Introduction
EYK, a herbal plant-based extract derived from
Epimedium koreanum Nakai, consists of multiple elements
including anhydroicaritin-3-O-α-L-rhamnopyranoside, icariin,
icariside II quercetin, epimedin A, epimedin B, ikarisoside,
b-sitosterol, daucosterol, campesterol, epimediphine and
chlorogenic acid (1). Icariin,
acting through the phosphoinositide 3-kinase and protein kinase B
(PI3/AKT) and nuclear factor-kappa B (NF-κB) pathways, inhibits
lipopolysaccharide (LPS)-induced lung inflammation (2). EYK decreases the abundance of
β-amyloid plaque (Aβ) in APP transgenic mice (3) and inhibits adipocyte differentiation
by downregulating adipogenic transcription factors in 3T3-L1, a
murine preadipocyte cell line (4).
Another constituent, icariside II, promotes terminal
deoxynucleotidyl transferase dUTP nick end labeling
(TUNEL)-positive apoptosis in PC-3 prostate carcinoma cells
(5) and exerts antitumor activity
in human adenocarcinoma alveolar basal epithelial A549 cells via
the mitochondria apoptotic pathway, including bcl2-related X
protein/B-cell lymphoma 2 (BAX/Bcl-2) as well as the caspase
signaling pathway (6). EYK also has
antiviral activity against porcine epidemic diarrhea virus (PEDV),
which induces diarrhea in adult pigs and suppresses viral
replication in both PEDV-infected cell lines and piglets (7). Thus, EYK extracts confer beneficial
effects in various diseases, including human lung cancer (6), prostate carcinoma (5), myeloid acute leukemia (8), angiogenesis (9), Alzheimers disease (3) and inflammatory conditions (2). However, it is still not clear whether
EYK can affect cancer cell metastasis in human glioblastoma.
Human glioblastoma multiforme (GBM) is a type of
brain cancer that arises from astrocytes. Brain cancer is
accompanied by symptoms such as seizure, abnormal behavior and
memory deficits. Currently, brain tumors are treated by surgery,
radiation therapy and chemotherapy with temozolomide (TMZ).
However, this regimen can also damage healthy tissues (10) and some cases of GBM are resistant to
TMZ therapy (11,12). TMZ is an anti-neoplastic agent that
delivers methyl groups to purine bases (guanine at N7 and O6 and
adenine at N3) in DNA of cancer cells, inducing DNA damage and
ultimately leading to apoptosis and cytotoxicity. Clinical studies
showed that TMZ induces side-effects such as myelosuppression,
nausea and vomiting (13). Thus, a
new anti-brain cancer drug is necessary for the successful
treatment and cure of GBM.
In the present study, we examined the effect of EYK
on brain cancer cell migration and invasion. We found that pre- or
post-treatment with EYK inhibited cancer cell migration and
invasionin monomorphic malignant human glioma cells. We observed
that EYK required matrix metalloprotease-9 (MMP-9) activity to
inhibit phorbol 12-myristate 13-acetate (PMA)-induced cancer cell
migration. In addition, pre- or post-treatment with EYK regulated
NF-κB nuclear translocation in PMA-induced A172 cells. Moreover,
treatment with NF-κB inhibitor followed by EYK significantly
suppressed cell migration in PMA-induced A172 cells in comparison
with treatment with PMA and NF-κB inhibitor or PMA and EYK. Taken
together, our results suggest that EYK inhibits cell migration in
monomorphic malignant human glioma cells by downregulating the
NF-κB nuclear translocation and thereby decreasing MMP-9
activity.
Materials and methods
Preparation of Epimedium koreanum
Nakai
Epimedium koreanum Nakai (EYK) was as
described in our previous studies (7). Briefly, the dried bark of the plant
was obtained from a domestic Korean market (Kyung Dong Crude Drugs
Market, Seoul, Korea) and boiled in distilled water for 2.5 h at
105°C; the resultant suspension was filtered and lyophilized. The
powder was stored at 4°C and the extract was dissolved in
phosphate-buffered saline (PBS) and diluted with medium before each
experiment.
Cell lines and culture conditions
A172, U373MG and T98G cells (human brain cancer
cells; Korean Cell Line Bank, Seoul, Korea) were maintained in
RPMI-1640 medium (HyClone Laboratories, Logan, UT, USA)
supplemented with 10% fetal bovine serum (FBS; HyClone
Laboratories) in a 5% CO2 incubator.
Antibodies and inhibitors
We used the following primary antibodies: mouse
β-actin (Santa Cruz Biotechnology, Dallas, TX, USA), mouse
anti-p-IκBα (B-9, Ser32; Santa Cruz Biotechnology), rabbit
anti-IκBα (Santa Cruz Biotechnology), rabbit anti-NF-κB (P65; Santa
Cruz Biotechnology), PCNA (Abcam, Cambridge, MA, USA), rabbit
anti-ERK (Cell Signaling Technology, Danvers, MA, USA), rabbit
anti-p-ERK (Thr202/Tyr204; Cell Signaling Technology) and rabbit
anti-p-NF-κB (Ser536; Cell Signaling Technology). The following
secondary antibodies and dilutions were used: horseradish
peroxidase (HRP)-conjugated anti-mouse and rabbit IgG (Bethyl
Laboratories, Montgomery, TX, USA). Phorbol 12-myristate 13-acetate
(PMA), MMP-9 inhibitor (CAS1177749-58-4) and NF-κB inhibitor (BAY
11-7085) were purchased from Sigma-Aldrich (St. Louis, MO,
USA).
Western blotting
Western blot analysis was performed as previously
described (14). Briefly, A172,
U373MG and T98G cells were harvested in ice-cold lysis buffer (50
mM Tris-Cl, 150 mM NaCl, 1% NP-40) containing protease and
phosphatase inhibitor (Roche Diagnostics, Indianapolis, IN, USA).
Proteins from cell extracts were separated under denatured and
reduced conditions using Tris-glycine polyacrylamide gel
electrophoresis. Separated proteins were transferred onto
polyvinylidene difluoride membrane (PVDF; EMD Millipore, Temecula,
CA, USA) at 110 V for 1 h and blocked with 5% skim milk or 5%
bovine serum albumin (BSA) in 0.1% TBS-T (Tris-buffered saline with
0.1% Tween-20) for 1 h at room temperature. The blots were
incubated overnight at 4°C with specific primary antibodies: mouse
β-actin (1:5,000; Santa Cruz Biotechnology), mouse anti-p-IκBα
(Ser32, 1:500; Santa Cruz Biotechnology), rabbit anti-IκBα
(1:1,000; Santa Cruz Biotechnology), rabbit anti-NF-κB (P65,
1:1,000; Santa Cruz Biotechnology), rabbit anti-ERK (Cell Signaling
Technology), rabbit anti-p-ERK (Thr202/Tyr204; Cell Signaling
Technology), and PCNA (1:1,000; Abcam). HRP-conjugated secondary
antibody was visualized by enhanced chemiluminescence detection
reagents (ECL; ATTO Corp., Tokyo, Japan) and image were captured
using chemiluminescence imaging system (Vilber Lourmat Deutschland
GmbH, Eberhardzell, Germany). Equal amount of protein (10 mg) were
used for immunoblotting and all blot images were analyzed using
either Fusion software or ImageJ.
MTT assays
To measure cell viability, A172, U373MG and T98G
cells were seeded in 96-well plates and treated for 24 h with
vehicle or EYK at various concentrations (1, 10, 100, 200, or 400
µg/ml) in the absence of serum. After 24 h, cells were treated with
0.5 mg/ml MTT and incubated for 3 h at 37°C in a 5% CO2
incubator. Plates were read by measuring absorbance at 580 nm.
Cell migration (wound healing
assay)
A172, U373MG and T98G cells were 80% confluence, and
then the monolayer of cells was scratched using a fine pipette tip
and immediately imaged (0 h). The cells were pre-treated with
vehicle or PMA (75 nM) for 45 min, and then treated with vehicle or
EYK (200 µg/ml) for 24 h. Images of the wound gap were acquired
immediately (0 h) and 24 h after (24 h) scratching and gap width
was measured using ImageJ.
Transwell invasion assay
Corning Matrigel® was loaded onto the
membrane of a culture insert (Corning Transwell System; Corning
Inc., Corning, NY, USA) and incubated at 37°C for 30 min in a 5%
CO2 incubator. A172 cells (1×105/ml) were
plated onto the Matrigel-coated insert in the upper chamber, which
contained serum-free RPMI-1640, and then treated with vehicle, EYK
(200 µg/ml) or PMA (75 nM). RPMI-1640 media containing 10% FBS was
added to the lower chamber. After 24 h, the cells were fixed in
methanol (100%) and stained with hematoxylin-eosin (Sigma-Aldrich).
The membrane was cut and mounted on a glass microscope slide.
Images were randomly acquired (two or three images from each
sample) on aninverted microscope (Carl Zeiss, Oberkochen,
Germany).
Gelatin zymography assay
To measure MMP activity, A172 cells were pre-treated
with vehicle or PMA (75 nM) for 45 min, and then treated for 24 h
with vehicle or EYK (200 µg/ml). After 24 h, the conditioned medium
was collected and analyzed on a 10% acrylamide gel containing 0.1%
(w/v) gelatin. Following electrophoresis, the gel was washed three
times for 10 min each with 2.5% Triton X-100, and then incubated
overnight in zymography incubation buffer (2.5 mM CaCl2,
200 mM NaCl, 50 mM Tris-HCl, pH 7.5). The next day, the gel was
stained with 0.2% Coomassie brilliant blue solution R-250 for 1 h,
and then washed three times for 10 min each in destaining buffer
(10% MeOH, 10% acetic acid in distilled water). Gel images were
acquired on a light box (Vilber Lourmat, Seoul, Korea) and analyzed
using Image Gauge V4.0 (Fujifilm, Tokyo, Japan).
Cytosolic and nuclear
fractionation
A172 cells were lysed on ice for 5 min in buffer A
[10 mM HEPES (pH 8.0), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 300 mM
sucrose, 0.1% NP-40 and 0.5 mM PMSF]. After 5 min, the cell lysates
were centrifuged at 10,000 rpm at 4°C for 1 min, and the
supernatant was collected as the cytosolic fraction. Then, the cell
pellet was lysed on ice for 15 min in buffer B [20 mM HEPES (pH
8.0), 20% glycerol, 100 mM KCl, 100 mM NaCl, 0.2 mM EDTA, 0.5 mM
DTT and 0.5 mM PMSF] and the cell lysates were centrifuged at
15,000 rpm at 4°C for 20 min. The supernatant was collected as the
nuclear fraction.
RT-PCR
To examine the effects of EYK on MMP-9 and
TIMP-1 mRNA levels, A172 cells were pre-treated with vehicle
or PMA (75 nM) for 45 min, and then treated for 24 h with vehicle
or EYK (200 µg/ml). After 24 h, RNA was extracted using TRIzol
(Ambion, Inc., Austin, TX, USA). RT-PCR was performed using the
following primers: MMP-9: 5-CGG AGC ACG GAG ACG GGT AT and
3-TGA AGG GGA AGA CGC ACA GC; TIMP-1: 5-AGC GCC CAG AGA GAC
ACC and 3-CCA CTC CGG GCA GGA TT; GAPDH: 5-GTT ACC AGG GCT
GCC TTC TC and 3-GTG ATG GCA TGG ACT GTG GT. Image analyses were
performed using the Fusion or ImageJ software to measure average
band intensities.
Immunocytochemistry
A172 and U373MG cells were fixed for 8 min using ice
cold 100% methanol, washed with 1X PBS, and then incubated with the
following primary antibodies overnight: rabbit anti-p-NF-κB
(Ser536, 1:100; Cell Signaling Technology), mouse anti-β-actin
(1:200; Santa Cruz Biotechnology) in GDB buffer (0.1% gelatin, 0.3%
Triton X-100, 16 mM sodium phosphate pH 7.4 and 450 mM NaCl)
overnight at 4°C. The next day, cells were washed with 1X PBS three
times and incubated with the following secondary antibodies in GDB
solution for 1 h at room temperature: anti-rabbit-AlexaFluor 488
and anti-mouse-AlexaFluor 555 (1:200; Molecular Probes, Eugene, OR,
USA) and cell covered using DAPI containing mount solution (Vector
Laboratories, Burlingame, CA, USA). Images were taken on a single
plane using a confocal microscope (Nikon) and analyzed using ImageJ
software.
Statistical analyses
All data were analyzed by either two-tailed t-test
or ANOVA using the GraphPad Prism 4 software. Post hoc analyses
were completed with the Tukeys multiple comparison test with
P<0.05 considered to represent significance (P<0.05,
P<0.01, P<0.001).
Results
EYK inhibits PMA-induced cell
migration in A172 cells
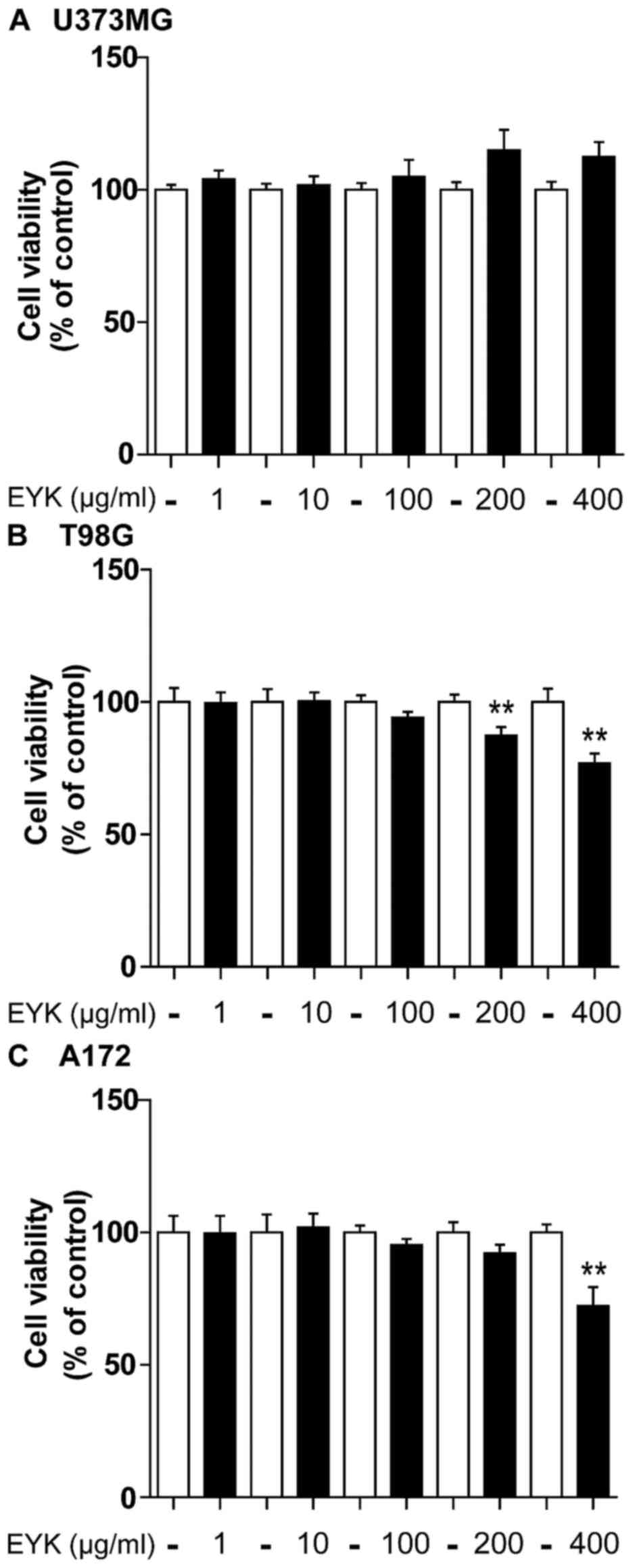
To test the effects of EYK on cell viability in
malignant human tumor cells, we treated U373MG, T98G and A172 cells
with vehicle or EYK (1, 10, 100, 200 or 400 µg/ml) for 24 h and
then conducted MTT assays. In U373MG cells, EYK did not influence
cell viability at any doses (Fig.
1A). In T98G cells, EYK slightly decreased cell viability at
the highest doses, by 13 and 23% at 200 and 400 µg/ml, respectively
(Fig. 1B, P<0.01; two-tailed
t-test). In A172 cells, EYK caused no toxicity up to a
concentration of 200 µg/ml, but had some toxicity at 400 µg/ml
(Fig. 1C, P<0.01; two-tailed
t-test). Based on these findings, we selected a working
concentration of 200 µg/ml for the following experiments.
To determine whether EYK can regulate PMA-induced
brain tumor cell migration, we initially optimized the dose and
timing for treatment with PMA, a cancer inducer (15), in our culture system. For these
experiments, U373MG, T98G and A172 cells were grown in a monolayer,
scratched with a pipette tip, and treated with vehicle or PMA (25,
50, 75, 100 or 125 nM) for 24 h. At 0 h (i.e., immediately after
scratching) and 24 h, we acquired images of the wound gap at both
time-points and measured the proportion of cells migrated from 0 h
to 24 h at different PMA concentrations. Subtracting the surface
area at 0–24 h with vehicle treatment were listed as 100%, and
difference between the areas at 0–24 h at various PMA
concentrations were compared with vehicle treatment. PMA
dramatically increased cell migration relative to the vehicle at
all doses tested in U373MG (Fig. 2A and
D, P<0.01; P<0.001; ANOVA with post hoc Tukeys test),
T98G (Fig. 2B and E, P<0.001,
ANOVA with post hoc Tukeys test), and A172 cells (Fig. 2C and F, P<0.01, P<0.001, ANOVA
with post hoc Tukeys test), with an optimal concentration at 75 nM
in all three cell lines.
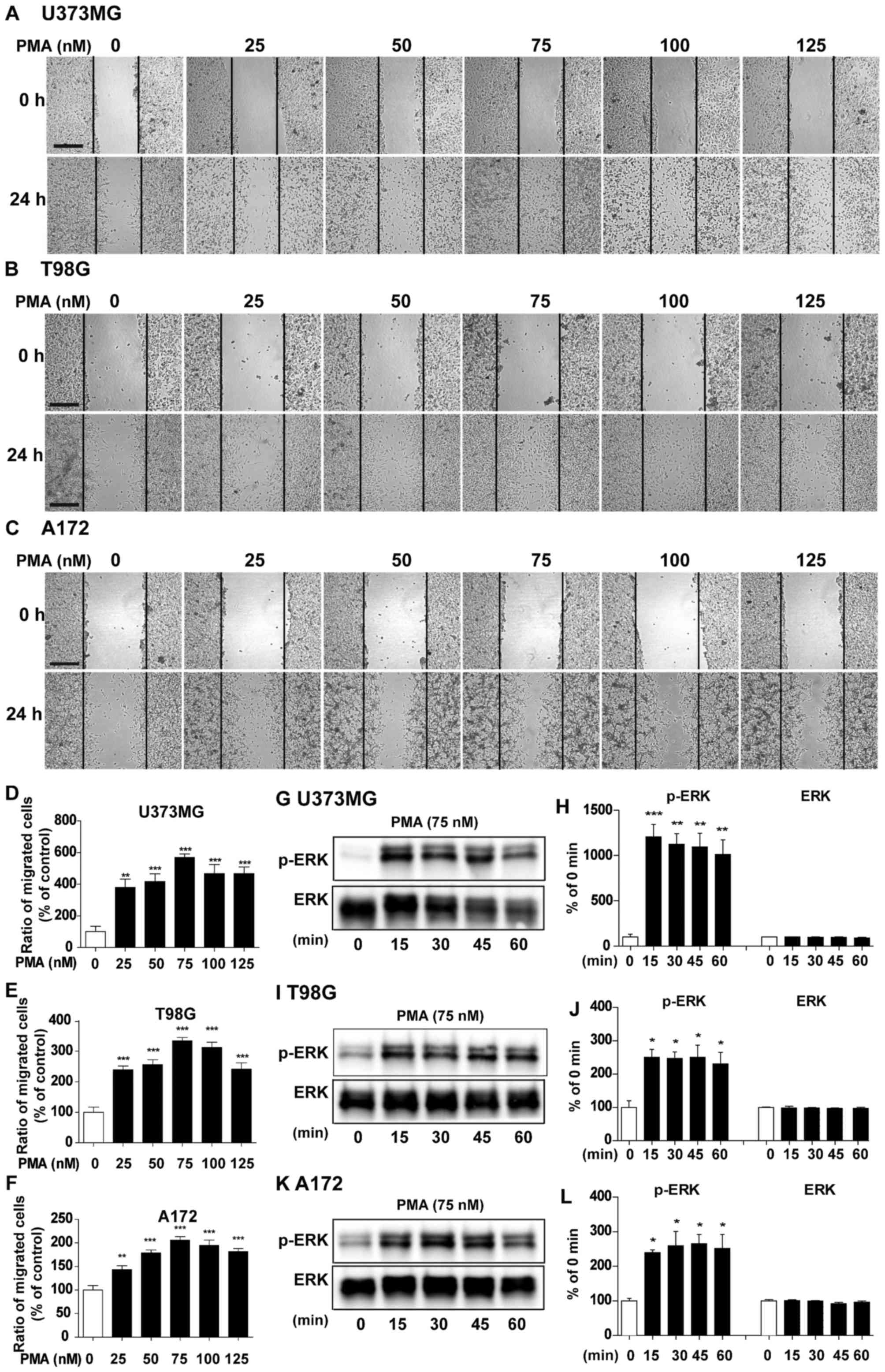 | Figure 2.Dose- and time-dependent effects of
PMA on cell migration and signaling. (A) U373MG, (B) T98G and (C)
A172 cell monolayers were scratched with a pipette tip and then
treated with vehicle or PMA (25, 50, 75, 100 or 125 nM) for 24 h.
Images of the wound gap were acquired at 0 h (i.e., immediately
after scratching) and after 24 h. (D-F) Quantification of data from
(A) (U373MG: PMA 0 nM, n=9; PMA 25 nM, n=7; PMA 50 nM, n=9; PMA 75
nM, n=12; PMA 100 nM, n=10; PMA 125 nM, n=12; **P<0.01;
***P<0.001; ANOVA with post hoc Tukeys test), (B) (T98G: PMA 0
nM, n=11; PMA 25 nM, n=11; PMA 50 nM, n=9; PMA 75 nM, n=14; PMA 100
nM, n=13; PMA 125 nM, n=11; ***P<0.001, ANOVA with post hoc
Tukeys test), and (C) (A172: PMA 0 nM, n=11; PMA 25 nM, n=10; PMA
50 nM, n=9; PMA 75 nM, n=9; PMA 100 nM, n=9; PMA 125 nM, n=10;
**P<0.01, ***P<0.001, ANOVA with post hoc Tukeys test). (G)
U373MG, (I) T98G and (K) A172 cells were treated with PMA (75 nM)
or vehicle for 15, 30, 45 or 60 min, and western blotting was
performed with anti-p-ERK and ERK antibodies. (H, J and L)
Quantification of data from (G) (U373MG; 0, 15, 30, 45 and 60 min,
n=3; **P<0.01; ***P<0.001, ANOVA with post hoc Tukeys test),
(I) (T98G; 0, 15, 30, 45 and 60 min, n=3; *P<0.05, ANOVA with
post hoc Tukeys test), (K) (A172; 0, 15, 30, 45 and 60 min, n=3;
*P<0.05, ANOVA with post hoc Tukeys test). (A-C) Scale bar, 200
µm. |
To determine the optimal time for PMA treatment, we
treated U373MG, T98G and A172 cells with vehicle or PMA at various
times (15, 30, 45 and 60 min) and performed western blotting to
detect anti-p-ERK and total ERK, an important signaling for cancer
migration (16). PMA significantly
increased p-ERK at all time-points in U373MG (Fig. 2G and H, P<0.01, P<0.001, ANOVA
with post hoc Tukeys test), T98G (Fig.
2I and J, P<0.05, ANOVA with post hoc Tukeys test), and A172
cells (Fig. 2K and L, P<0.05,
ANOVA with post hoc Tukeys test). Based on our findings and
literature, we selected a dose of 75 nM and a duration of 45 min
for PMA treatment in the following experiments (15).
To determine whether EYK can alter cell migration in
human malignant cells, we performed wound healing assays on U373MG,
T98G and A172 cells pre-treated with PMA (75 nM) or vehicle for 45
min, and then treated with EYK (200 µg/ml) or vehicle for 24 h. As
expected, PMA alone significantly increased cell migration in all
three cell lines (Fig. 3A-F). EYK
did not alter PMA-induced cell migration in comparison with PMA in
U373MG or T98G cells (Fig. 3A-D,
P<0.001; ANOVA with post hoc Tukeys test). By contrast, EYK
significantly suppressed PMA-mediated cell migration in A172 cells
(Fig. 3E and F, P<0.001; ANOVA
with post hoc Tukeys test).
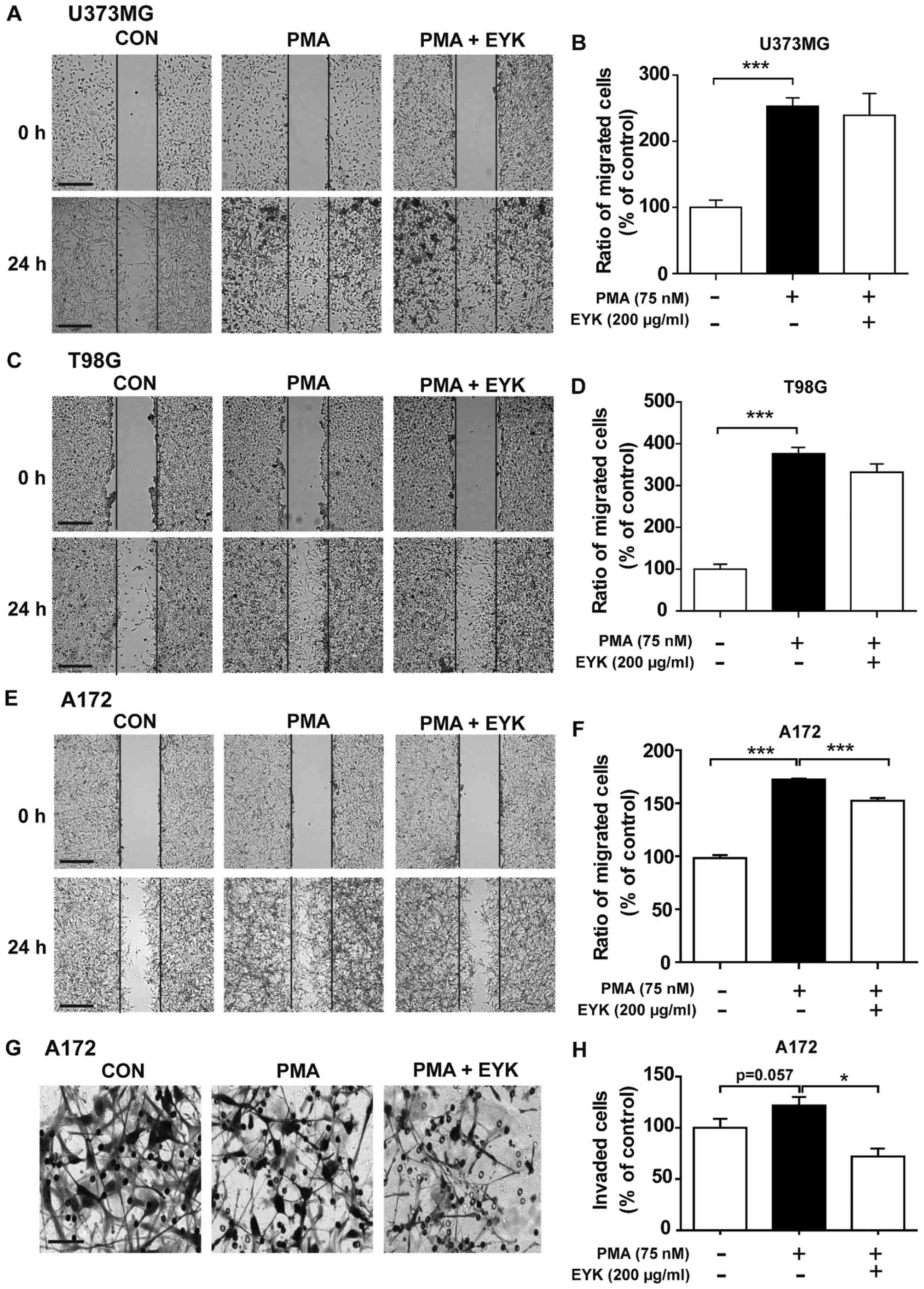 | Figure 3.Pre-treatment with PMA and EYK
treatment inhibits PMA-induced cell migration and invasion. (A)
U373MG, (C) T98G or (E) A172 cell monolayers were scratched with a
pipette tip, pre-treated with vehicle or PMA (75 nM) for 45 min,
and then treated with vehicle or EYK (200 µg/ml) for 24 h. Images
of the wound gap were acquired at 0 h (i.e., immediately after
scratching) and after 24 h. (B, D and F) Quantification of data
from (A) (U373MG: con, n=19; PMA, n=27; PMA+EYK, n=15;
***P<0.001; ANOVA with post hoc Tukeys test), (C) (T98G: con,
n=13; PMA, n=18; PMA+EYK, n=10; ***P<0.001; ANOVA with post hoc
Tukeys test), and (E) (A172: con, n=26; PMA, n=26; PMA+EYK, n=28;
***P<0.001; ANOVA with post hoc Tukeys test). (G) A172 cells
were pre-treated with PMA (75 nM) or vehicle for 45 min, treated
with vehicle or EYK (200 µg/ml) for 24 h, and then subjected to
Transwell invasion assays. (H) Quantification of data from (G)
(A172: con, n=7; PMA, n=7; PMA+EYK, n=7; *P<0.05; ANOVA with
post hoc Tukeys test, con vs. PMA, P=0.057; two-tailed t-test). (A,
C and E) Scale bar, 200 µm, and (G) 100 µm. |
Next, we performed Transwell assays to investigate
whether EYK can regulate cell invasion. To this end, A172 cells
were pre-treated with PMA or vehicle for 45 min, treated with
vehicle or EYK (200 µg/ml) for 24 h, and then subjected to cell
invasion assays. PMA increased cell invasion in comparison with
vehicle (Fig. 3G and H, P<0.05;
ANOVA with post hoc Tukeys test, CON vs. PMA, P=0.057; two-tailed
t-test). However, EYK inhibited PMA-induced cell invasion (Fig. 3G and H). Together, these data
indicate that EYK can inhibit PMA-mediated cell migration and
invasion, at least in monomorphic malignant human glioma cells.
Pre-treatment with PMA followed by EYK
decreased MMP-9 activity in A172 cells
To determine whether EYK regulates activity and
expression of MMPs, which are involved in cell migration and
invasion (15,17), we pre-treated A172 cells with PMA
(75 nM) or vehicle for 45 min, treated with EYK (200 µg/ml) or
vehicle for 24 h, and then conducted gelatin zymography assays. PMA
significantly increased MMP-9 activity compared to vehicle
(Fig. 4A and B, P<0.001; ANOVA
with post hoc Tukeys test). EYK significantly decreased PMA-induced
MMP-9 activity (Fig. 4A and B).
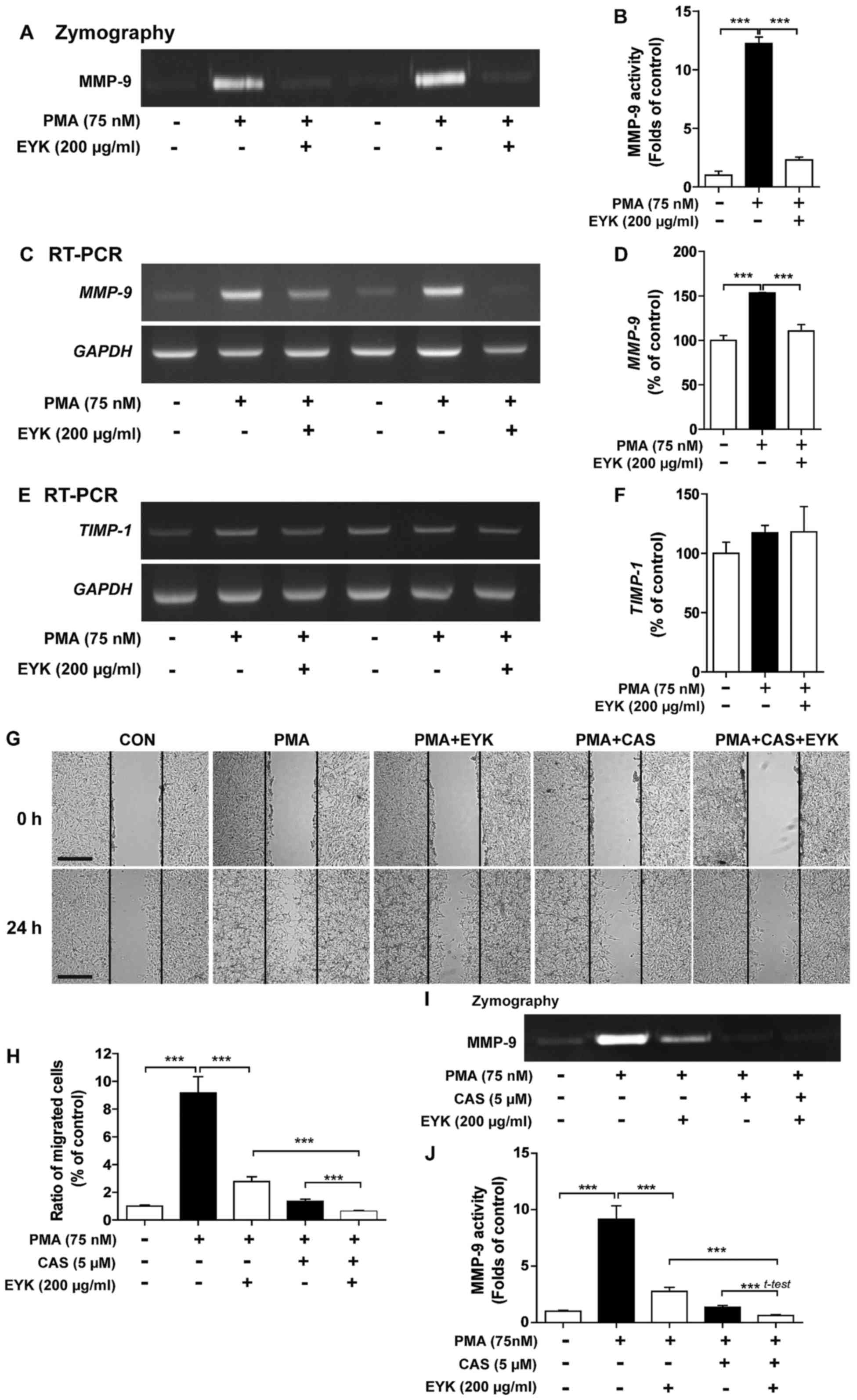 | Figure 4.Pre-treatment with PMA followed by
EYK treatment decreases MMP-9 activity in A172 cells. (A) A172
cells were pre-treated with vehicle or PMA (75 nM) for 45 min, and
then treated with EYK (200 µg/ml) or vehicle for 24 h. After 24 h,
gelatin zymography assays were conducted on conditioned medium to
measure MMP-9 activity. (B) Quantification of data from A (A172:
con, n=6; PMA, n=6; PMA+EYK, n=6; ***P<0.001; ANOVA with post
hoc Tukeys test). (C and E) A172 cells were pre-treated with
vehicle or PMA (75 nM) for 45 min and treated with EYK (200 µg/ml)
or vehicle for 24 h. After 24 h, mRNA levels of MMP-9 (C),
TIMP-1 (E) and GAPDH were measured by RT-PCR. (D and
F) Quantification of data from (C) (A172: con, n=8; PMA, n=8;
PMA+EYK, n=8; ***P<0.001; ANOVA with post hoc Tukeys test) and
(E) (A172: con, n=4; PMA, n=4; PMA+EYK, n=4). (G) A172 cell
monolayers were scratched with a pipette tip, pre-treated with
vehicle or PMA (75 nM) for 45 min, treated with vehicle or CAS
1177749-58-4 (5 µM, MMP-9 inhibitor) for 1 h, and then treated with
vehicle or EYK (200 µg/ml) for 24 h. Images of the wound gap were
acquired at 0 h (i.e., immediately after scratching) and after 24
h. (H) Quantification of data from (G) (A172: con, n=25; PMA, n=25;
PMA+EYK, n=28; PMA+CAS, n=40; PMA+CAS+EYK, n=37; ***P<0.001;
ANOVA with post hoc Tukeys test). (I) A172 cells were pre-treated
with vehicle or PMA (75 nM) for 45 min, treated with vehicle or CAS
1177749-58-4 (5 µM, MMP-9 inhibitor) for 1 h, and treated with
vehicle or EYK (200 µg/ml) for 24 h. After 24 h, gelatin zymography
assays were conducted. (J) Quantification of data from I (A172:
con, n=28; PMA, n=28; PMA+EYK, n=28; PMA+CAS, n=28; PMA+CAS+EYK,
n=28; ***P<0.001; ANOVA with post hoc Tukeys test, PMA+CAS vs.
PMA+CAS+EYK; two-tailed t-test). (G) Scale bar, 200 µm. |
In order to examine the effects of EYK on
MMP-9 and TIMP-1 mRNA levels, we conducted RT-PCR
under the same conditions as described above. We found that PMA
significantly increased MMP-9 and TIMP-1 mRNA levels
compared to vehicle (Fig. 4C-F).
EYK significantly decreased PMA-mediated MMP-9 mRNA levels
(Fig. 4C and D, P<0.001; ANOVA
with post hoc Tukeys test), but had no effect on TIMP-1 mRNA
levels (Fig. 4E and F). These data
suggest that EYK can regulate PMA-induced MMP-9 activity as well as
its mRNA levels.
We then investigated whether EYK requires MMP-9
activity to alter cell migration. For this purpose, A172 cells were
pre-treated with vehicle or PMA (75 nM), treated with
CAS1177749-58-4 (an MMP-9 inhibitor, 5 µM) for 1 h, exposed to
vehicle or EYK (200 µg/ml) for 24 h, and then subjected to the
wound healing assay. Consistent with our findings above, PMA
significantly increased cell migration but EYK significantly
decreased PMA-induced cell migration (Fig. 4G and H, P<0.001; ANOVA with post
hoc Tukeys test). In addition, pretreatment with PMA, MMP-9
inhibitor, and EYK treatment further decreased cell migration
compared to treatment with PMA and EYK (Fig. 4G and H). Moreover, we found that
treatments with PMA, MMP-9 inhibitor, and EYK significantly reduced
MMP-9 activity compared to PMA and EYK treatment (Fig. 4I and J; P<0.001; ANOVA with post
hoc Tukeys test, PMA+CAS vs. PMA+CAS+EYK; two-tailed t-test).
EYK significantly decreased
PMA-induced NF-κB translocation to the nucleus
A transcriptional factor NF-κB plays an important
role in cell migration and invasion by regulating MMP activity
(18). Hence, we initially
investigated whether EYK could alter PMA-induced NF-κB levels in
the cytosol vs. the nucleus. For these experiments, A172 cells were
pre-treated with PMA (75 nM) or vehicle for 45 min, treated with
EYK (200 µg/ml) or vehicle for 45 min, and then subjected to
subcellular fractionation. PMA treatment showed a trend toward
increased p-IκBα and NF-κB levels, but decreased IκBα levels in the
cytosol (Fig. 5A-D; P<0.01;
ANOVA with post hoc Tukeys test). EYK did not alter PMA-induced
p-IκBα and IκBα levels, but significantly decreased PMA-mediated
NF-κB levels in the cytosol (Fig.
5A-D). In addition, EYK did not alter the PMA-induced NF-κB
levels in the nucleus (Fig. 5E and
F, P<0.01; ANOVA with post hoc Tukeys test).
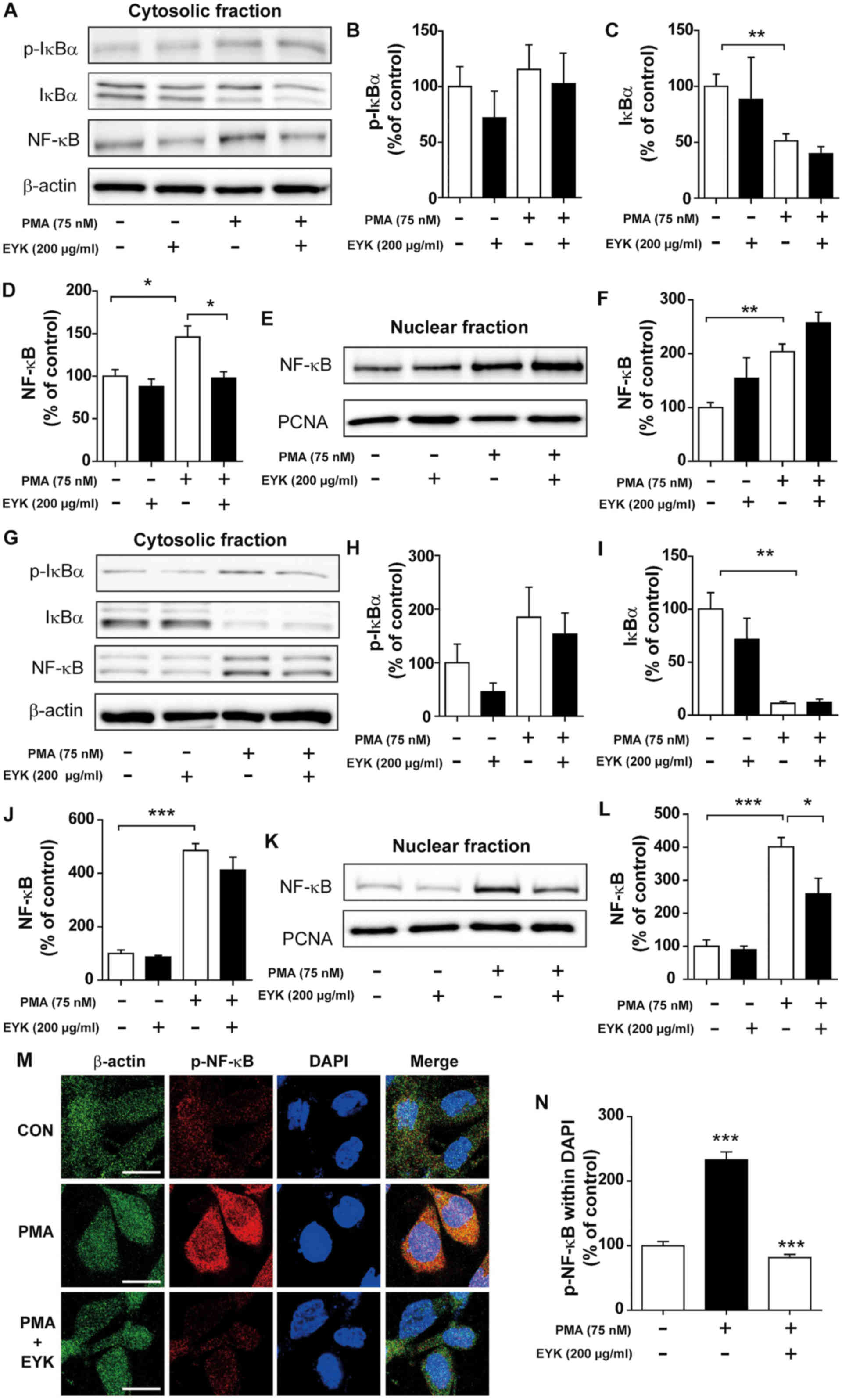 | Figure 5.Pre-treatment with PMA followed by
EYK treatment decreased NF-κB levels in the nucleus. (A) A172 cells
were pre-treated with vehicle or PMA (75 nM) for 45 min, treated
with EYK (200 µg/ml) or vehicle for 45 min, and then subjected to
subcellular fractionation (nucleus vs. cytosol). Western blotting
was performed on the cytosolic fraction using antibodies against
p-IκBα, IκBα, NF-κB and β-actin. (B-D) Quantification of data from
A (A172: con, n=5; EYK, n=2; PMA, n=5; PMA+EYK, n=5; *P<0.05;
**P<0.01; ANOVA with post hoc Tukeys test). (E) Western blotting
was performed on the nuclear fraction using antibodies against
NF-κB and PCNA. (F) Quantification of data from E (A172: con, n=5;
EYK, n=2; PMA, n=5; PMA+EYK, n=5; **P<0.01; ANOVA with post hoc
Tukeys test). (G) A172 cells were pre-treated with vehicle or PMA
(75 nM) for 45 min, treated with EYK (200 µg/ml) or vehicle for 24
h, and then subjected to subcellular fractionation (nucleus vs.
cytosol). Western blotting was performed on the cytosolic fraction
using antibodies against p-IκBα, IκBα, NF-κB and β-actin. (H-J)
Quantification of data from (G) (A172: con, n=4; EYK, n=4; PMA,
n=4; PMA+EYK, n=4; **P<0.01; ***P<0.001; ANOVA with post hoc
Tukeys test). (K) Western blotting was performed on the nuclear
fraction using antibodies against NF-κB and PCNA. (L)
Quantification of data from (K) (A172: con, n=4; EYK, n=4; PMA,
n=4; PMA+EYK, n=4; *P<0.05; ***P<0.001; ANOVA with post hoc
Tukeys test). (M) A172 cells were pre-treated with vehicle or PMA
(75 nM) for 45 min, treated with EYK (200 µg/ml) or vehicle for 24
h, and immunostaing were performed using antibodies against p-NF-κB
and β-actin. (N) Quantification of data from M (A172: con, n=63
cells; PMA, n=61 cells; PMA+EYK, n=68 cells; ***P<0.01; ANOVA
with post hoc Tukeys test). (M) Scale bar, 20 µm. |
We then asked whether EYK could alter NF-κB
subcellular localization when administered for longer periods of
time. To answer this question, A172 cells were pre-treated with PMA
(75 nM) or vehicle for 45 min, treated with EYK (200 µg/ml) or
vehicle for 24 h, and then subjected to subcellular fractionation.
EYK slightly decreased PMA-induced p-IκBα and NF-κB levels, but did
not alter IκBα levels in the cytosol (Fig. 5G-J, P<0.001; ANOVA with post hoc
Tukeys test). In addition, EYK significantly decreased PMA-mediated
NF-κB levels in the nucleus (Fig. 5K
and L, P<0.05; P<0.001; ANOVA with post hoc Tukeys
test).
To examine whether EYK can alter PMA-induced p-NF-κB
levels in the nucleus, A172 cells were pre-treated with PMA (75 nM)
or vehicle for 45 min, treated with EYK (200 µg/ml) or vehicle for
24 h, and then immunostaining were performed. PMA significantly
increased p-NF-κB levels in the nucleus (Fig. 5M and N, P<0.001; ANOVA with post
hoc Tukeys test). EYK significantly decreased PMA-induced p-NF-κB
levels in the nucleus (Fig. 5M and
N). These data suggest that EYK can alter NF-κB nuclear
translocation between subcellular compartments.
EYK suppresses PMA-mediated cell
migration via the NF-κB pathway
To determine whether EYK requires NF-κB to suppress
cancer cell migration, we performed wound healing assays on A172
cells pre-treated with PMA (75 nM) or vehicle for 45 min, treated
with BAY 11–7085 (10 µM, a NF-κB inhibitor) or vehicle for 1 h, and
then treated with EYK (200 µg/ml) or vehicle for 24 h (Fig. 6A and B, P<0.05; P<0.001; ANOVA
with post hoc Tukeys test). Treatments with PMA, NF-κB inhibitor,
and EYK significantly inhibited cell migration in comparison with
treatments with PMA and EYK, suggesting that NF-κB is necessary for
EYK to regulate cell migration.
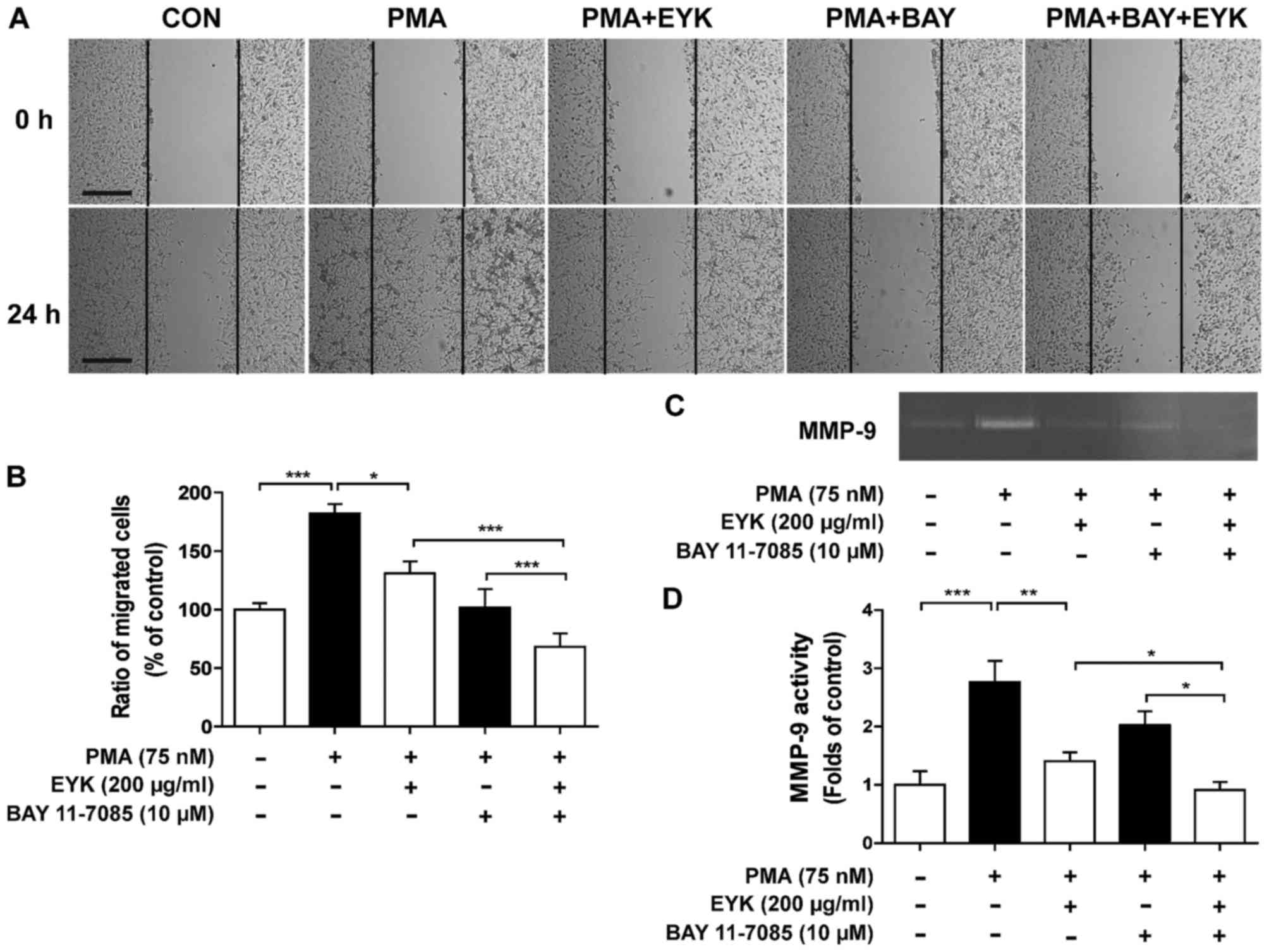 | Figure 6.Pre-treatment with PMA and EYK
treatment inhibits PMA-induced cancer cell migration via NF-κB. (A)
A172 cell monolayers were scratched with a scraper, pre-treated
with PMA (75 nM) or vehicle for 45 min, treated with BAY 11–7085
(NF-κB inhibitor, 10 µM) or vehicle for 1 h, and then treated with
EYK (200 µg/ml) or vehicle for 24 h. Images of the wound gap were
acquired at 0 h (i.e., immediately after scratching) and after 24
h. (B) Quantification of data from (A) (A172: con, n=20; PMA, n=17;
PMA+EYK, n=17; PMA+BAY, n=17; PMA+BAY+EYK, n=21; *P<0.05;
***P<0.001; ANOVA with post hoc Tukeys test). (C) A172 cells
were pre-treated with PMA (75 nM) or vehicle for 45 min, treated
with BAY 11–7085 (NF-κB inhibitor, 10 µM) or vehicle for 1 h,
treated with EYK (200 mg/ml) or vehicle for 24 h, and then
subjected to gelatin zymography. (D) Quantification of data from
(C) (A172: con, n=8; PMA, n=8; PMA+EYK, n=8; PMA+BAY, n=8;
PMA+BAY+EYK, n=8; *P<0.05; **P<0.01; ***P<0.001; ANOVA
with post hoc Tukeys test). (A) Scale bar, 200 µm. |
Next, we examined the effects of EYK on MMP-9
activity by treating cells with PMA and NF-κB inhibitor and found
that inhibition of NF-κB in combination with EYK treatment further
decreased PMA-induced MMP-9 activity in comparison with PMA and EYK
treatments (Fig. 6C and D,
P<0.05; P<0.01; P<0.001; ANOVA with post hoc Tukeys
test).
Pre-treatment with EYK and PMA
treatment suppresses cell migration
To determine whether pre-treatment with EYK can
modulate cell migration, we performed wound healing assays on A172
cells pre-treated with vehicle or EYK (200 µg/ml) for 45 min,
treated with PMA (75 nM) or vehicle for 24 h, and then wound
healing assays were conducted. PMA significantly increased cancer
cell migration, but this effect was suppressed by pre-treatment
with EYK (Fig. 7A and B,
P<0.001; ANOVA with post hoc Tukeys test).
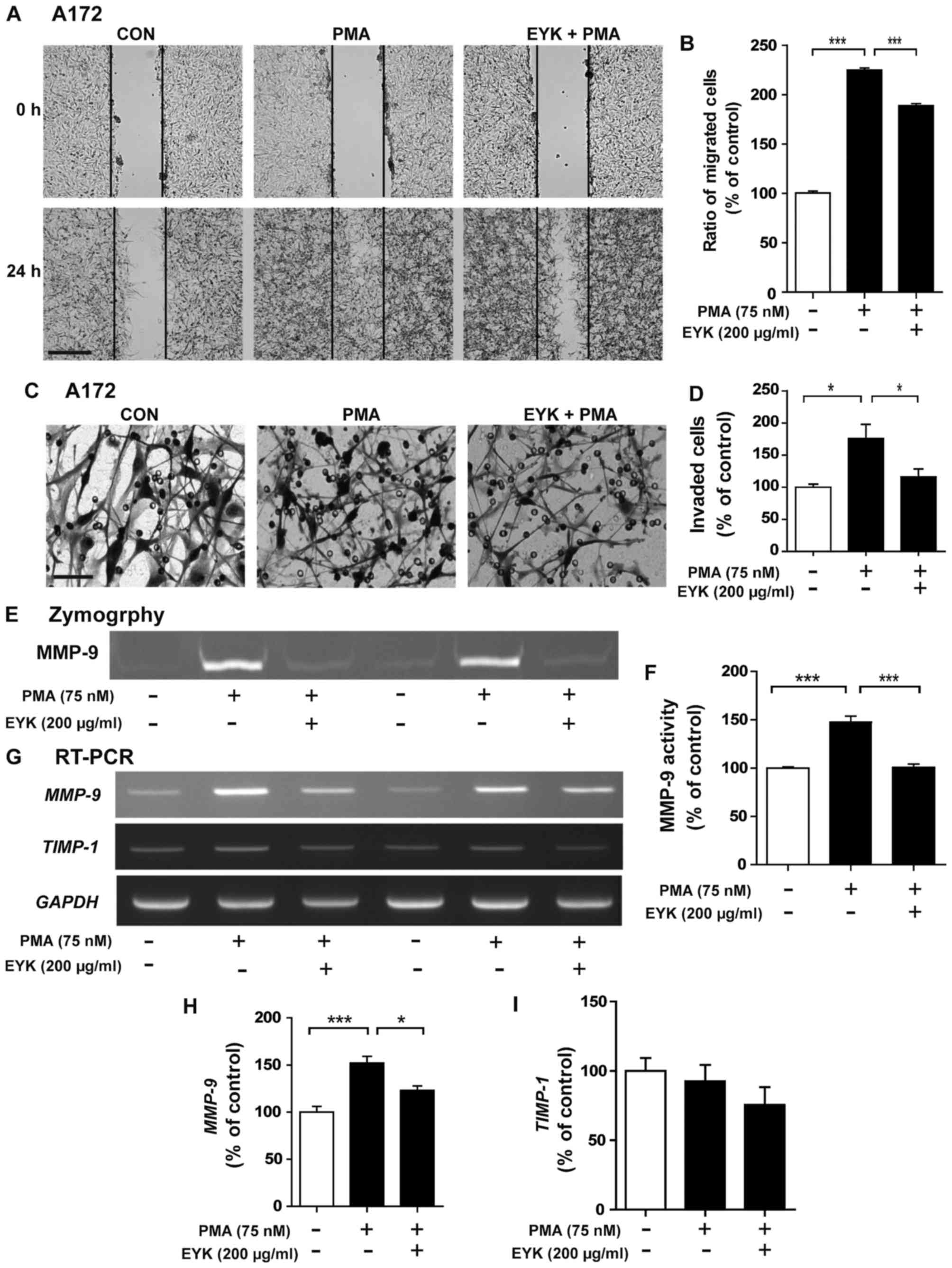 | Figure 7.Pre-treatment with EYK and PMA
inhibits cancer cell migration and invasion in A172 cells. (A) A172
cell monolayers were scratched with a pipette tip and immediately
imaged (0 h), pre-treated with vehicle or EYK (200 µg/ml) for 45
min, and then treated with vehicle or PMA (75 nM) for 24 h. Images
of the wound gap were acquired at 0 h (i.e., immediately after
scratching) and after 24 h. (B) Quantification of data from (A)
(A172: con, n=23; PMA, n=31; PMA+EYK, n=35; ***P<0.001; ANOVA
with post hoc Tukeys test). (C) A172 cells were pre-treated with
vehicle or EYK (200 µg/ml) for 45 min, and then treated with
vehicle or PMA (75 nM) for 24 h. After 24 h, Transwell invasion
assays were conducted. (D) Quantification of data from (C) (A172:
con, n=4; PMA, n=4; PMA+EYK, n=4; *P<0.05; ANOVA with post hoc
Tukeys test). (E) A172 cells were pre-treated with EYK (200 µg/ml)
or vehicle for 45 min, and treated with vehicle or PMA (75 nM) for
24 h, and then a gelatin zymography assay was conducted on
conditioned medium to measure MMP-9 activity. (F) Quantification of
data from (E) (A172: con, n=4; PMA, n=4; PMA+EYK, n=4;
***P<0.001; ANOVA with post hoc Tukeys test). (G) A172 cells
were pre-treated with vehicle or EYK (200 µg/ml) for 45 min, and
then treated with PMA (75 nM) or vehicle for 24 h. mRNA levels of
MMP-9, TIMP1 and GAPDH were measured by
RT-PCR. (H and I) Quantification of data from (G) (MMP-9: con, n=8;
PMA, n=8; PMA+EYK, n=8; *P<0.05; ***P<0.001; ANOVA with post
hoc Tukeys test; TIMP1: con, n=4; PMA, n=4; PMA+EYK, n=4). (A)
Scale bar, 200 µm and (C) 100 µm. |
Next, we performed the Transwell assays to
investigate the effects of pre-treatment with EYK on cell invasion
and found that PMA significantly increased cell invasion while
pre-treatment with EYK significantly inhibited PMA-induced cell
invasion (Fig. 7C and D, P<0.05;
ANOVA with post hoc Tukeys test).
We then investigated whether pre-treatment with EYK
could alter MMP-9 activity and found that pre-treatment with EYK
significantly decreased PMA-induced MMP-9 activity (Fig. 7E and F, P<0.001; ANOVA with post
hoc Tukeys test). Moreover, pre-treatment with EYK significantly
decreased PMA-induced MMP-9 mRNA levels, but had no effect
on TIMP-1 mRNA levels (Fig.
7G-I, P<0.05; P<0.001; ANOVA with post hoc Tukeys
test).
Pre-treatment with EYK alters cell
migration through NF-κB pathway
To determine whether pre-treatment with EYK can
regulate PMA-induced NF-κB subcellular translocation, we
pre-treated A172 cells with EYK (200 µg/ml) or vehicle for 45 min,
treated with PMA (75 nM) or vehicle for 24 h, and then performed
subcellular fractionation. Pre-treatment with EYK followed by PMA
treatment showed a trend toward decreased p-IκBα levels and did not
alter IκBα and NF-κB levels in the cytosol (Fig. 8A-D, P<0.05; ANOVA with post hoc
Tukeys test). In the nuclear fraction, pre-treatment with EYK
significantly decreased PMA-induced NF-κB levels (Fig. 8E and F, P<0.01; P<0.001, ANOVA
with post hoc Tukeys test). Moreover, pre-treatment with EYK
significantly decreased PMA-induced p-NF-κB levels in the nucleus
(Fig. 8G and H, P<0.001; ANOVA
with post hoc Tukeys test).
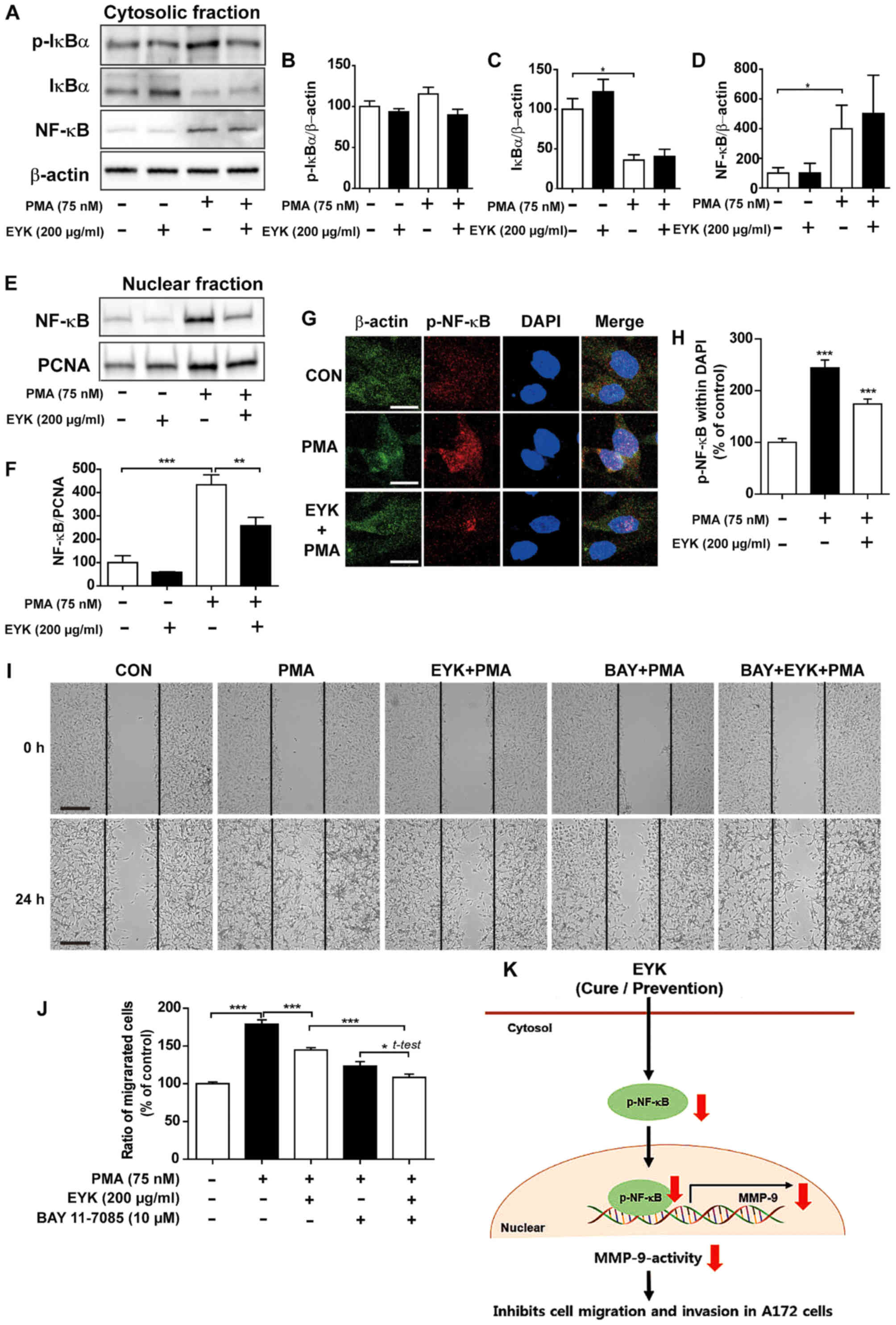 | Figure 8.Pre-treatment with EYK and PMA
decreases translocation of NF-κB to the nucleus. A172 cells were
pre-treated with EYK (200 µg/ml) or vehicle for 45 min, treated
with vehicle or PMA (75 nM) for 24 h and subjected to subcellular
fractionation (nucleus vs. cytosol). (A) Western blotting was
performed on the cytosolic fraction using antibodies against
p-IκBα, IκBα, NF-κB and β-actin. (B-D) Quantification of data from
(A) (A172: con, n=4; EYK, n=4; PMA, n=4; EYK+PMA, n=4; *P<0.05;
ANOVA with post hoc Tukeys test). (E) Western blotting was
performed on the nuclear fraction using antibodies against NF-κB
and PCNA. (F) Quantification of data from (E) (A172: con, n=4; EYK,
n=4; PMA, n=4; EYK+PMA, n=4; **P<0.01; ***P<0.001, ANOVA with
post hoc Tukeys test). (G) A172 cells were pre-treated with EYK
(200 µg/ml) or vehicle for 45 min, treated with vehicle or PMA (75
nM) for 24 h, and immunostaing were performed using antibodies
against p-NF-κB and β-actin. (H) Quantification of data from (G)
(A172: con, n=126 cells; PMA, n=106 cells; PMA+EYK, n=122 cells;
***P<0.001; ANOVA with post hoc Tukeys test). (I) A172 cell
monolayers were scratched with a scraper, pre-treated with vehicle
or BAY 11–7085 (10 µM, NF-κB inhibitor) for 1 h, treated with
vehicle or EYK (200 µg/ml) for 45 min, and then treated with PMA
(75 nM) or vehicle for 24 h. Images of the wound gap were acquired
at 0 h (i.e., immediately after scratching) and after 24 h. (J)
Quantification of data from (I) (A172: con, n=68; PMA, n=53;
EYK+PMA, n=69; BAY+EYK, n=45; BAY+EYK+PMA, n=58; *P<0.05;
***P<0.001, ANOVA with post hoc Tukeys test; EYK+PMA vs.
BAY+EYK+PMA, two-tailed t-test). (K) Schematic model of suppression
of brain cancer cell migration and invasion by EYK. (G) Scale bar,
20 µm and (I) 200 µm. |
We then investigated whether pre-treatment with EYK
could alter cell migration via NF-κB pathways. To test this, we
performed wound healing assays on A172 cells pre-treated with
vehicle or BAY11-7085 (10 µM, an NF-κB inhibitor) for 1 h, treated
with EYK (200 µg/ml) or vehicle for 45 min, and then treated with
PMA 75 nM) or vehicle for 24 h. Pre-treatment with EYK decreased
PMA-induced cell migration (Fig. 8I and
J, P<0.05; P<0.001, ANOVA with post hoc Tukeys test;
EYK+PMA vs. BAY+EYK+PMA, two-tailed t-test). In addition,
pre-treatment with NF-κB inhibitor followed by treatments with EYK
and PMA further inhibited cell migration in comparison with
treatments with EYK and PMA (Fig. 8I
and J). Based on these findings, we conclude that EYK could be
used as a drug to prevent cancer cell migration by decreasing the
nuclear localization of NF-κB and MMP-9 activity in A172 cells
(Fig. 8K).
Discussion
Glioblastoma multiforme (GBM) is characterized by
its aggressive cell proliferation and invasive infiltration into
the surrounding brain tissue (19),
and these features are associated with very poor prognosis
(20). Temozolomide (TMZ) in
combination with radiation therapy is the typical chemotherapy used
to treat GBM (21). However, this
approach has several problems, including drug resistance and
induction of proliferation and metastasis (21–23).
Therefore, it is necessary to develop a novel antitumor agent in
order to successfully treat GBM.
In the present study, we discovered that EYK
inhibitsmonomorphic malignant human glioma cell migration and
invasion by downregulating MMP-9 activity and nuclear localization
of NF-κB, the transcription factor primarily responsible for
expression of MMP-9. Several studies have demonstrated that EYK has
antitumor effects including reduction of drug resistance-causing
gene mutations in lung cancer cells (24), induction of apoptosis in human
prostate cancer cells (5) and
non-small cell lung cancer (6), and
downregulation of cell proliferation in osteosarcoma cells
(25). However, this is the first
study to demonstrate the ability of EYK to inhibit brain cancer
cell migration and invasion in human malignant cells.
Cancer cell migration and invasion are the main
biological characteristics associated with tumor malignancy
(26). In a previous study of
Epimedium species, icaritin, a bioactive compound in
Epimedium extract, inhibited adhesion, migration and
invasion of glioblastoma cells via downregulation of extracellular
matrix (ECM) and MMPs mediated by the PTEN/AKT/HIF-1a pathway
(27). In addition, consistently
with those previously published results, our present findings
demonstrated that pre-treatment with PMA followed by EYK treatment
effectively inhibited migration and invasion (Fig. 3) in A172 cells, but not in two other
cell lines, T98G and U373MG. A172 cells are monomorphic and
fibroblast-like whereas T98G cells are polymorphic, fibroblast-like
and polygonal (28). U373MG cells
have pleomorphic features (29).
Thus, EYK may selectively alter cell migration and invasion in
specific cell types of human brain cancer cells. Indeed, we
confirmed that cell migration and invasion were inhibited when EYK
was used as a pre-treatment prior to PMA exposure (Fig. 7).
Degradation of ECM is an important process in cancer
cell migration and invasion (30).
MMPs induce cell migration and invasion by degrading ECM proteins
on and around surrounding normal brain tissue (31). One of the MMPs, MMP-9, is
overexpressed in experimental glioma models and brain tumor patient
tissue samples (32), and plays a
major role in invasive infiltration and migration of brain cancer
cells (31,33). Accordingly, downregulation of MMP-9
levels prevents tumor growth and invasion in glioblastoma cells
lines (34). Quercetin, one of the
elements of EYK, inhibits expression of MMP-9 via the AKT/ERK
signaling pathway in human glioma cells (35). Our results showed that pre-treatment
with PMA followed by EYK treatment dramatically downregulated
PMA-induced MMP-9 activity and its mRNA levels in A172 cells
(Fig. 4). These results were
confirmed in wound healing assays and gelatin zymography assays by
treating with CAS 1177749-58-4, MMP-9 inhibitor. Similar to our
findings shown in Fig. 4,
pre-treatment with EYK and PMA inhibited MMP-9 activity compared to
PMA treatment (Fig. 7).
Regulation of MMP-9 is the major anti-oncogenic
effect of TIMP-1 (tissue inhibitor of metalloproteinases), an
endogenous inhibitor. For instance, B16F10 melanoma-expressing-mice
followed by injection of recombinant TIMP-1 decreases pulmonary
metastases (36). Hence, we
investigated whether EYK regulates MMP-9 activity directly or by
modulating levels of TIMP-1. To this end, we asked whether EYK
affects TIMP-1 mRNA levels. In this study, neither
pre-treatment with PMA followed by EYK nor exposure to the
compounds in the opposite order altered TIMP-1 mRNA levels
(Figs. 4 and 7). Therefore, we concluded that EYK
directly regulates MMP-9 activity to alter cancer cell
migration.
Mitrogen-activated protein kinases (MAPK) including
ERK, JNK and p38 are involved in cancer cell migration and invasion
(37–40) and regulate MMP-9 activity and its
expression (41). Notably, we found
that PMA in combination with EYK did not alter phosphorylation of
ERK, JNK, or p38 (data not shown). We then examined another
potential signaling pathways, focal adhesion kinase (FAK)
signaling, that are targeted by EYK to regulate cancer cell
migration. It is well-established that FAK signaling plays an
important role in cancer cell migration as well as invasion
(42). For these reasons, we
investigated whether EYK can modulate FAK signaling pathway and
found that treatment with PMA and EYK in either temporal order did
not alter the phosphorylation of FAK (data not shown). Therefore,
we concluded that EYK inhibits cell migration independent of
PMA-induced MAP kinases and FAK signaling.
To elucidate the molecular mechanism by which EYK
inhibits human brain cancer cell migration and invasion, we
examined the effect of EYK on NF-κB subcellular localization, which
is primarily responsible for regulating MMP-9 expression. In
addition, NF-κB activation contributes to PMA-induced MMP-9
activity and glioma cell migration (18). In this study, pre- or post-treatment
with EYK decreased PMA-mediated nuclear levels of NF-κB (Figs. 5 and 8). Surprisingly, our results indicated
that EYK mainly affects nuclear levels of NF-κB without changing
the levels of IκBα, a cellular inhibitor of NF-κB. Thus, EYK may
directly regulate subcellular localization of NF-κB or modulate its
translocation to the nucleus via an unknown inhibitory factor,
thereby altering cancer cell migration. Accordingly, future studies
should further investigate the molecular mechanism underlying
regulation of NF-κB by EYK in monomorphic malignant human glioma
cells.
In summary, this study provides for the first time
demonstration that EYK exerts its anticancer effect in monomorphic
human glioblastoma by inhibiting MMP-9 activity, mediated by a
reduction in nuclear localization of NF-κB. This effect could be
achieved by pre-treating with PMA followed by EYK treatment (as a
cure condition) or by pre-treating with EYK followed by PMA
exposure (as a prevention condition) (Fig. 8K). Taken together, our results
suggest that EYK could be used as a drug for the cure and
prevention of monomorphic malignant human glioma.
Acknowledgements
The present study was supported by the KBRI Basic
Research Program through the Korea Brain Research Institute funded
by the Ministry of Science, ICT, & Future Planning [grant
number: 17-BR-03] (H.S.H.), by the National Research Foundation of
the Korean government (H.S.H., grant no. 2016R1A2B4011393) and BH
community. This study was supported by the Korea Institute of
Oriental Medicine (KIOM) funded by the Ministry of Science,
ICT,& Future Planning (MISP) (grant no. K17281). The authors
declare no competing financial interests. Bright-field microscopy
(Carl Zeiss) data were acquired in the Advanced Neural Imaging
Center at the Korea Brain Research Institute (KBRI). We used an
English language service-BIOEDIT (https://www.bioedit.com) to prepare our manuscript. We
thank So Yeon Koo for writing, editing and valuable comments in our
manuscript.
References
|
1
|
Jiang F, Wang XL, Wang NL and Yao XS: Two
new flavonol glycosides from Epimedium koreanum Nakai. J
Asian Nat Prod Res. 11:401–409. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu CQ, Liu BJ, Wu JF, Xu YC, Duan XH, Cao
YX and Dong JC: Icariin attenuates LPS-induced acute inflammatory
responses: Involvement of PI3K/Akt and NF-kappaB signaling pathway.
Eur J Pharmacol. 642:146–153. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang L, Shen C, Chu J, Zhang R, Li Y and
Li L: Icariin decreases the expression of APP and BACE-1 and
reduces the β-amyloid burden in an APP transgenic mouse model of
Alzheimer's disease. Int J Biol Sci. 10:181–191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Han YY, Song MY, Hwang MS, Hwang JH, Park
YK and Jung HW: Epimedium koreanum Nakai and its main
constituent icariin suppress lipid accumulation during adipocyte
differentiation of 3T3-L1 preadipocytes. Chin J Nat Med.
14:671–676. 2016.PubMed/NCBI
|
|
5
|
Lee KS, Lee HJ, Ahn KS and Kim SH, Nam D,
Kim DK, Choi DY, Ahn KS, Lu J and Kim SH:
Cyclooxygenase-2/prostaglandin E2 pathway mediates icariside II
induced apoptosis in human PC-3 prostate cancer cells. Cancer Lett.
280:93–100. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Song J, Shu L, Zhang Z, Tan X, Sun E, Jin
X, Chen Y and Jia X: Reactive oxygen species-mediated mitochondrial
pathway is involved in Baohuoside I-induced apoptosis in human
non-small cell lung cancer. Chem Biol Interact. 199:9–17. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cho WK, Kim H, Choi YJ, Yim NH, Yang HJ
and Ma JY: Epimedium koreanum Nakai water extract exhibits
antiviral activity against porcine epidermic diarrhea virus in
vitro and in vivo. Evid Based Complement Alternat Med.
2012:9851512012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kang SH, Jeong SJ and Kim SH, Kim JH, Jung
JH, Koh W, Kim JH, Kim DK, Chen CY and Kim SH: Icariside II induces
apoptosis in U937 acute myeloid leukemia cells: Role of
inactivation of STAT3-related signaling. PLoS One. 7:e287062012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chung BH, Kim JD, Kim CK, Kim JH, Won MH,
Lee HS, Dong MS, Ha KS, Kwon YG and Kim YM: Icariin stimulates
angiogenesis by activating the MEK/ERK- and PI3K/Akt/eNOS-dependent
signal pathways in human endothelial cells. Biochem Biophys Res
Commun. 376:404–408. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang J, Stevens MF and Bradshaw TD:
Temozolomide: Mechanisms of action, repair and resistance. Curr Mol
Pharmacol. 5:102–114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Johannessen TC and Bjerkvig R: Molecular
mechanisms of temozolomide resistance in glioblastoma multiforme.
Expert Rev Anticancer Ther. 12:635–642. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Munoz JL, Rodriguez-Cruz V, Greco SJ,
Ramkissoon SH, Ligon KL and Rameshwar P: Temozolomide resistance in
glioblastoma cells occurs partly through epidermal growth factor
receptor-mediated induction of connexin 43. Cell Death Dis.
5:e11452014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Armstrong TS, Cao Y, Scheurer ME,
Vera-Bolaños E, Manning R, Okcu MF, Bondy M, Zhou R and Gilbert MR:
Risk analysis of severe myelotoxicity with temozolomide: The
effects of clinical and genetic factors. Neuro Oncol. 11:825–832.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nam JH, Cho HJ, Kang H, Lee JY, Jung M,
Chang YC, Kim K and Hoe HS: A Mercaptoacetamide-based class II
histone deacetylase inhibitor suppresses cell migration and
invasion in monomorphic malignant human glioma cells by inhibiting
FAK/STAT3 Signaling. J Cell Biochem. May 12–2017.(Epub ahead of
print). doi: 10.1002/jcb.26133. View Article : Google Scholar
|
|
15
|
Cho HJ, Kang JH, Kwak JY, Lee TS, Lee IS,
Park NG, Nakajima H, Magae J and Chang YC: Ascofuranone suppresses
PMA-mediated matrix metalloproteinase-9 gene activation through the
Ras/Raf/MEK/ERK- and Ap1-dependent mechanisms. Carcinogenesis.
28:1104–1110. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen H, Zhu G, Li Y, Padia RN, Dong Z, Pan
ZK, Liu K and Huang S: Extracellular signal-regulated kinase
signaling pathway regulates breast cancer cell migration by
maintaining slug expression. Cancer Res. 69:9228–9235. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hong S, Park KK, Magae J, Ando K, Lee TS,
Kwon TK, Kwak JY, Kim CH and Chang YC: Ascochlorin inhibits matrix
metalloproteinase-9 expression by suppressing activator
protein-1-mediated gene expression through the ERK1/2 signaling
pathway: Inhibitory effects of ascochlorin on the invasion of renal
carcinoma cells. J Biol Chem. 280:25202–25209. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee CS, Cho HJ, Jeong YJ, Shin JM, Park
KK, Park YY, Bae YS, Chung IK, Kim M, Kim CH, et al:
Isothiocyanates inhibit the invasion and migration of C6 glioma
cells by blocking FAK/JNK-mediated MMP-9 expression. Oncol Rep.
34:2901–2908. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Swiatek-Machado K, Mieczkowski J,
Ellert-Miklaszewska A, Swierk P, Fokt I, Szymanski S, Skora S,
Szeja W, Grynkiewicz G, Lesyng B, et al: Novel small molecular
inhibitors disrupt the JAK/STAT3 and FAK signaling pathways and
exhibit a potent antitumor activity in glioma cells. Cancer Biol
Ther. 13:657–670. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shankar A, Kumar S, Iskander AS, Varma NR,
Janic B, de Carvalho A, Mikkelsen T, Frank JA, Ali MM, Knight RA,
et al: Subcurative radiation significantly increases cell
proliferation, invasion, and migration of primary glioblastoma
multiforme in vivo. Chin J Cancer. 33:148–158. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stepanenko AA, Andreieva SV, Korets KV,
Mykytenko DO, Baklaushev VP, Huleyuk NL, Kovalova OA, Kotsarenko
KV, Chekhonin VP, Vassetzky YS, et al: Temozolomide promotes
genomic and phenotypic changes in glioblastoma cells. Cancer Cell
Int. 16:362016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kumar DM, Patil V, Ramachandran B, Nila
MV, Dharmalingam K and Somasundaram K: Temozolomide-modulated
glioma proteome: Role of interleukin-1 receptor-associated kinase-4
(IRAK4) in chemosensitivity. Proteomics. 13:2113–2124. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sun S, Wong TS, Zhang XQ, Pu JK, Lee NP,
Day PJ, Ng GK, Lui WM and Leung GK: Protein alterations associated
with temozolomide resistance in subclones of human glioblastoma
cell lines. J Neurooncol. 107:89–100. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Song J, Zhong R, Huang H, Zhang Z, Ding D,
Yan H, Sun E and Jia X: Combined treatment with Epimedium
koreanum Nakai extract and gefitinib overcomes drug resistance
caused by T790M mutation in non-small cell lung cancer cells. Nutr
Cancer. 66:682–689. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Geng YD, Yang L, Zhang C and Kong LY:
Blockade of epidermal growth factor receptor/mammalian target of
rapamycin pathway by Icariside II results in reduced cell
proliferation of osteosarcoma cells. Food Chem Toxicol. 73:7–16.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jiang Y, Han Y, Sun C, Han C, Han N, Zhi W
and Qiao Q: Rab23 is overexpressed in human bladder cancer and
promotes cancer cell proliferation and invasion. Tumour Biol.
37:8131–8138. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu B, Jiang C, Han H, Liu H, Tang M, Liu
L, Ji W, Lu X, Yang X, Zhang Y, et al: Icaritin inhibits the
invasion and epithelial-to-mesenchymal transition of glioblastoma
cells by targeting EMMPRIN via PTEN/AKt/HIF-1α signalling. Clin Exp
Pharmacol Physiol. 42:1296–1307. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kiseleva LN, Kartashev AV, Vartanyan NL,
Pinevich AA and Samoilovich MP: A172 and T98G cell lines
characteristics. Cell Tissue Biol. 58:349–355. 2016.
|
|
29
|
Zhao Y, Xiao A, di Pierro CG, Carpenter
JE, Abdel-Fattah R, Redpath GT, Lopes MB and Hussaini IM: An
extensive invasive intracranial human glioblastoma xenograft model:
Role of high level matrix metalloproteinase 9. Am J Pathol.
176:3032–3049. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shi C, Zhang N, Feng Y, Cao J, Chen X and
Liu B: Aspirin inhibits IKK-β-mediated prostate cancer cell
invasion by targeting matrix metalloproteinase-9 and urokinase-type
plasminogen activator. Cell Physiol Biochem. 41:1313–1324. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Choe G, Park JK, Jouben-Steele L, Kremen
TJ, Liau LM, Vinters HV, Cloughesy TF and Mischel PS: Active matrix
metalloproteinase 9 expression is associated with primary
glioblastoma subtype. Clin Cancer Res. 8:2894–2901. 2002.PubMed/NCBI
|
|
32
|
Liu MF, Hu YY, Jin T, Xu K, Wang SH, Du
GZ, Wu BL, Li LY, Xu LY, Li EM, et al: Matrix
metalloproteinase-9/neutrophil Gelatinase-Associated lipocalin
complex activity in human glioma samples predicts tumor presence
and clinical prognosis. Dis Markers. 2015:1389742015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Veeravalli KK and Rao JS: MMP-9 and uPAR
regulated glioma cell migration. Cell Adhes Migr. 6:509–512. 2012.
View Article : Google Scholar
|
|
34
|
Gondi CS, Lakka SS, Dinh DH, Olivero WC,
Gujrati M and Rao JS: Downregulation of uPA, uPAR and MMP-9 using
small, interfering, hairpin RNA (siRNA) inhibits glioma cell
invasion, angiogenesis and tumor growth. Neuron Glia Biol.
1:165–176. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pan HC, Jiang Q, Yu Y, Mei JP, Cui YK and
Zhao WJ: Quercetin promotes cell apoptosis and inhibits the
expression of MMP-9 and fibronectin via the AKT and ERK signalling
pathways in human glioma cells. Neurochem Int. 80:60–71. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shoji T, Kobori M, Shinmoto H, Tanabe M
and Tsushida T: Progressive effects of phloridzin on melanogenesis
in B16 mouse melanoma cells. Biosci Biotechnol Biochem.
61:1963–1967. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen CM, Hsieh YH, Hwang JM, Jan HJ, Hsieh
SC, Lin SH and Lai CY: Fisetin suppresses ADAM9 expression and
inhibits invasion of glioma cancer cells through increased
phosphorylation of ERK1/2. Tumour Biol. 36:3407–3415. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang W, Murao K, Zhang X, Matsumoto K,
Diah S, Okada M, Miyake K, Kawai N, Fei Z and Tamiya T: Resveratrol
represses YKL-40 expression in human glioma U87 cells. BMC Cancer.
10:5932010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang XJ, Wang YH, Gao S, Guo LY, Li HH
and Song MY: Relationship between gestational glucose, lipid
metabolism parameters and fetal distress. Zhonghua Liu Xing Bing
Xue Za Zhi. 37:876–879. 2016.(In Chinese). PubMed/NCBI
|
|
40
|
Zhou X, Hua L, Zhang W, Zhu M, Shi Q, Li
F, Zhang L, Song C and Yu R: FRK controls migration and invasion of
human glioma cells by regulating JNK/c-Jun signaling. J Neurooncol.
110:9–19. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hao XN, Wang WJ, Chen J, Zhou Q, Qu YX,
Liu XY and Xu W: Effects of resveratrol on ARPE-19 cell
proliferation and migration via regulating the expression of
proliferating cell nuclear antigen, P21, P27 and p38MAPK/MMP-9. Int
J Ophthalmol. 9:1725–1731. 2016.PubMed/NCBI
|
|
42
|
Lin CC, Chen JT, Yang JS, Lu HF, Hsu SC,
Tan TW, Lin YT, Ma YS, Ip SW, Wu JJ, et al: Danthron inhibits the
migration and invasion of human brain glioblastoma multiforme cells
through the inhibition of mRNA expression of focal adhesion kinase,
Rho kinases-1 and metalloproteinase-9. Oncol Rep. 22:1033–1037.
2009.PubMed/NCBI
|