USA) was used to identify and collect the CD133-positive PC9 cells
(CD133-FITC antibody was purchased from Miltenyi Biotec GmbH,
Bergisch Gladbach, Germany). To estimate the purity of CSC
population in PC9, flow cytometry was performed.
Transfection
Negative control oligonucleotides (miR-NC),
microRNA-23a mimics (miR-23a), miR-23a inhibitors (anti-miR-23a)
and PTEN siRNA were purchased from Guangzhou RiboBio, Co., Ltd.
(Guangzhou, China). For transfection, 50 pmol/ml miR-NC, miR-23a,
anti-miR-23a or PTEN siRNA were packaged with Lipofectamine 2000
(Invitrogen, Waltham, MA, USA). Then, PC9 CSCs were incubated with
RNAs in serum-free medium (Gibco). Six hours later, the culture
medium was changed to 10% FBS RPMI-1640 medium for 24 h.
Cell viability assay
PC9 CSCs (5×103) or PC9 non-CSCs
(5×103) were plated on 96-well plates with 200 µl
RPMI-1640 medium. Then, cells were transfected with RNAs followed
by treating with EGFR-TKIs for 48 h. After incubation, 20 µl
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
(5 mg/ml; Sigma-Aldrich, St. Louis, MO, USA) was added into the
medium for 4 h. Subsequently, we replaced the culture medium with
150 µl dimethyl sulfoxide (DMSO) for 30 min. Cell viability of PC9
CSCs and PC9 non-CSCs was evaluated according to the absorbance at
570 nm using a microplate reader. Half maximal inhibitory
concentration (IC50) of EGFR-TKIs to PC9 CSCs and PC9
non-CSCs was calculated according to the results of cell viability
assays.
Cell proliferation assays
PC9 CSCs (5×105) or PC9 non-CSCs
(5×105) were plated on 6-well plates with 2 ml RPMI-1640
medium. Then, cells were transfected with RNAs followed by treating
with 0.04 µM erlotinib for 48 h. After incubation, 3H-
thymidine was added into the culture medium for 6 h. Then,
3H-thymidine incorporation assays were performed to
determine the cell proliferation of PC9 CSCs and PC9 non-CSCs.
Colony forming assay
PC9 CSCs and PC9 non-CSCs were seeded at a density
of 50 cells/cm2 (185 cells/well) in a 12-well plate
format. Subsequently, cells were treated with 0.04 µM erlotinib for
10 days. After treatment, cell colonies were stained with crystal
violet/ethanol mixture and then were counted under a microscope.
The proportion of colonies formed is shown as a percentage of its
untreated control (18).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the PC9 CSCs and PC9
non-CSCs using TRIzol reagent (Invitrogen). Expression of miR-23a,
Oct4 and Sox2 was measured by One-Step RT-qPCR with SYBR Premix Ex
Taq (Takara Bio Inc., Otsu, Japan) according to the manufacturers
instructions. U6 snRNA was used as the internal control to
calculate the relative expression of miR-23a. GAPDH was used as the
internal control to calculate the relative expression of Oct4 and
Sox2. 2−∆∆CT method was used to analyze the results of
RT-qPCR (19).
Western blot analysis
Total proteins were extracted from the PC9 CSCs and
PC9 non-CSCs using RIPA buffer (Cell Signaling Technology, Beverly,
MA, USA) and then quantified by bicinchoninic acid assay kit
(Beyotime Institute of Biotechnology, Haimen, China). Proteins were
then separated by 12% sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE) and were transferred onto a PVDF
membrane (Millipore, Billerica, MA, USA). Membranes were then
blocked in 5% skim milk for 1 h at room temperature and then
incubated overnight at 4°C with the following antibodies:
anti-PTEN, anti-EGFR, anti-phosphorylated EGFR, anti-PI3K,
anti-phosphorylated PI3K, anti-AKT, anti-phosphorylated AKT,
anti-pro-caspase-9, anti-cleaved caspase-9, anti-pro-caspase-3,
anti-cleaved caspase-3 and anti-β-actin (Cell Signaling
Technology). After washing the membranes with Tris-buffered saline,
they were incubated with goat anti-rabbit secondary antibody (Cell
Signaling Technology) for 1 h at room temperature. The blots were
visualized using an enhanced chemiluminescence detection kit
(Pierce, Rockford, IL, USA).
Apoptosis analysis
PC9 CSCs (5×105) or PC9 non-CSCs
(5×105) were plated on 6-well plates with 2 ml RPMI-1640
medium. Then, cells were transfected with RNAs followed by treating
with 0.04 µM erlotinib for 48 h. After incubation, cells were
collected and were stained with PI and Annexin V (Sigma-Aldrich)
for 20 min at room temperature. Subsequently, percentage of
apoptotic cells was quantified using flow cyto-metry.
Luciferase reporter assay
3′UTR of PTEN mRNA was amplified and inserted into
pGL3 Luciferase reporter vectors (Promega, Madison, WI, USA)
downstream of the luciferase gene. PC9 CSCs (1×105) were
plated on 24-well plates with 500 µl RPMI-1640 medium. Then, cells
were transfected with 50 pmol/ml miR-23a mimics, 2 µg/ml firefly
luciferase reporters and 100 ng/ml Renilla luciferase pRL-TK
vector (Promega) for 24 h. After transfection, cells were collected
and washed with phosphate-buffered saline (PBS). Luciferase
reporter assays were then performed using the Dual-Luciferase
reporter assay system (Promega) according to the manufacturers
protocol.
Statistical analysis
Quantitative data are presented as the mean ± SD.
All of the experiments were repeated in triplicate. For comparison
analysis, the statistical analysis was performed by the Students
t-test using SPSS 15.0 software (SPSS; Chicago, IL, USA). A value
of P<0.05 was considered to indicate a statistically significant
difference.
Results
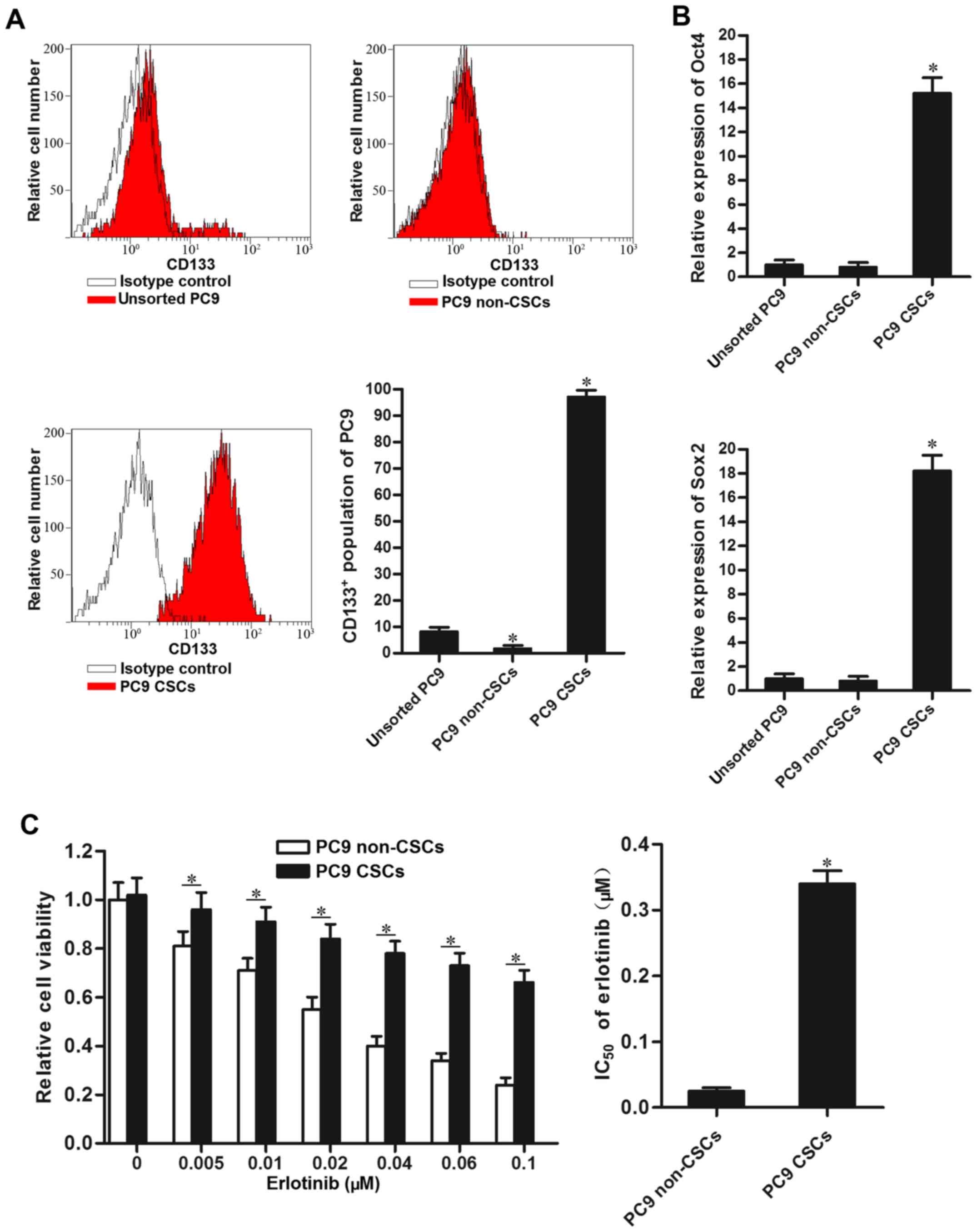
PC9 cancer stem cells show resistance
to erlotinib
To study the effect of EGFR-TKIs on lung cancer stem
cells, we isolated the CSCs population of PC9 using CD133 antibody.
Our separated PC9 CSCs expressed high level of CD133, and CD133
negative PC9 cell population was considered as the non-CSCs
(Fig. 1A). As Oct4 and Sox2 were
identified as the markers of CSCs (20), we detected their expression by using
RT-qPCR analysis. As shown in Fig.
1B, the CD133 positive PC9 cell population expressed
significantly higher level of Oct4 and Sox2 compared to the PC9
non-CSCs. Notably, we found that PC9 CSCs showed significant
resistance to erlotinib compared to the PC9 non-CSCs.
IC50 of erlotinib to PC9 CSCs is 12.6-fold higher than
the PC9 non-CSCs (Fig. 1C). The
results suggested that lung cancer stem cells are resistant to
EGFR-TKIs-induced cell death.
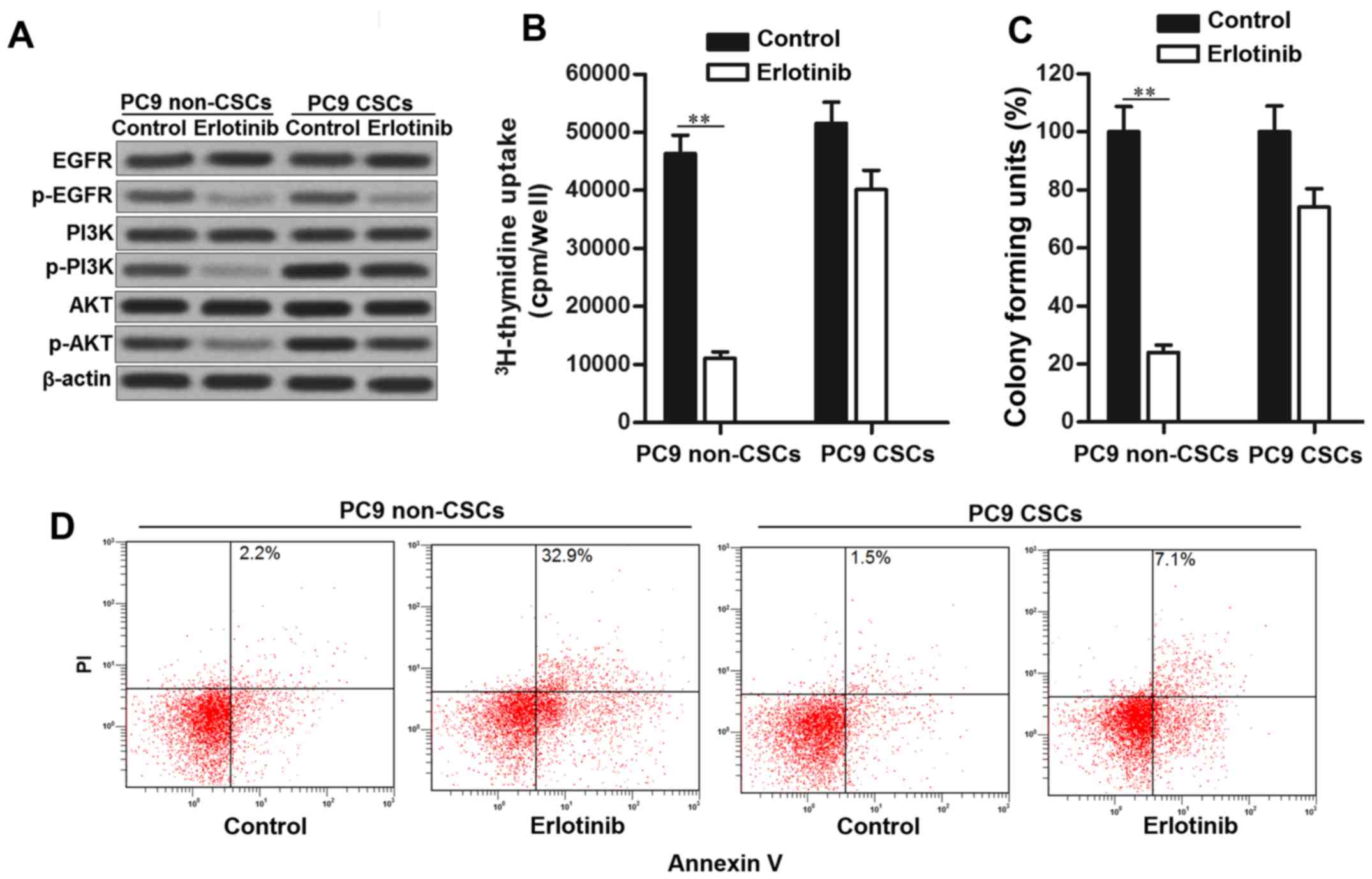
Erlotinib fails to suppress the
phosphorylation of PI3K and AKT in PC9 CSCs
Suppression of EGFR/PI3K/AKT pathway is the
mechanism of EGFR-TKIs in killing the NSCLC cells (21). Consistent with this, phosphorylation
of EGFR, PI3K and AKT was significantly inhibited in PC9 non-CSCs
treated with erlotinib. However, in PC9 CSCs, we observed that
erlotinib failed to suppress the activation of PI3K and AKT
obviously, despite inhibiting the EGFR phosphorylation. High
activation of PI3K and AKT promotes cell proliferation and inhibits
apoptosis in cancer cells (22,23).
Since erlotinib failed to suppress the PI3K/AKT pathway in PC9
CSCs, the results of 3H-thymidine incorporation assays
showed that PC9 CSCs still exhibited high proliferative activity
even when they were treated with erlotinib (Fig. 2B). Similarly, the effect of
erlotinib on suppressing the colony formation of PC9 CSCs was
significantly less than the PC9 non-CSCs (Fig. 2C). Furthermore, PC9 CSCs showed
obvious resistance to erlotinib-induced apoptosis rather than the
PC9 non-CSCs (Fig. 2D). Taken
together, we demonstrated that erlotinib failed to suppress the
activation of PI3K and AKT in lung cancer stem cells.
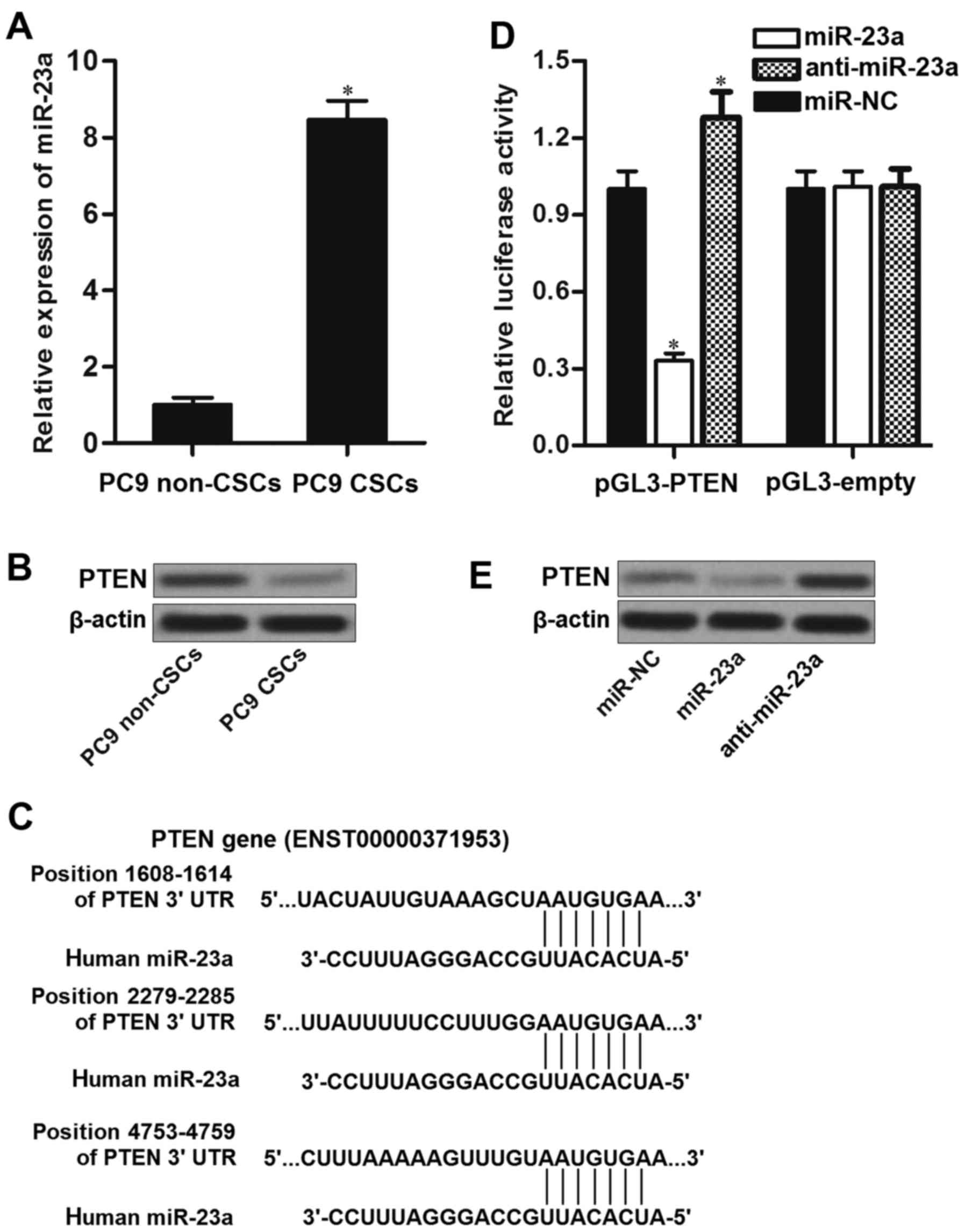
PTEN is the target of miR-23a in PC9
cancer stem cells
To investigate the mechanism by which erlotinib
failed to inhibit the PI3K/AKT pathway in PC9 CSCs, we detected the
miRNA expression in PC9 cell line. We found that the expression of
miR-23a was significantly upregulated in PC9 CSCs rather than PC9
non-CSCs (Fig. 3A). Furthermore,
PTEN, which is an effective suppressor of PI3K and AKT (22), was downregulated in PC9 CSCs
compared to the corresponding non-CSCs (Fig. 3B). Negative correlation between
miR-23a and PTEN was observed. As the public miRNA TargetScan
database (http://www.targetscan.org) as well as
miRanda database (http://www.microrna.org/) predicted that PTEN mRNA
3′UTR contained putative binding sites to miR-23a (Fig. 3C), we inferred that PTEN is the
target of miR-23a, and overexpression of miR-23a in PC9 CSCs was
responsible for their erlotinib resistance. To confirm the
relationship between miR-23a and PTEN, we performed luciferase
reporter assays. The results of these assays showed that
transfection with miR-23a in PC9 CSCs significantly decreased the
activities of luciferase in pGL3-PTEN plasmid. In contrast,
introduction with anti-miR-23a was able to increase the activities
of luciferase in pGL3-PTEN vector (Fig.
3D). These results implied the effect of miR-23a on regulating
the PTEN gene. In addition, we observed that transfection with
miR-23a decreased the protein level of PTEN, whereas the miR-23a
antisense oligonucleotides (anti-miR-23a) significantly increased
the expression level of PTEN in PC9 CSCs (Fig. 3E). Taken together, we proved that
miR-23a regulated the expression of PTEN in lung cancer stem
cells.
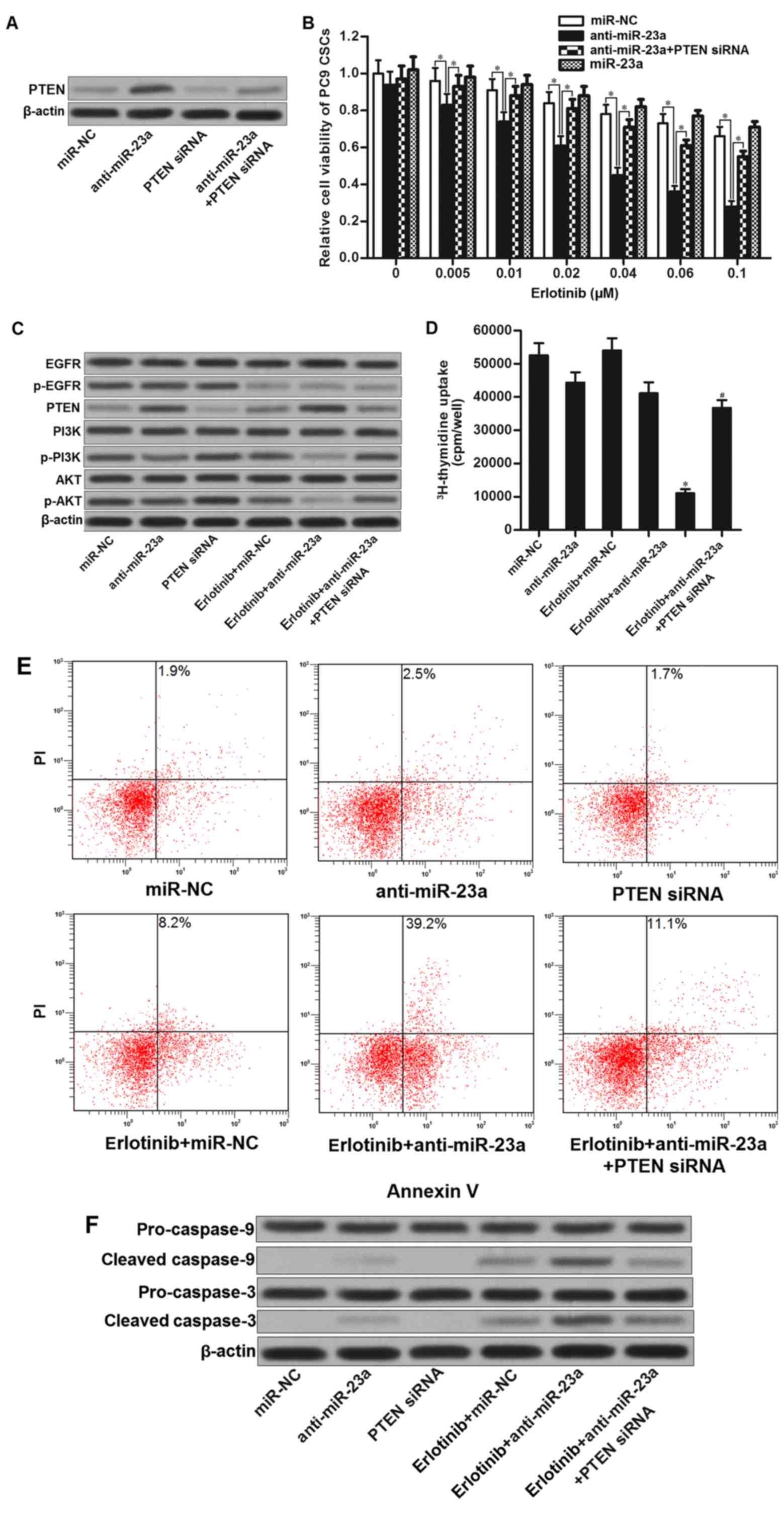
miR-23a antisense oligonucleotides
resensitize PC9 CSCs to erlotinib by increasing the expression of
PTEN
The preceding results demonstrated that miR-23a
which was overexpressed in PC9 CSCs decreased the PTEN expression.
We therefore investigated whether knockdown of miR-23a enhances the
antitumor effect of erlotinib on PC9 CSCs. Results of western blot
analysis illustrated that transfection with miR-23a antisense
oligonucleotides (anti-miR-23a) significantly increased the
expression of PTEN, whereas the PTEN siRNA abolished the effect of
anti-miR-23a (Fig. 4A). Next, we
found that miR-23a antisense oligonucleotides significantly
enhanced the effect of erlotinib on killing PC9 CSCs. However,
co-transfection with PETN siRNA obviously impaired the effect of
anti-miR-23a (Fig. 4B). These
results proved the synergistic effects of miR-23a antisense
oligonucleotides on EGFR-TKIs. Mechanically, although erlotinib
alone treatment inhibited the EGFR signaling, absence of PTEN in
PC9 CSCs still induced the activation of PI3K and AKT. By contrast,
combination with erlotinib and anti-miR-23a inhibited the
phosphorylation of EGFR, and upregulated the expression of PTEN.
Phosphorylation of PI3K and AKT was significantly suppressed
(Fig. 4C). As cell proliferation
and apoptosis were regulated by PI3K/AKT pathway (22–24),
we observed that combination with miR-23a antisense
oligonucleotides and erlotinib induced significant inhibition of
cell proliferation in PC9 CSCs (Fig.
4D). Moreover, anti-miR-23a promoted erlotinib-induced
apoptosis and cleavage of caspase-9 and caspase-3 by upregulating
the expression of PTEN in PC9 CSCs (Fig. 4E and F). Taken together, we
demonstrated that miR-23a antisense oligonucleotides resensitize
lung cancer stem cells to erlotinib by increasing the expression of
PTEN.
miR-23a antisense oligonucleotides
enhance the antitumor effect of other EGFR-TKIs
To investigate the effect of anti-miR-23a on other
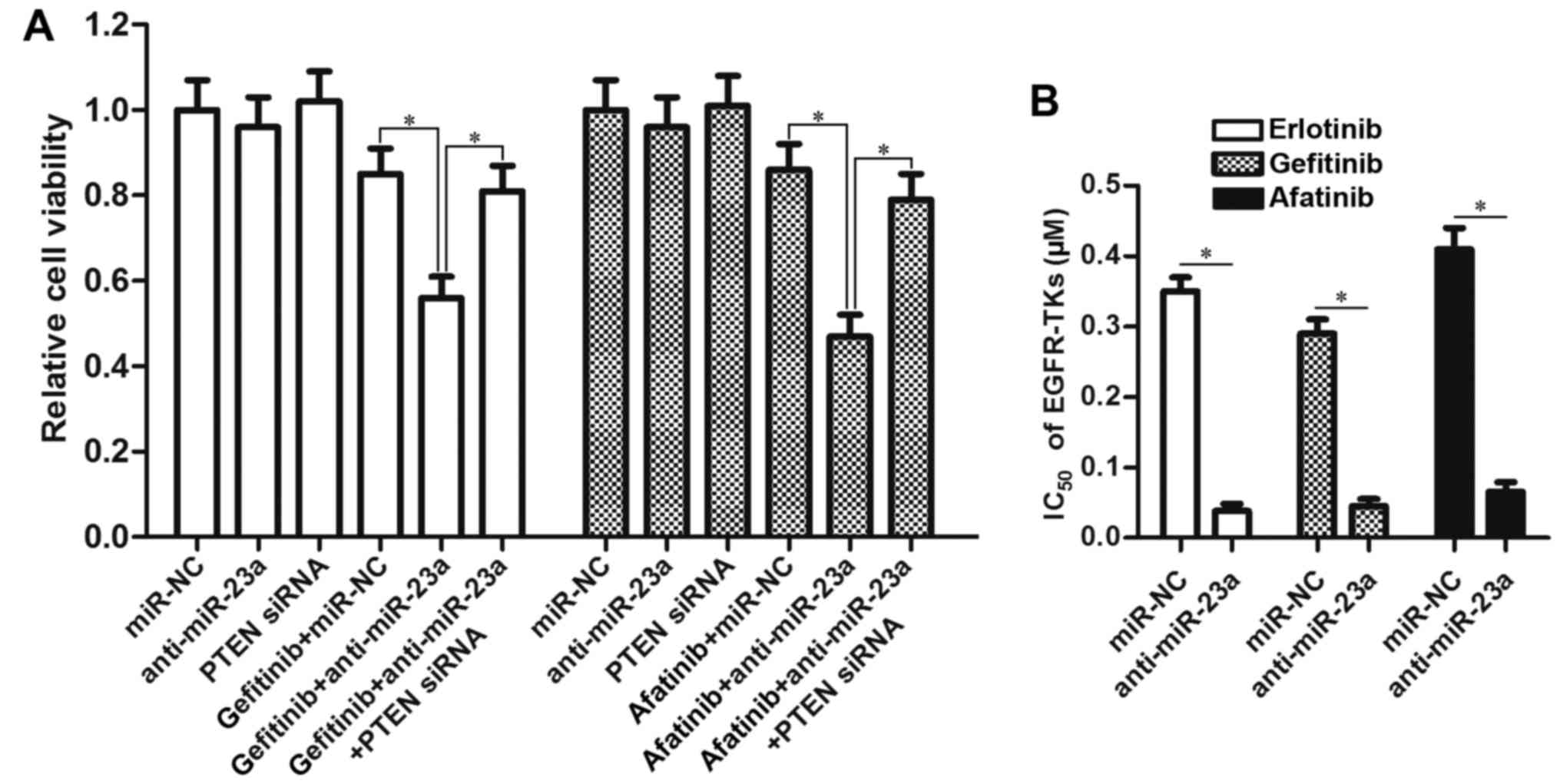
EGFR-TKIs, we treated the PC9 CSCs with gefitinib and afatinib
after transfection with anti-miR-23a and PTEN siRNA. The results of
MTT demonstrated that knockdown of miR-23a also enhanced the
antitumor effect of gefitinib and afatinib by upregulating the
expression of PTEN (Fig. 5A).
Intuitively, transfection with miR-23a antisense oligonucleotides
significantly decreased the IC50 of all these EGFR-TKIs
(gefitinib, erlotinib and afatinib) to PC9 CSCs (Fig. 5B). We therefore demonstrated the
effect of miR-23a antisense oligonucleotides on resensitizing lung
cancer stem cells to EGFR-TKIs.
Discussion
Approximately 25% NSCLC patients exhibit mutation
for epidermal growth factor receptor (EGFR). For these NSCLC
patients, the cancer cells are sensitive to EGFR-TKIs such as
erlotinib, gefitinib and afatinib. As the treatment of EGFR-TKIs is
the targeted molecular therapy without serious side-effects for
NSCLC patients, EGFR-TKIs have become the first-line therapy for
NSCLC (4,5,25,26).
However, almost all patients become resistant to EGFR-TKIs
following 8–10 months of treatment (27,28).
Recent research has emphasized the importance of cancer stem cells
(CSCs) for the acquired drug-resistance in cancers. Due to the high
tumorigenicity and self-renewal, surviving CSCs in NSCLC inevitably
become EGFR-TKIs resistant (29).
In the present study, we found that the IC50 of
erlotinib to PC9 CSCs was significantly higher than the PC9
non-CSCs. We demonstrated that lung cancer stem cells are
insensitive to EGFR-TKIs.
MicroRNA-23a is reported to function as an oncogene
in various human cancers. In NSCLC, high expression level of
miR-23a shows poor prognosis (30).
Furthermore, increasing evidence has declared the relationship
between miRNAs and drug-resistance in cancer cells (31,32).
In the present study, we found that the expression level of miR-23a
was significantly upregulated in PC9 CSCs rather than their
corresponding non-CSCs. For reducing the drug-resistance of lung
cancer stem cells to erlotinib, we knocked down the miR-23a by its
antisense oligonucleotides in PC9 CSCs. Interestingly, we found the
miR-23a antisense oligonucleotides can resensitize PC9 CSCs to
erlotinib-induced cell death and apoptosis. Thus, for the first
time, we reported the effect of anti-miR-23a on reversing the
resistance of EGFR-TKIs by targeting lung cancer stem cells.
Phosphatase and tensin homologue (PTEN) is an
important regulator for suppressing tumorigenesis and cancer
development. PTEN mutation or loss induced uncontrolled cell cycle
(33,34). Among the pathways downstream of
PTEN, PI3K/AKT whose phosphorylation is inhibited by PTEN regulates
cell proliferation and apoptosis in cancer cells (35,36).
Recent studies demonstrate that expression of PTEN is regulated by
certain miRNAs in cancers (37). In
this study, we found that erlotinib failed to inhibit the
phosphorylation of PI3K and AKT in PC9 CSCs. We indicate that this
phenomenon is caused by the low-expression of PTEN in lung cancer
stem cells. However, introduction with miR-23a antisense
oligonucleotides increase the expression of PTEN in lung cancer
stem cells. Therefore, the PC9 CSCs recover the sensitivity to
erlotinib.
In conclusion, we demonstrated that lung cancer stem
cells express low level of PTEN. As PTEN is targeted by miR-23a, we
proved that introduction with miR-23a antisense oligonucleotides
resensitize lung cancer stem cells to EGFR-TKIs by suppressing
PI3K/AKT pathway. Combination of miR-23a antisense oligonucleotides
and EGFR-TKIs could be attractive strategy for treatment of
NSCLC.
Acknowledgements
Thanks are due to all the contributors who assisted
with this study.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Allemani C, Weir HK, Carreira H, Harewood
R, Spika D, Wang XS, Bannon F, Ahn JV, Johnson CJ, Bonaventure A,
et al CONCORD Working Group, : Global surveillance of cancer
survival 1995–2009: Analysis of individual data for 25,676,887
patients from 279 population-based registries in 67 countries
(CONCORD-2). Lancet. 385:977–1010. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sequist LV, Martins RG, Spigel D, Grunberg
SM, Spira A, Jänne PA, Joshi VA, McCollum D, Evans TL, Muzikansky
A, et al: First-line gefitinib in patients with advanced
non-small-cell lung cancer harboring somatic EGFR mutations. J Clin
Oncol. 26:2442–2449. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Larsen JE and Minna JD: Molecular biology
of lung cancer: Clinical implications. Clin Chest Med. 32:703–740.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Riely GJ, Pao W, Pham D, Li AR, Rizvi N,
Venkatraman ES, Zakowski MF, Kris MG, Ladanyi M and Miller VA:
Clinical course of patients with non-small cell lung cancer and
epidermal growth factor receptor exon 19 and exon 21 mutations
treated with gefitinib or erlotinib. Clin Cancer Res. 12:839–844.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jiang J, Feng X, Zhou W, Wu Y and Yang Y:
MiR-128 reverses the gefitinib resistance of the lung cancer stem
cells by inhibiting the c-met/PI3K/AKT pathway. Oncotarget.
7:73188–73199. 2016.PubMed/NCBI
|
|
7
|
Sharma SV, Lee DY, Li B, Quinlan MP,
Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach
MA, et al: A chromatin-mediated reversible drug-tolerant state in
cancer cell subpopulations. Cell. 141:69–80. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dean M, Fojo T and Bates S: Tumour stem
cells and drug resistance. Nat Rev Cancer. 5:275–284. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kobayashi I, Takahashi F, Nurwidya F, Nara
T, Hashimoto M, Murakami A, Yagishita S, Tajima K, Hidayat M,
Shimada N, et al: Oct4 plays a crucial role in the maintenance of
gefitinib-resistant lung cancer stem cells. Biochem Biophys Res
Commun. 473:125–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gargalionis AN and Basdra EK: Insights in
microRNAs biology. Curr Top Med Chem. 13:1493–1502. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun Y, He N, Dong Y and Jiang C:
MiR-24-BIM-Smac/DIABLO axis controls the sensitivity to doxorubicin
treatment in osteosarcoma. Sci Rep. 6:342382016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ye Z, Hao R, Cai Y, Wang X and Huang G:
Knockdown of miR-221 promotes the cisplatin-inducing apoptosis by
targeting the BIM-Bax/Bak axis in breast cancer. Tumour Biol.
37:4509–4515. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chai S, Ng KY, Tong M, Lau EY, Lee TK,
Chan KW, Yuan YF, Cheung TT, Cheung ST, Wang XQ, et al: Octamer
4/microRNA-1246 signaling axis drives Wnt/β-catenin activation in
liver cancer stem cells. Hepatology. 64:2062–2076. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang H, Meng Y, Cui Q, Qin F, Yang H, Chen
Y, Cheng Y, Shi J and Guo Y: MiR-101 targets the EZH2/Wnt/β-catenin
the pathway to promote the osteogenic differentiation of human bone
marrow-derived mesenchymal stem cells. Sci Rep. 6:369882016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Feng X, Jiang J, Shi S, Xie H, Zhou L and
Zheng S: Knockdown of miR-25 increases the sensitivity of liver
cancer stem cells to TRAIL-induced apoptosis via PTEN/PI3K/Akt/Bad
signaling pathway. Int J Oncol. 49:2600–2610. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee SO, Yang X, Duan S, Tsai Y, Strojny
LR, Keng P and Chen Y: IL-6 promotes growth and
epithelial-mesenchymal transition of CD133+ cells of
non-small cell lung cancer. Oncotarget. 7:6626–6638. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
French R, Hayward O, Jones S, Yang W and
Clarkson R: Cytoplasmic levels of cFLIP determine a broad
susceptibility of breast cancer stem/progenitor-like cells to
TRAIL. Mol Cancer. 14:2092015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Murakami A, Takahashi F, Nurwidya F,
Kobayashi I, Minakata K, Hashimoto M, Nara T, Kato M, Tajima K,
Shimada N, et al: Hypoxia increases gefitinib-resistant lung cancer
stem cells through the activation of insulin-like growth factor 1
receptor. PLoS One. 9:e864592014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gadgeel SM and Wozniak A: Preclinical
rationale for PI3K/Akt/mTOR pathway inhibitors as therapy for
epidermal growth factor receptor inhibitor-resistant non-small-cell
lung cancer. Clin Lung Cancer. 14:322–332. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang T, Gong X, Jiang R, Li H, Du W and
Kuang G: Ferulic acid inhibits proliferation and promotes apoptosis
via blockage of PI3K/Akt pathway in osteosarcoma cell. Am J Transl
Res. 8:968–980. 2016.PubMed/NCBI
|
|
23
|
Wang R, Zhang Q, Peng X, Zhou C, Zhong Y,
Chen X, Qiu Y, Jin M, Gong M and Kong D: Stellettin B induces G1
arrest, apoptosis and autophagy in human non-small cell lung cancer
A549 cells via blocking PI3K/Akt/mTOR pathway. Sci Rep.
6:270712016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu YR, Qi HJ, Deng DF, Luo YY and Yang SL:
MicroRNA-21 promotes cell proliferation, migration, and resistance
to apoptosis through PTEN/PI3K/AKT signaling pathway in esophageal
cancer. Tumour Biol. 37:12061–12070. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sharma SV, Bell DW, Settleman J and Haber
DA: Epidermal growth factor receptor mutations in lung cancer. Nat
Rev Cancer. 7:169–181. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu HA and Pao W: Targeted therapies:
Afatinib - new therapy option for EGFR-mutant lung cancer. Nat Rev
Clin Oncol. 10:551–552. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chong CR and Jänne PA: The quest to
overcome resistance to EGFR-targeted therapies in cancer. Nat Med.
19:1389–1400. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Murakami A, Takahashi F, Nurwidya F,
Kobayashi I, Minakata K, Hashimoto M, Nara T, Kato M, Tajima K,
Shimada N, et al: Hypoxia increases gefitinib-resistant lung cancer
stem cells through the activation of insulin-like growth factor 1
receptor. PLoS One. 9:e864592014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qu WQ, Liu L and Yu Z: Clinical value of
microRNA-23a upregulation in non-small cell lung cancer. Int J Clin
Exp Med. 8:13598–13603. 2015.PubMed/NCBI
|
|
31
|
Zhou S, Huang Q, Zheng S, Lin K, You J and
Zhang X: miR-27a regulates the sensitivity of breast cancer cells
to cisplatin treatment via BAK-SMAC/DIABLO-XIAP axis. Tumour Biol.
37:6837–6845. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yin W, Nie Y, Zhang Z, Xie L and He X:
miR-193b acts as a cisplatin sensitizer via the caspase-3-dependent
pathway in HCC chemotherapy. Oncol Rep. 34:368–374. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang YS, Wang YH, Xia HP, Zhou SW,
Schmid-Bindert G and Zhou CC: MicroRNA-214 regulates the acquired
resistance to gefitinib via the PTEN/AKT pathway in EGFR-mutant
cell lines. Asian Pac J Cancer Prev. 13:255–260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Darido C, Georgy SR, Wilanowski T, Dworkin
S, Auden A, Zhao Q, Rank G, Srivastava S, Finlay MJ, Papenfuss AT,
et al: Targeting of the tumor suppressor GRHL3 by a
miR-21-dependent proto-oncogenic network results in PTEN loss and
tumorigenesis. Cancer Cell. 20:635–648. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Carnero A, Blanco-Aparicio C, Renner O,
Link W and Leal JF: The PTEN/PI3K/AKT signalling pathway in cancer,
therapeutic implications. Curr Cancer Drug Targets. 8:187–198.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ma Y, Zhang P, Gao Y, Fan H, Zhang M and
Wu J: Evaluation of AKT phosphorylation and PTEN loss and their
correlation with the resistance of rituximab in DLBCL. Int J Clin
Exp Pathol. 8:14875–14884. 2015.PubMed/NCBI
|
|
37
|
Wang J, Xu J, Fu J, Yuan D, Guo F, Zhou C
and Shao C: MiR-29a regulates radiosensitivity in human intestinal
cells by targeting PTEN gene. Radiat Res. 186:292–301. 2016.
View Article : Google Scholar : PubMed/NCBI
|