Introduction
Originating in the epithelial cells lining the
biliary tree, cholangiocarcinoma (CCC) is an aggressive malignancy
with a poor prognosis. CCC represents 10–15% of total hepatobiliary
tumors (1) and its incidence and
mortality are continuously increasing worldwide (2). Although the 5-year survival rate of
patients receiving curative resection for CCC is 30–40%,
individuals with unresectable CCC generally survive less than 12
months after diagnosis (3). Thus,
the establishment of effective clinical molecular markers for early
diagnosis and targeted molecular therapies are urgently needed for
CCC.
Among the numerous inhibitors of tyrosine kinase,
sorafenib (BAY 43-9006) has attracted considerable attention.
Sorafenib inhibited Raf-1 (4) and
BRAF (5), both members of the
RAF/mitogen-activated protein kinase (MEK)/extracellular
signal-related kinase (ERK) signaling pathway, and suppressed the
proliferation and growth of several human tumor cell lines and
xenograft models. The drug also exhibited significant activity
against multiple receptor tyrosine kinases involved in tumor
progression, including vascular endothelial growth factor receptor
(VEGFR)-2, VEGFR-3, c-KIT, Flt-3, and platelet-derived growth
factor receptor β (5).
Sorafenib is effective for hepatocellular carcinoma
(HCC) by virtue of prolonged median survival in advanced-stage
patients (6). The effects of
sorafenib, however, are far less understood for CCC. Sorafenib
exerted low activity in a phase II CCC trial (7), and not even a combination of sorafenib
and erlotinib could exhibit clinical activity in patients with
biliary cancers in a phase II trial (8). Thus, sorafenib is the first
molecular-targeted therapy to be approved for HCC, but not for CCC,
despite the fact that the liver and bile duct are derived from the
same embryological origin.
The in vitro antitumor activity of sorafenib
in human CCC has been assessed in several signaling pathways.
Blockage of the MAPK pathway by sorafenib inhibited cell
proliferation through cell cycle arrest (9), and sorafenib accelerated STAT3
dephosphorylation and induced TRAIL-mediated apoptosis (10).
Recently, the phosphoinositide 3-kinase
(PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway was
implicated in sorafenib-resistant HCC, whereby constitutive
activation of the mTOR pathway was present in drug-resistant HCC
cells (11). Increased AKT
phosphorylation was also witnessed in established
sorafenib-resistant HCC cells (12). We therefore focused our attention on
the AKT/mTOR pathway as a CCC escape mechanism from
RAF/MEK/ERK-mediated cell death under sorafenib based on
comparisons of HCC and CCC.
Two forms of mTOR protein complexes exist. mTORC1,
defined by the Raptor scaffolding protein (13), is sensitive to rapamycin. Activation
of mTORC1 triggers mitochondrial oxidative metabolism and
lipogenesis, which are critically important in tumorigenesis
(14). mTORC1 is also a negative
regulator of autophagy (15).
Characterized by Rictor, mTORC2 phosphorylates AKT on Ser 473,
regulates forkhead box protein (FOXO) activation, and modifies the
actin cytoskeleton with oaxilin and Rho GTPases (16). Inhibition of mTORC2 by Rictor
disruption decreases AKT-dependent tumor progression in
lapatinib-resistant HER2-amplified breast cancers (17). Accordingly, we examined the
influence of sorafenib on the AKT/mTOR pathway to observe how
dissociation of the mTORC2 component by Rictor knockdown altered
this pathway in CCC. Since activated mTORC1 has been reported in
CCC, we also combined everolimus with sorafenib to simultaneously
suppress mTORC1 under mTORC2 disassembly.
Materials and methods
Cell lines and culture
The human HCC cell lines HLF, Huh7, and PLC/PRF/5
and human intrahepatic CCC cell line Huh28 were obtained from the
Japanese Collection of Research Bioresources (JCRB) Cell Bank
(Osaka, Japan). The human intrahepatic CCC cell lines RBE and YSCCC
were procured from the RIKEN Cell Bank (Ibaraki, Japan). HCC cells
were maintained in Dulbecco's modified Eagle's medium and CCC cells
were maintained in RPMI-1640. All cultures were supplemented with
10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 µg/ml
streptomycin in a humidified atmosphere of 5% CO2 at
37°C.
Analysis of cell proliferation and
anchorage-independent growth assay
The MTT assay was used for analyzing cell
proliferation. At 24 h after inoculation of HCC and CCC cells, DMSO
(0.1% in culture medium) or 5 or 10 µM sorafenib were administered
and the cells were cultured for an additional 24 h. Ten microliters
WST-1 reagent (Roche Diagnostics GmbH, Mannheim, Germany) was added
to each well and the absorbance was assessed at 450 nm after 1 h of
incubation using an Epoch microplate reader (BioTek Instruments,
Inc., Winooski, VT, USA). We performed anchorage-independent assays
according to a previously described method (18). Briefly, cells were mixed in 0.36%
agar-containing medium with 10% FBS containing DMSO or 5 or 10 µM
sorafenib. The mixture was placed on a bed of 0.72% agar-containing
medium with 10% FBS and DMSO or 5 or 10 µM sorafenib in 35-mm
dishes. Three weeks after the inoculation, the colony areas were
assessed using NIH ImageJ software (Rockville, MD, USA).
Apoptosis assay
One day after inoculation, RBE cells were treated
with DMSO or 5 or 10 µM sorafenib for 24 h and cellular apoptosis
was examined by Annexin V and propidium iodide (PI) staining using
an Annexin V-FITC Apoptosis kit (BioVision, Inc., Milpitas, CA,
USA) according to the manufacturer's protocol. Following staining,
the cells were analyzed using flow cytometry (FACS) on a
FACSCalibur device (BD Biosciences, Franklin Lakes, NJ, USA).
Western blot analysis
Cells were lysed in lysis buffer containing 50 mM
Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium
deoxycholate, 0.1% SDS, 1X EDTA-free proteinase inhibitor cocktail
and 1X phosphatase inhibitor cocktail (both from Roche Diagnostics
GmbH) for 15 min at 4°C. The lysates were separated by 15% SDS-PAGE
and the proteins were transferred onto nitrocellulose membranes.
After blocking with 5% skim milk for 1 h, the membranes were
incubated with primary antibodies overnight at 4°C, and then
secondary antibodies conjugated with horseradish peroxidase were
applied for 1 h. Immune complexes were developed with ECL Select™
Western blotting detection reagent (Amersham, GE Healthcare Life
Sciences, Chicago, IL USA). The results were photographed with a
Molecular Imager ChemiDoc XRS device (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The integrated density of the immunoblots was
analyzed by Image Lab Software (Bio-Rad Laboratories, Inc.) and
results were expressed as a percentage of the immunoblots of the
internal control, β-actin. The anti-human antibodies used were as
follows: rabbit monoclonal antibodies: ERK (cat. no. 4695), p-ERK
(cat. no. 4376), AKT (cat. no. 4691), 0020P-AKT Thr 308 (cat. no.
2965), p-AKT Ser473 (cat. no. 4060), p-mTOR s2448 (cat. no. 5536),
Rictor (cat. no. 9476) and FOXO1 (cat. no. 2880) (all from Cell
Signaling Technology, Inc., Danvers, MA, USA); rabbit polyclonal
antibodies: p-mTOR s2481 (cat. no. 2974), p-FOXO1/3 (cat. no. 9464)
(Cell Signaling Technology, Inc.), LC3 (cat. no. PM036; MBL,
Nagoya, Japan); and mouse monoclonal antibody: β-actin (cat. no.
A5441; Sigma-Aldrich, St. Louis, MO, USA). The anti-β-actin
antibody was used at a 1:3,000 dilution and the other antibodies
were used at a 1:1,000 dilution.
Knockdown of Rictor via siRNA
transfection for disassembly of the mTORC2 complex in RBE
cells
Silencer Select siRNA was purchased from Life
Technologies/Thermo Fisher Scientific (Carlsbad, CA, USA) and
modified to target human Rictor by reverse transfection (19). Either 10 µM Silencer Select
non-targeting negative control or 10 µM Rictor siRNA was mixed with
Lipofectamine RNAiMAX (Life Technologies/Thermo Fisher Scientific)
according to the manufacturer's instructions and added to 35-mm
tissue culture plates. The cells were then plated onto
siRNA/Lipofectamine RNAiMAX complexes at a density of
1×105 cells/well in RPMI-1640 containing 5 mM glucose
and 10% FBS. At 48 h after transfection, Rictor knockdown was
confirmed by western blotting and the cells were subjected to
ensuing experiments.
Statistical analysis
Statistical significance was evaluated using the
Student's t-test on the data of 3–6 experiments for each assay.
P<0.05 was accepted as statistically significant. All values
were expressed as the mean ± standard error of the mean.
Results
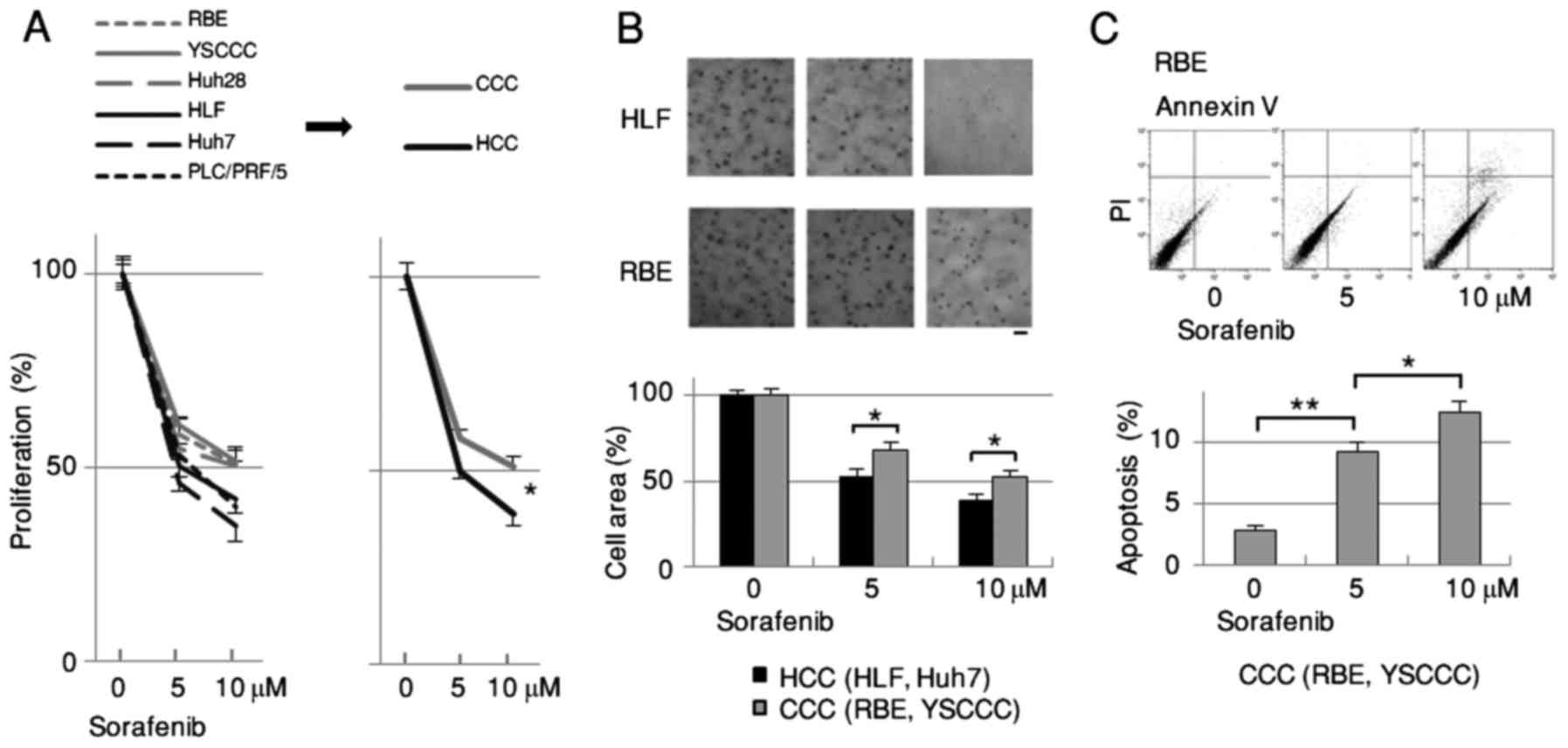
Sorafenib inhibits growth
significantly less in CCC than in HCC
Since sorafenib is the first molecular-targeted drug
approved for HCC but not for CCC, we compared its effects on the
proliferation of HCC and CCC cells. The degree of growth inhibition
by sorafenib was significantly less in CCC (RBE, YSCCC, Huh28) than
in HCC (HLF, Huh7, PLC/PRF/5) cells as assessed by MTT assay
(Fig. 1A). These results were
supported by the anchorage-independent assay comparing HCC (HLF,
Huh7) and CCC (RBE, YSCCC) cells 3 weeks after cell plating
(Fig. 1B). The apoptosis assay
using FACS revealed that the population of Annexin V-positive and
PI-negative cells increased dose-dependently by sorafenib in RBE
and YSCCC cells (Fig. 1C).
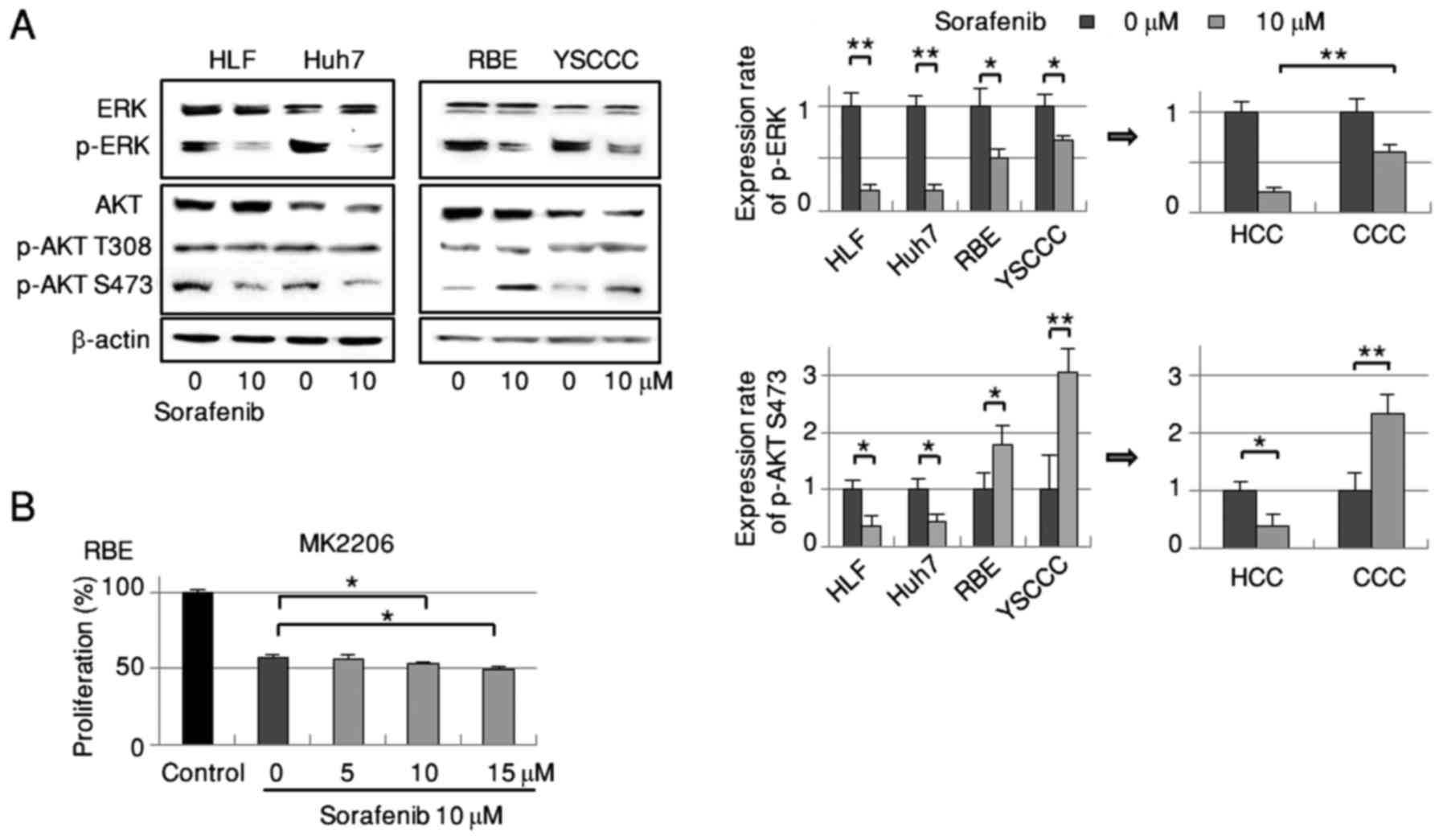
Phosphorylation of AKT Ser473 is
decreased in HCC but increased in CCC by sorafenib
Based on sorafenib's known inhibitory effects of
sorafenib on the RAF/MEK/ERK signaling pathway, we examined the
drug's impact on this signal transduction pathway in HCC and CCC
cells. Administration of sorafenib markedly suppressed ERK
phosphorylation in both cell types, with a significantly lower
suppression in CCC cells (Fig. 2A).
Regarding AKT Ser473, a signaling molecule in the PI3 kinase
pathway, phosphorylation was significantly decreased in HCC cells
by sorafenib treatment but significantly increased in CCC cells
(Fig. 2A). We observed no marked
alterations in AKT Thr308 phosphorylation in either cell type.
These findings raised the biochemical possibility of an escape
mechanism from the major RAF/MEK/ERK signaling pathway elicited by
activation of the AKT/mTOR signaling cascade in sorafenib treatment
for CCC.
We next inhibited the sorafenib-dependent increase
of AKT Ser473 phosphorylation in RBE cells using the selective
allosteric AKT inhibitor MK2206 to clarify the drug's growth
inhibitory effect. High dose (10 and 15 µM) administration of
MK2206 for 72 h significantly enhanced the suppression of cell
growth caused by 10 µM sorafenib-treated RBE cells in the MTT assay
(Fig. 2B).
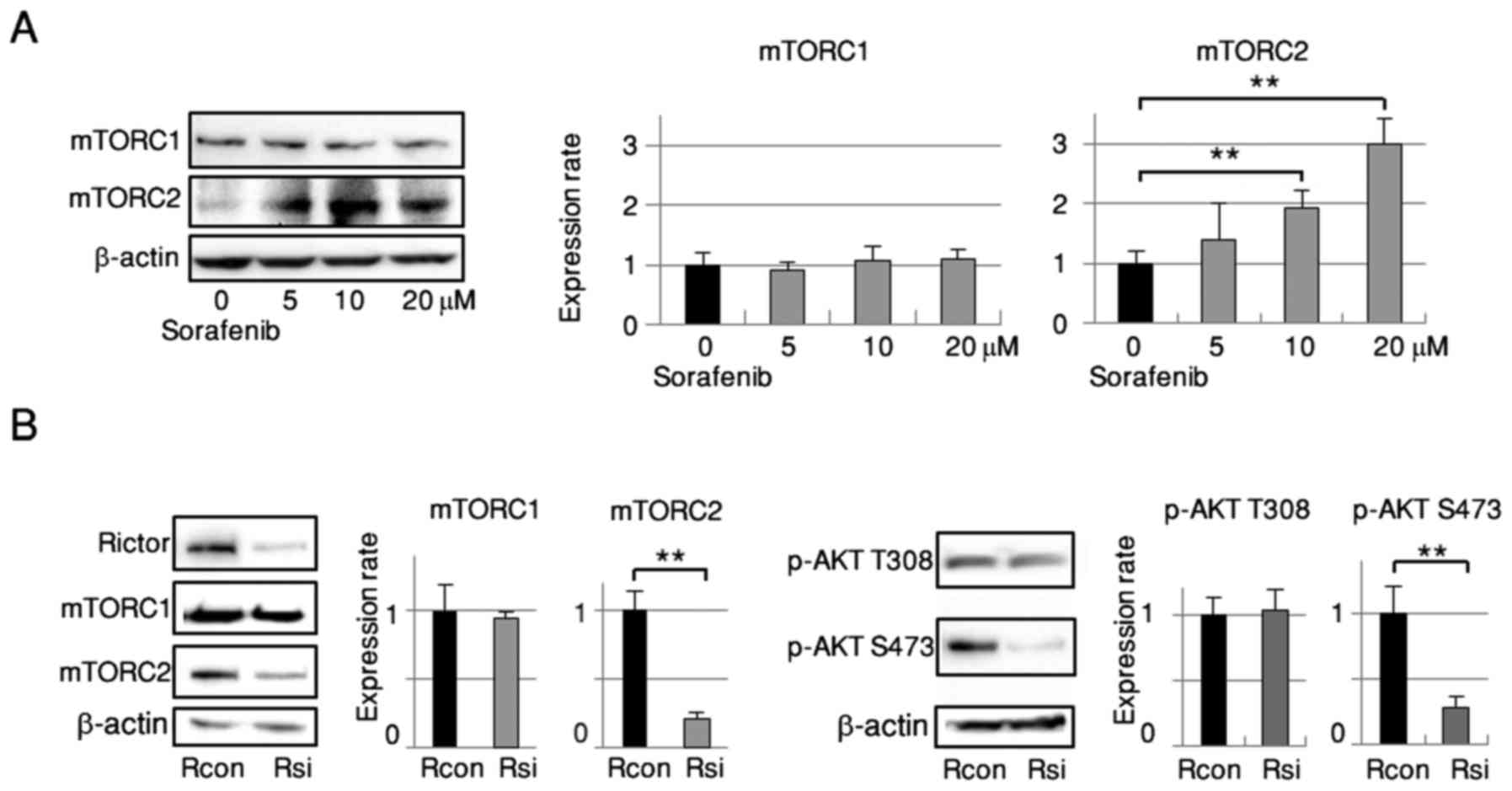
Downregulation of AKT Ser473
phosphorylation is obtained via disassembly of the mTORC2 complex
induced by Rictor silencing in RBE cells
Twenty-four hours after treatment with sorafenib,
dose-dependent activation of mTORC2 (mTOR Ser2481 phosphorylation)
was detected by western blot analysis (Fig. 3A) with no apparent alteration in
mTORC1 activation (mTOR Ser 2448 phosphorylation) (20). Since mTORC2 is located upstream of
AKT Ser473, we silenced it by means of siRNA to abrogate AKT Ser473
phosphorylation. RBE cells were transfected with control siRNA or
siRNA targeting Rictor, an essential and specific component of
mTORC2. As shown in Fig. 3B, Rictor
expression was markedly decreased and the phosphorylation of mTORC2
was significantly reduced. Phosphorylation of AKT Ser473 was
significantly suppressed as well. Rictor knockdown did not affect
mTORC1 or AKT Thr308 phosphorylation.
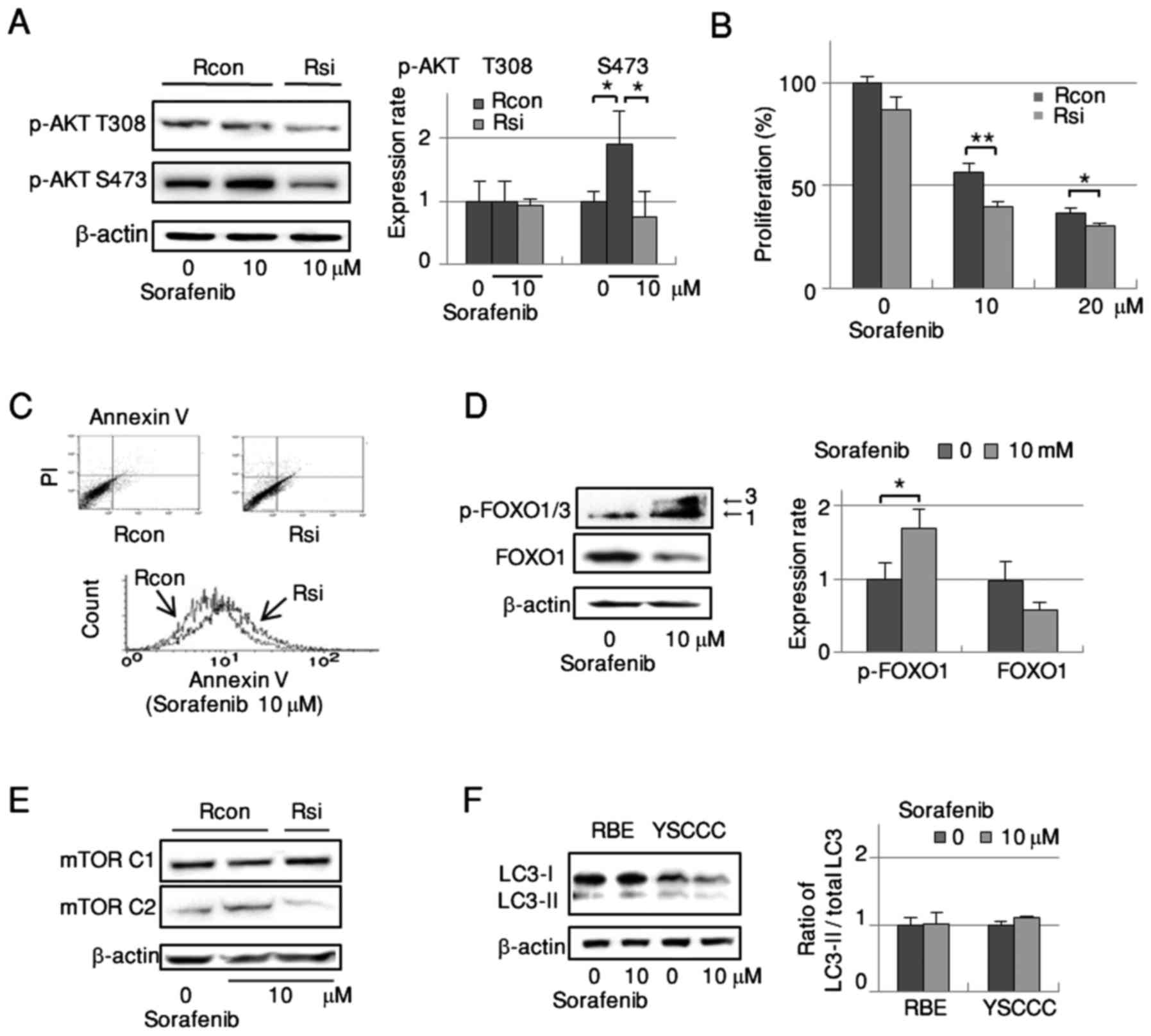
Disassembly of mTORC2 prevents
sorafenib-dependent activation of the AKT/mTOR pathway and enhances
the antitumor efficacy of sorafenib in RBE cells without affecting
autophagy
The increase of AKT Ser473 phosphorylation by
sorafenib was significantly abrogated in mTORC2-disassembled RBE
cells as detected by western blotting (Fig. 4A), while mTORC2 disassembly did not
affect the phosphorylation of AKT Thr308. In cell growth assays,
RBE cell proliferation was dose-dependently suppressed by sorafenib
treatment. This suppression was significantly stronger in
mTORC2-disassembled cells than in controls (Fig. 4B).
The growth suppression induced by sorafenib
treatment under mTORC2 disassembly corresponded with an increase in
apoptosis (Fig. 4C) as detected by
FACS. Therefore, we examined the effect of sorafenib on FOXO1 as an
inducer of cell death. Sorafenib treatment elicited marked
upregulation of FOXO1/3 phosphorylation along with a reduction of
FOXO1 expression (Fig. 4D).
Collectively, sorafenib appeared to enhance mTORC2 and AKT Ser473
phosphorylation and decrease FOXO1, which may have suppressed
apoptosis and consequently facilitated cell survival. Since mTORC1
is a negative regulator of autophagy, we searched for alterations
in mTORC1 and autophagy by sorafenib. Disassembly of mTORC2 with
sorafenib did not alter mTORC1 phosphorylation in RBE cells
(Fig. 4E). Sorafenib did not affect
autophagy in CCC cells as determined by western blot analysis of
LC3-II/total LC3 expression (Fig.
4F). Thus, sorafenib played no apparent role in the
autophagy-related pathway of CCC cells.
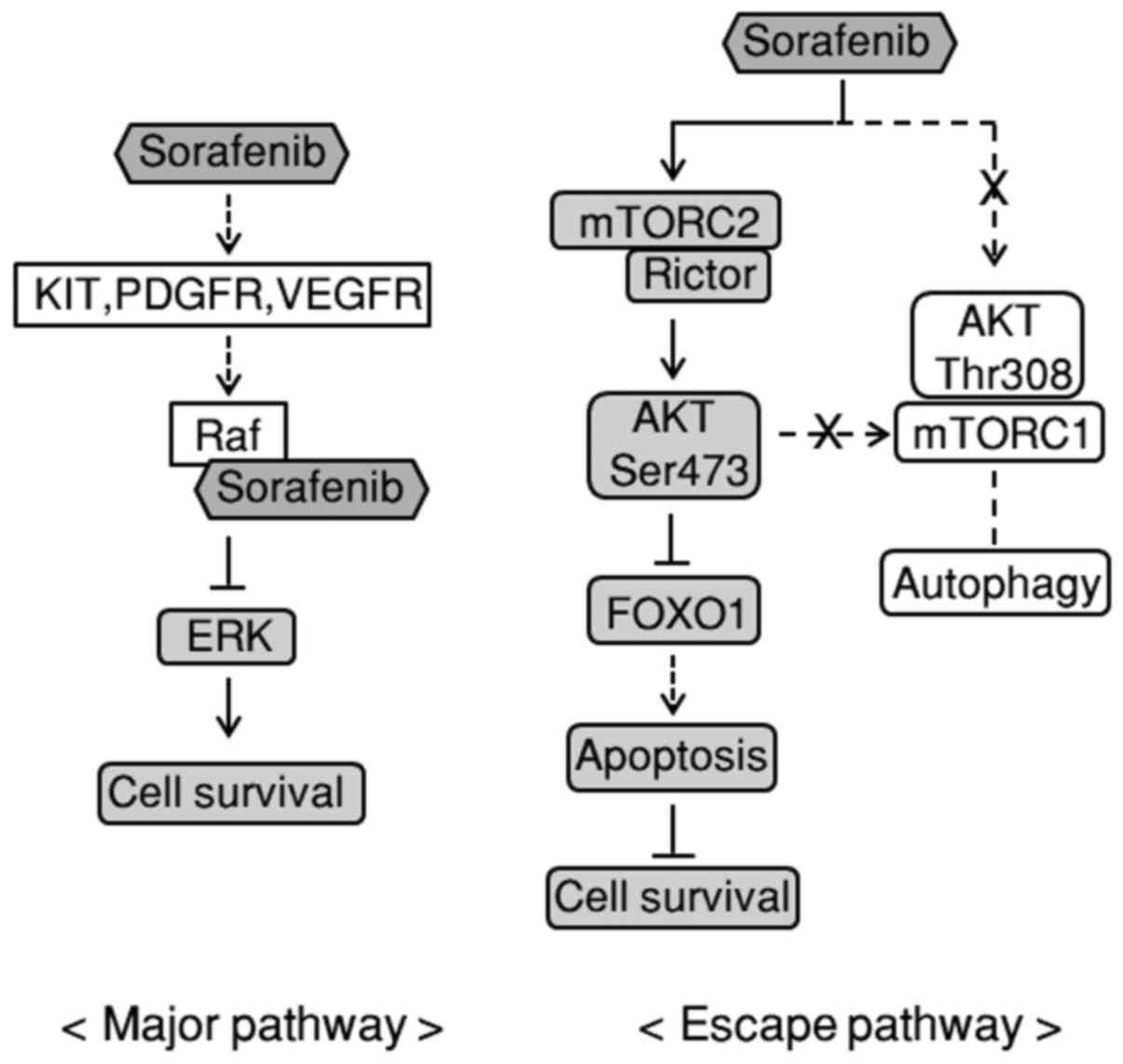
Schematic representation of an escape
mechanism via AKT/mTOR signaling from the RAF/MEK/ERK pathway
evoked by sorafenib in RBE cells
Based on the aforementioned findings, we speculated
that the AKT/mTOR pathway activated by sorafenib represented an
escape mechanism (Fig. 5, right)
from the RAF/MEK/ERK signaling pathway by which sorafenib normally
exerted its cell-death properties (Fig.
5, left). In the RAF/MEK/ERK pathway, inhibited ERK
phosphorylation and cell proliferation by sorafenib were lower in
CCC cells than in HCC cells. In the AKT/mTOR escape pathway,
sorafenib upregulated the phosphorylation of AKT Ser473 via mTORC2
activation without influencing mTORC1 or AKT Thr308 phosphorylation
through a yet unknown initial receptor. The upregulated AKT Ser473
by sorafenib decreased the expression level of FOXO1, presumably
leading to a decrease in apoptosis and consequently facilitating
cell survival. As disassembled mTORC2 with sorafenib did not
influence the phosphorylation of mTORC1, the cascade from AKT
Ser473 to mTORC1 by sorafenib was unaffected, and thus sorafenib
did not alter autophagy in CCC cells.
Combination of everolimus with
sorafenib under mTORC2 disassembly enhances the inhibitory effects
on cell growth in RBE cells
Everolimus is a potent inhibitor of mTORC1.
Disassembly of mTORC2 suppressed the everolimus-dependent
phosphorylation of mTORC2 and AKT Ser473 (Fig. 6A). Everolimus was then combined with
sorafenib under mTORC2 disassembly in RBE cells. As shown in
Fig. 6B, everolimus or sorafenib
alone suppressed cell growth, with the latter being enhanced by
mTORC2 dissociation. Furthermore, under mTORC2 disassembly,
combined treatment with everolimus and sorafenib more strongly
suppressed cell growth than did sorafenib alone. This enhanced
growth suppression corresponded with evident downregulation of
mTORC1, mTORC2, and AKT Ser473 phosphorylation (Fig. 6C). Unexpectedly, the phosphorylation
of AKT Ser473 was strongly suppressed by combined
everolimus/sorafenib treatment under mTORC2 disassembly.
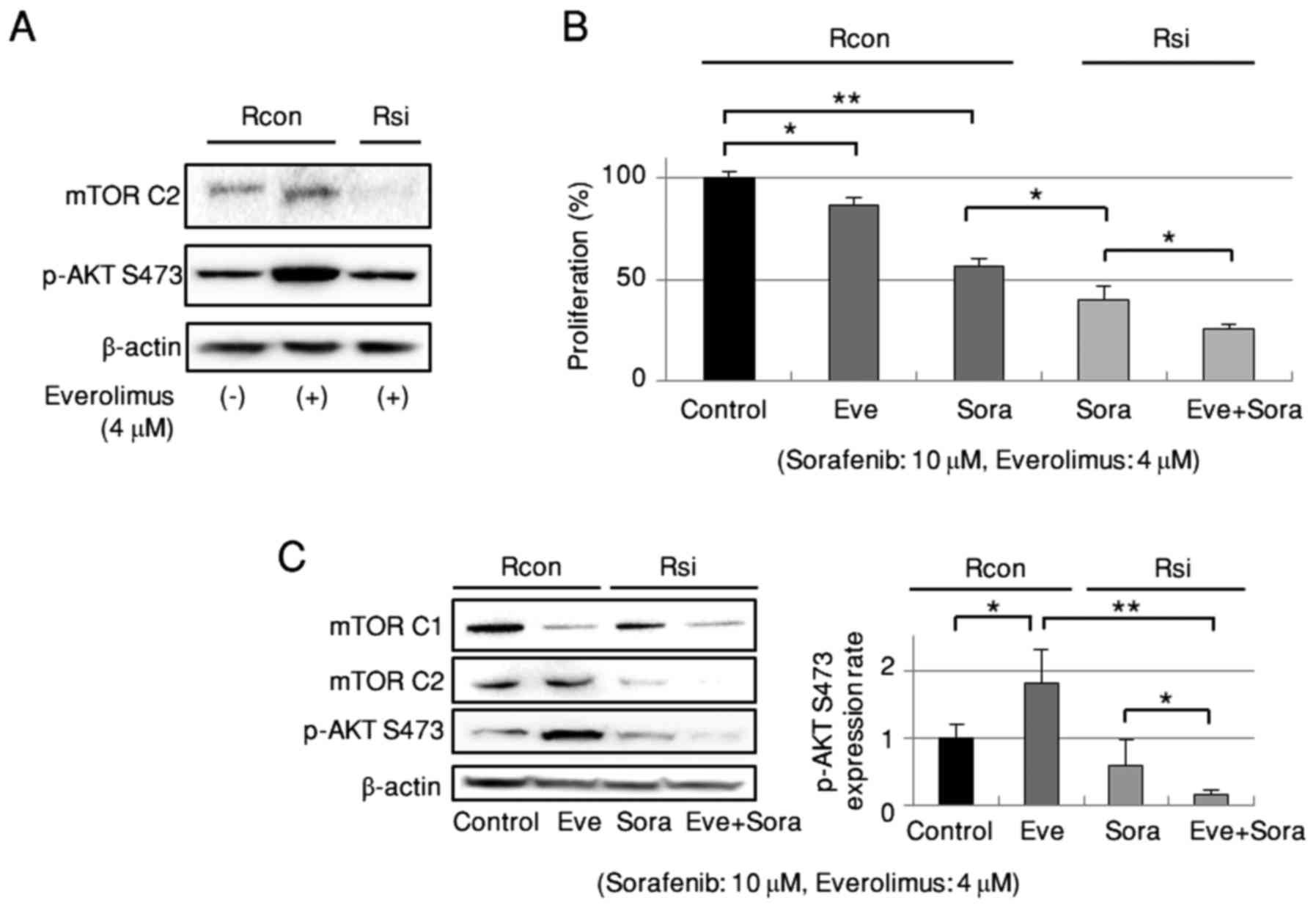 | Figure 6.Combined everolimus and sorafenib
treatment under disassembly of mTORC2 more effectively inhibits
proliferation of RBE cells. (A) Effect of mTORC2 disassembly on
everolimus-dependent AKT Ser473 phosphorylation. Representative
data of triplicate experiments. (B) Inhibitory effect on cell
proliferation of everolimus alone, sorafenib alone, sorafenib under
mTORC2 disassembly, and combined everolimus and sorafenib under
mTORC2 disassembly (n=6). (C) Phosphorylation of mTORC1, mTORC2,
and AKT Ser473 by everolimus alone, sorafenib under mTORC2
disassembly, and combined everolimus and sorafenib under mTORC2
disassembly (n=5). Rcon, control siRNA; Rsi, siRNA targeting
Rictor; Sora, sorafenib; Eve, everolimus. *P<0.05,
**P<0.01. |
Discussion
The present investigation uncovered a possible
escape mechanism of CCC from the RAF/MEK/ERK pathway by AKT/mTOR
signaling during sorafenib treatment. Since disassembly of the
mTORC2 complex led to an inhibition of AKT Ser473 phosphorylation
and suppressed cell growth, the prevention of AKT/mTOR pathway
function by suppressing mTORC2 during sorafenib treatment may be a
promising therapeutic option for CCC.
Constitutive activation of the AKT/mTOR pathway was
recently reported in sorafenib-resistant HCC cells (11). Decreases in AKT Ser473
phosphorylation in sorafenib-sensitive HCC cells vs. increases in
sorafenib-resistant HCC cells with sorafenib treatment have been
documented as well (21). In the
present study, the increased phosphorylation of AKT Ser473 in CCC
by sorafenib was similar to that in sorafenib-resistant HCC cells.
The inhibitory effects of the drug on the RAF/MEK/ERK signaling
pathway (9) and STAT3 pathway
(10) in CCC are well known.
However, the AKT/mTOR pathway has not yet been addressed, and thus
we focused on this cascade as a possible escape mechanism from cell
death via the RAF/MEK/ERK signaling pathway in sorafenib treatment
for CCC and searched for ways to abrogate the increase in AKT
Ser473 phosphorylation.
Our initial attempts to prevent sorafenib-dependent
AKT Ser473 phosphorylation employed the AKT inhibitor MK2206, which
could inhibit endogenous (22) and
everolimus-elicited (23)
phosphorylation of AKT in CCC. Similarly, MK2206 administration
with sorafenib significantly enhanced the suppression of cell
growth at high concentrations and prolonged treatment. We therefore
searched for a more effective method than MK2206 to suppress the
sorafenib-dependent increase of AKT Ser473 phosphorylation.
Sorafenib administration to RBE cells significantly
increased mTORC2 without altering mTORC1. Therefore, we
disassembled the mTORC2 complex using siRNA that targeted Rictor
(19) to effectively suppress the
phosphorylation of AKT Ser473 in RBE cells. mTORC2 regulates the
phosphorylation of AKT Ser473 (13). Our results demonstrated that
sorafenib administration following siRNA treatment significantly
reduced the phosphorylation of AKT Ser473 and more strongly
suppressed cell proliferation as compared with sorafenib treatment
alone in RBE cells. This finding was consistent with a study
revealing that the depletion of Rictor decreased AKT Ser473
phosphorylation and tumor cell survival in multiple amplified human
breast cancer cell lines, including those with acquired resistance
to lapatinib (17).
Since mTORC2 disassembly increased apoptosis, we
examined the involvement of the transcription factor FOXO1 in
cell-death activity. Sorafenib upregulated FOXO1/3 phosphorylation
and downregulated FOXO1 in RBE cells. According to Salazar et
al, increased phosphorylation of AKT on Ser473 enhanced the
phosphorylation and inactivation of FOXO3 (24). Phosphorylated FOXO exits the nucleus
for degradation. Moreover, mTORC2 inhibition increased the
expression level of FOXO1/3, and knockdown of FOXO3 abrogated
rhein-induced apoptosis (25).
Thus, we considered that the suppression of sorafenib-dependent AKT
Ser473 phosphorylation by mTORC2 disassembly induced apoptosis via
increased FOXO1.
Autophagy is a double-edged sword that depends on
its cellular context. Sorafenib treatment led to autophagy in HCC
cells, while pharmacological inhibition of this autophagy increased
apoptosis and decreased cell viability (26). In addition, the drug activated AKT
in sorafenib-resistant HCC cells, and inhibition of this activation
reversed the acquired resistance to sorafenib by switching
autophagy from cell survival to cell death (27). In CCC cells, however, the negative
autophagy regulator mTORC1 was not affected by sorafenib or mTORC2
disassembly. Sorafenib did not influence autophagy in CCC as
evidenced by LC3-II, although autophagy was increased in HCC (data
not shown). This indicated the existence of an escape mechanism
from cell death with sorafenib in CCC that was unrelated to
autophagy.
Our body of findings enabled the elucidation of a
possible escape mechanism via the AKT/mTOR pathway from the major
RAF/MEK/ERK pathway under sorafenib treatment in an RBE cell line
(Fig. 5). Sorafenib activated
mTORC2 and AKT Ser473 and inhibited apoptosis via suppressed FOXO1,
which consequently increased cell growth independently of
autophagy.
Lastly, since we detected constitutively
phosphorylated mTORC1 in RBE cells, we attempted to suppress mTORC1
by everolimus. The combined administration of sorafenib and
everolimus under mTORC2 disassembly produced additional growth
inhibitory effects by abrogating both sorafenib- and
everolimus-dependent AKT Ser473 phosphorylation in RBE cells.
In HCC, the efficacy of combined therapy with
rapamycin analogs and sorafenib has been demonstrated by the
suppression of mTORC1 activation and cell growth in
sorafenib-less-sensitive lines (27). Moreover, increased AKT
phosphorylation observed not only with sorafenib, but also with
rapamycin, in sorafenib-resistant HCC cells implied that feedback
activation of AKT may limit the rapamycin-mediated antitumor
effects (17). mTORC2 has been
proposed to be rapamycin insensitive (28). However, a recent study described
that everolimus induced mTORC2-mediated AKT Ser473 activation in
ovarian carcinoma and that inhibition of mTORC2 during treatment
enhanced the antitumor effects (29). Moreover, Pignochino et al
demonstrated that everolimus increased mTORC2 activity with mTORC1
suppression, and the combination of sorafenib and everolimus
potentiated the antiproliferative effect of each drug with
decreased phosphorylation of mTORC2 and AKT Ser473 in osteosarcoma
(30). Therefore, we hypothesized
that a feedback increase of AKT Ser473 phosphorylation elicited by
both sorafenib and everolimus would be simultaneously blocked by
disassembly of the mTORC2 component in RBE cells. In our
experiments, disassembly of mTORC2 suppressed everolimus-dependent
AKT Ser473 activation, and the combination of everolimus and
sorafenib under mTORC2 disassembly significantly prevented AKT
Ser473 phosphorylation and synergistically exerted a regulatory
effect on RBE cell proliferation. Thus, dissociation of mTORC2 may
disable the escape mechanism from sorafenib and permit the mTORC1
inhibitory effect of everolimus without a feedback increase of AKT.
We examined the phosphorylation of mTORC2 and AKT treated with a
combination of everolimus and sorafenib. Both phosphorylations were
slightly reduced compared with those produced by everolimus alone
(data not shown), although the degree was smaller than a study on
osteosarcoma by Pignochino et al (30). As they reported, this combination
may dissociate mTORC2 to some degree and contribute to the markedly
suppressed phosphorylation of AKT Ser473 by combination treatment
under mTORC2 disassembly in RBE cells shown in Fig. 6C.
In conclusion, although RAF kinases are one of the
main molecules targeted by sorafenib, intervention of active
AKT/mTOR signaling in the RAF/MEK/ERK pathway may be one of the
mechanisms responsible for the resistance of CCC to sorafenib.
AKT/mTOR pathway regulation is exceedingly complex due to multiple
feedback loops and direct activation mechanisms. Nonetheless,
suppression of mTORC2 activity by microRNA targeting of Rictor
should be considered in potential therapies combining sorafenib and
everolimus for CCC malignancies.
Acknowledgements
Sorafenib was kindly provided by Bayer Health Care
Pharmaceuticals (Berlin, Germany). This study was supported by a
grant-in-aid for General Scientific Research (JP26293300) from the
Ministry of Education, Culture, Sports, Science and Technology of
Japan.
References
|
1
|
Lazaridis KN and Gores GJ:
Cholangiocarcinoma. Gastroenterology. 128:1655–1667. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Patel T: Worldwide trends in mortality
from biliary tract malignancies. BMC Cancer. 2:102002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gores GJ: Cholangiocarcinoma: Current
concepts and insights. Hepatology. 37:961–969. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lyons JF, Wilhelm S, Hibner B and Bollag
G: Discovery of a novel Raf kinase inhibitor. Endocr Relat Cancer.
8:219–225. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wilhelm SM, Carter C, Tang L, Wilkie D,
McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al:
BAY 43-9006 exhibits broad spectrum oral antitumor activity and
targets the RAF/MEK/ERK pathway and receptor tyrosine kinases
involved in tumor progression and angiogenesis. Cancer Res.
64:7099–7109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al SHARP Investigators Study Group, : Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:378–390. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bengala C, Bertolini F, Malavasi N, Boni
C, Aitini E, Dealis C, Zironi S, Depenni R, Fontana A, Del Giovane
C, et al: Sorafenib in patients with advanced biliary tract
carcinoma: A phase II trial. Br J Cancer. 102:68–72. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
El-Khoueiry AB, Rankin C, Siegel AB, Iqbal
S, Gong IY, Micetich KC, Kayaleh OR, Lenz HJ and Blanke CD: S0941:
A phase 2 SWOG study of sorafenib and erlotinib in patients with
advanced gallbladder carcinoma or cholangiocarcinoma. Br J Cancer.
110:882–887. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sugiyama H, Onuki K, Ishige K, Baba N,
Ueda T, Matsuda S, Takeuchi K, Onodera M, Nakanuma Y, Yamato M, et
al: Potent in vitro and in vivo antitumor activity of sorafenib
against human intrahepatic cholangiocarcinoma cells. J
Gastroenterol. 46:779–789. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Blechacz BR, Smoot RL, Bronk SF, Werneburg
NW, Sirica AE and Gores GJ: Sorafenib inhibits signal transducer
and activator of transcription-3 signaling in cholangiocarcinoma
cells by activating the phosphatase shatterproof 2. Hepatology.
50:1861–1870. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Masuda M, Chen WY, Miyanaga A, Nakamura Y,
Kawasaki K, Sakuma T, Ono M, Chen CL, Honda K and Yamada T:
Alternative mammalian target of rapamycin (mTOR) signal activation
in sorafenib-resistant hepatocellular carcinoma cells revealed by
array-based pathway profiling. Mol Cell Proteomics. 13:1429–1438.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhai B, Hu F, Jiang X, Xu J, Zhao D, Liu
B, Pan S, Dong X, Tan G, Wei Z, et al: Inhibition of Akt reverses
the acquired resistance to sorafenib by switching protective
autophagy to autophagic cell death in hepatocellular carcinoma. Mol
Cancer Ther. 13:1589–1598. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chiarini F, Evangelisti C, McCubrey JA and
Martelli AM: Current treatment strategies for inhibiting mTOR in
cancer. Trends Pharmacol Sci. 36:124–135. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Laplante M and Sabatini DM: mTOR signaling
at a glance. J Cell Sci. 122:3589–3594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dunlop EA and Tee AR: mTOR and autophagy:
A dynamic relationship governed by nutrients and energy. Semin Cell
Dev Biol. 36:121–129. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Morrison Joly M, Hicks DJ, Jones B,
Sanchez V, Estrada MV, Young C, Williams M, Rexer BN, Sarbassov D,
Muller WJ, et al: Rictor/mTORC2 drives progression and therapeutic
resistance of HER2-amplified breast cancers. Cancer Res.
76:4752–4764. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Horiuchi A, Nikaido T, Taniguchi S and
Fujii S: Possible role of calponin h1 as a tumor suppressor in
human uterine leiomyosarcoma. J Natl Cancer Inst. 91:790–796. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Soares HP, Ming M, Mellon M, Young SH, Han
L, Sinnet-Smith J and Rozengurt E: Dual PI3K/mTOR Inhibitors Induce
Rapid Overactivation of the MEK/ERK Pathway in Human Pancreatic
Cancer Cells through Suppression of mTORC2. Mol Cancer Ther.
14:1014–1023. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Copp J, Manning G and Hunter T:
TORC-specific phosphorylation of mammalian target of rapamycin
(mTOR): phospho-Ser2481 is a marker for intact mTOR signaling
complex 2. Cancer Res. 69:1821–1827. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guan DX, Shi J, Zhang Y, Zhao JS, Long LY,
Chen TW, Zhang EB, Feng YY, Bao WD, Deng YZ, et al: Sorafenib
enriches epithelial cell adhesion molecule-positive tumor
initiating cells and exacerbates a subtype of hepatocellular
carcinoma through TSC2-AKT cascade. Hepatology. 62:1791–1803. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wilson JM, Kunnimalaiyaan S,
Kunnimalaiyaan M and Gamblin TC: Inhibition of the AKT pathway in
cholangiocarcinoma by MK2206 reduces cellular viability via
induction of apoptosis. Cancer Cell Int. 15:132015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ewald F, Grabinski N, Grottke A, Windhorst
S, Nörz D, Carstensen L, Staufer K, Hofmann BT, Diehl F, David K,
et al: Combined targeting of AKT and mTOR using MK-2206 and RAD001
is synergistic in the treatment of cholangiocarcinoma. Int J
Cancer. 133:2065–2076. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Salazar M, Lorente M, García-Taboada E,
Pérez Gómez E, Dávila D, Zúñiga-García P, María Flores J, Rodríguez
A, Hegedus Z, Mosén-Ansorena D, et al: Loss of Tribbles
pseudokinase-3 promotes Akt-driven tumorigenesis via FOXO
inactivation. Cell Death Differ. 22:131–144. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang J, Liu S, Yin Y, Li M, Wang B, Yang L
and Jiang Y: FOXO3-mediated up-regulation of Bim contributes to
rhein-induced cancer cell apoptosis. Apoptosis. 20:399–409. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shimizu S, Takehara T, Hikita H, Kodama T,
Tsunematsu H, Miyagi T, Hosui A, Ishida H, Tatsumi T, Kanto T, et
al: Inhibition of autophagy potentiates the antitumor effect of the
multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J
Cancer. 131:548–557. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huynh H, Ngo VC, Koong HN, Poon D, Choo
SP, Thng CH, Chow P, Ong HS, Chung A and Soo KC: Sorafenib and
rapamycin induce growth suppression in mouse models of
hepatocellular carcinoma. J Cell Mol Med. 13(8B): 1–2683. 2009.
View Article : Google Scholar
|
|
28
|
Jacinto E, Loewith R, Schmidt A, Lin S,
Rüegg MA, Hall A and Hall MN: Mammalian TOR complex 2 controls the
actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol.
6:1122–1128. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hisamatsu T, Mabuchi S, Matsumoto Y,
Kawano M, Sasano T, Takahashi R, Sawada K, Ito K, Kurachi H,
Schilder RJ, et al: Potential role of mTORC2 as a therapeutic
target in clear cell carcinoma of the ovary. Mol Cancer Ther.
12:1367–1377. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pignochino Y, Del l'Aglio C, Basiricò M,
Capozzi F, Soster M, Marchiò S, Bruno S, Gammaitoni L, Sangiolo D,
Torchiaro E, et al: The combination of sorafenib and everolimus
abrogates mTORC1 and mTORC2 upregulation in osteosarcoma
preclinical models. Clin Cancer Res. 19:2117–2131. 2013. View Article : Google Scholar : PubMed/NCBI
|