Introduction
Triple-negative breast cancer (TNBC) is a
heterogeneous disease group characterized by lack of estrogen
receptor (ER), progesterone receptor (PR) and human epidermal
growth factor 2 (Her2) expression. Contrary to ER-positive and
Her2-positive breast cancers, which are treated with targeted
therapies such as tamoxifen and herceptin, the management of TNBC
is not standardized and it is based on the use of traditional
cytotoxic drugs that induce a plethora of side-effects. In
addition, conventional chemotherapy is not always effective for the
treatment of these tumors and many patients relapse, normally with
fatal consequences. Therefore, there is an urgent requirement for
more specific therapies for TNBC (1).
Olaparib is an oral inhibitor of poly(ADP-ribose)
polymerase (PARP), which blocks base-excision repair by trapping
PARP at the site of DNA damage, thus leading to the collapse of DNA
replication forks and the accumulation of DNA double stranded
breaks (DSBs) (2). Therefore, PARP
inhibition has been identified as a targeted therapy that may
exploit intrinsic defects in numerous cancer cells, and has been
reported to be selectively cytotoxic in breast cancer harboring
germ line mutations in BRCA1, DNA repair-associated (BRCA1) and
BRCA2, DNA repair-associated (BRCA2) (3).
The phosphatidylinositol 3-kinase (PI3K) pathway is
an important signaling network that regulates essential cellular
functions, including cell growth, proliferation and survival
(4,5). NVP-BKM120 (BKM120) is a pan-class I
PI3K inhibitor currently in Phase I/II clinical trials (6,7), which
has been reported to exert antiproliferative, pro-apoptotic and
antitumor activity in various cell lines, as well as in xenograft
models of cancers with or without aberrant PI3K pathway activation
(8,9). The combination of the PI3K inhibitor,
BKM120, and the PARP inhibitor olaparib exhibits synergistic
therapeutic effects on a genetic mouse model of BRCA1-associated
breast cancer, as well as on the treatment of BRCA1-proficient TNBC
(10). The results from these
studies have prompted clinical investigations into the combined use
of inhibitors of PI3K and PARP, and phase I clinical trials are
currently enrolling patients with TNBC (11).
The single agent carboplatin (CBP) has been
extensively investigated, and its effects have been analyzed in a
trial on patients with TNBC enriched for patients with cancers
harboring BRCA mutations (12). CBP
crosslinks with purine bases in the DNA, interfering with DNA
repair mechanisms, thus leading to DNA damage and the induction of
apoptosis. However, DNA damage triggers intrinsic repair
mechanisms, which are associated with drug resistance (13). To ensure that cell cycle progression
takes places without errors and in an orderly fashion,
non-homologous end joining (NHEJ) repair begins in the
G1 phase and homologous recombination (HR) begins in the
S/G2 phase; in addition, the repair of DSBs is strictly
regulated by the cell cycle (14).
The targeting of DNA repairs with DNA DSB-inducing agents, such as
platinum compounds, may be beneficial for the treament of patients
with breast cancer that are BRCA1 or BRCA2 mutation carriers
(15). Therefore, blocking DNA
repair pathways is a logical strategy for the development of
therapeutic options.
The present study aimed to explore the effects of a
combination of CBP, olaparib and BKM120 on a TNBC cell model. The
results detected a strong synergistic effect, providing a strong
rationale for the use of this combination in further studies using
animal models and future clinical trials on patients with TNBC.
Materials and methods
Cell lines, culture conditions and
reagents
MDA-MB-231 and MCF-7 cells were obtained from the
American Type Culture Collection (Manassas, VA, USA). CAL51 cells
were obtained from the German Collection of Microorganisms and Cell
Cultures (Leibniz Institute DSMZ, Braunschweig, Germany). All cells
were maintained in RPMI-1640 medium (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) supplemented with 10% fetal bovine serum
(Corning Incorporated, Corning, NY, USA) and 1%
penicillin-streptomycin (Corning Incorporated) at 37°C in an
atmosphere containing 95% air and 5% CO2. The triple
negative phenotype was validated by immunoblotting (data not
shown). Cells were discarded 15 passages after initial culture. The
PARP inhibitor olaparib (AstraZeneca, Cambridge, UK) was dissolved
in dimethyl sulfoxide (DMSO) at a final concentration of 10 mM;
BKM120 was purchased from Selleck Chemicals (Houston, TX, USA) and
dissolved in DMSO at a final concentration of 1 mM; and CBP was
purchased from Qilu Pharmaceutical Co., Ltd. (Jinan, China) and
dissolved in phosphate-buffered saline (PBS) at a final
concentration of 40 µM. All stock solutions were stored at
−20°C.
Toxicity assay
Drug toxicity was determined as previously described
(16). Briefly, cells were seeded
in 96-well plates (3,000 cells/well) in triplicate and were treated
with a range of concentrations of the indicated drugs, as depicted
in Fig. 1, at 37°C for 72 h.
Controls were treated with vehicle (either PBS or DMSO). Drug
toxicity was assessed after staining with MTT (Beijing Solarbio
Science & Technology Co., Ltd., Beijing, China) by measuring
the production of formazan, which is directly proportional to the
number of viable cells. Briefly, to each well of a 96-well plate
containing 100 µl medium, 10 µl MTT solution (5 mg/ml MTT in PBS)
was added and incubated at 37°C for 2 h. Solubilization of formazan
was achieved following the addition of 100 µl solubilization
solution [40% dimethylformadide v/v in 2% glacial acetic acid v/v,
to which 16% sodium dodecyl sulphate (pH 4.7) had been added w/v]
and reading optical density at 570 nm. For drug combination
experiments, a combination index (CI) was calculated using CalcuSyn
2.0 software (Biosoft, Cambridge, UK) based on the Chou and Talalay
method (17). CI values between 0.1
and 0.9 refer to different grades of synergism, values between 0.9
and 1.1 refer to additive effects, whereas values >1.1 refer to
antagonistic effects.
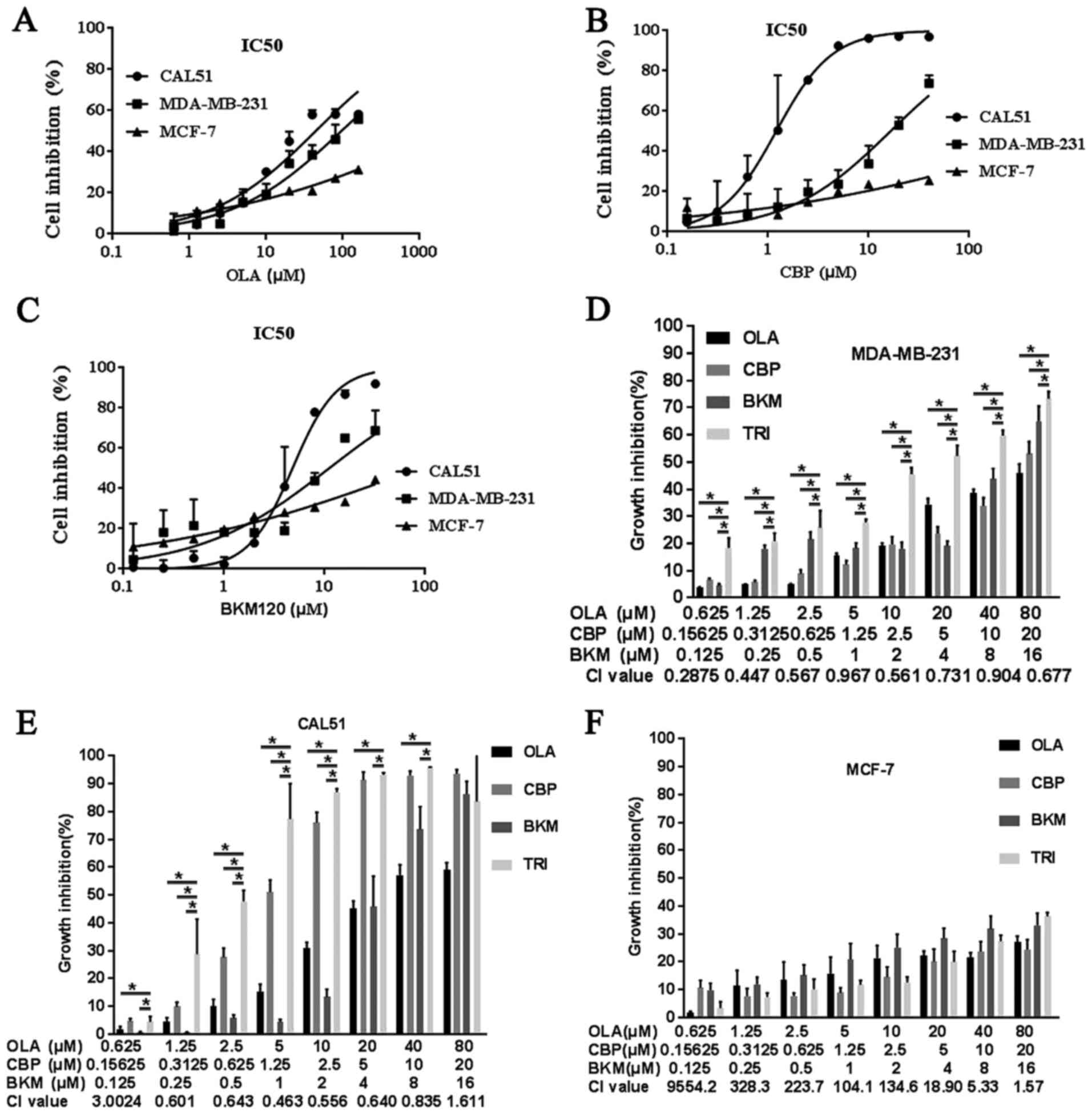 | Figure 1.OLA, CBP and BKM exert synergistic
toxicity on TNBC cells. MTT assays were used to determine toxicity
profiles. TNBC MDA-MB-231 and CAL51 cells, and the non-TNBC cell
line MCF-7, were treated with (A) CBP, (B) BKM or (C) OLA alone, or
(D-F) in combination for 72 h. Data are presented as the means ±
standard deviation from three independent experiments. *P<0.05.
BKM, NVP-BKM120; CBP, carboplatin; CI, combination index;
IC50, half maximal inhibitory concentration; OLA,
olaparib; TNBC, triple-negative breast cancer. |
Colony forming assay
The capacity of cells to survive and proliferate
following drug treatment was determined, according to a previously
described method (16). Briefly,
cells were seeded in 6-well plates (1×105 cells/well),
and were treated with 2.5 µM olaparib, 1 µM CBP and/or 1 µM BKM120,
after 1 day. After 10 days at 37°C, the surviving proliferating
clones were stained with 0.4 % (w/v) crystal violet for 30 min at
room temperature. The dye was solubilized using 33% (v/v) acetic
acid and the optical density was determined at 592 nm.
Cell cycle analysis
Flow cytometric analysis was used to evaluate
alterations in the cell cycle, as previously described (18). Cells (1×106) were fixed
in 70% ethanol overnight at 4°C, washed twice with PBS, treated
with RNase A (Sigma-Aldrich; Merck KGaA; final concentration, 50
µg/ml) for 15 min at 4°C and stained with propidium iodide
(Sigma-Aldrich; Merck KGaA; final concentration, 50 µg/ml)
overnight at 4°C. Samples were analyzed using a BD FACSCanto II
(Becton Dickinson, San Jose, CA, USA) flow cytometer, in order to
determine the proportion of cells at each stage of the cell cycle
using flow cytometry software (ModFit LT; Verity Software House,
Inc., Topsham, ME, USA).
Western blotting
Protein expression was determined as previously
described (16). Briefly, cells
were harvested and suspended in radioimmunoprecipitation assay
lysis buffer (Beijing Solarbio Science & Technology Co., Ltd.)
supplemented with protease and phosphatase inhibitors (1 mM sodium
fluoride, 2 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl
fluoride), placed on ice for 30 min, and centrifuged at 300 × g for
10 min at 4°C. The supernatant was collected and protein
concentration was determined using a Bicinchoninic Acid Protein
Assay kit (Pierce; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Proteins (50 µg for H2AX; 35 µg for all others) were
separated by 8% SDS-PAGE and were transferred onto a polvinylidene
fluoride membrane (EMD Millipore, Billerica, MA, USA).
Subsequently, the membrane, which was previously treated with 5%
(w/v) bovine serum albumin (BSA; Sigma-Aldrich; Merck KGaA) for 6 h
at 4°C to block non-specific antibody binding, was incubated
overnight at 4°C with the following specific primary antibodies:
Polyclonal anti-PARP (cat. no. 9542) and anti-53BP1 (cat. no.
4937); monoclonal anti-phosphorylated (p)-H2AX (Ser139; cat. no.
9718), anti-p-checkpoint kinase (Chk)1 (Ser345; cat. no. 2348),
anti-p-Chk2 (Thr68; cat. no. 2197), anti-p-p53 (Ser15; cat. no.
9286) anti-p-ATM serine/threonine kinase (ATM; Ser1981; cat. no.
5883), anti-p27 Kip1 (cat. no. 3686) and anti-p21 (cat. no. 2947)
(all at 1:1,000 dilution; all Cell Signaling Technology, Inc.,
Danvers, MA, USA); polyclonal anti-β-actin (cat. no. YT0099; at
1:2,000 dilution; ImmunoWay Biotechnology Company, Plano, TX, USA);
and monoclonal anti-PARP polymer (PADPR; cat. no. ab14459; at
1:1,000 dilution; Abcam, Cambridge, UK). After washing, the
membrane was incubated with anti-rabbit or anti-mouse secondary
antibodies (cat. nos. 7074 and 7076, respectively; Cell Signaling
Technology, Inc.) for 2 h at room temperature. Blots were
visualized using enhanced chemiluminiscence (Pierce; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Semi-quantification of blots was performed using ImageJ software
version 1.41o (National Institutes of Health, Bethesda, MD,
USA).
Immunofluorescence
Cells were washed with 1X PBS, fixed with methanol
and acetone (1:1, v:v) for 5 min, and then blocked with 3% BSA for
1 h at room temperature. Cells were incubated with the following
antibodies for 12 h at 4°C: Anti-BRCA1 (cat. no. YT0519),
anti-53BP1 (cat. no. YT0024) and anti-DNA repair protein RAD51
homolog 1 (RAD51; cat. no. YT3967) (all ImmunoWay Biotechnology
Company), and γ-H2AX (Ser139; cat. no. 9718; Cell Signaling
Technology, Inc.). Antibodies were used at a 1:500 dilution in 1%
BSA. After washing, samples were incubated with Alexa Fluor
488-conjugated secondary antibody (1:500 dilution; cat. no. 913921;
Invitrogen; Thermo Fisher Scientific, Inc.) for 1 h at room
temperature, after which, nuclei were stained with DAPI
(Sigma-Aldrich; Merck KGaA) for 5 min. Cells were washed with 1X
PBS and were observed under a fluorescence microscope (Carl Zeiss
AG, Oberkochen, Germany).
Statistical analysis
Data are presented as the means ± standard
deviation. Differences between groups were analyzed by one-way
analysis of variance and Dunnett's test using SPSS 22.0 software
(IBM Corp., Armonk, NY, USA). P<0.05 (two-tailed) was considered
to indicate a statistically significant difference.
Results
Treatment with a combination of
olaparib, CBP and BKM120 synergistically inhibits TNBC cell
growth
In order to assess the inhibitory effects of the
three drugs on the growth of MDA-MB-231 and CAL51 cells, the
present study determined the half maximal inhibitory concentration
(IC50) values after 72 h using MTT assays (Table I). Olaparib, CBP and BKM120
exhibited dose-dependent inhibition of MDA-MB-231 and CAL51 cells
(Fig. 1A-C); however, the
inhibitory effects of single drug treatment were limited. CBP and
BKM120 had relatively low IC50 values in CAL51 cells
(1.2 and 4.8 µM, respectively). Conversely, the toxicity of
olaparib in CAL51 and MDA-MB-231 cells was modest (IC50:
44.1 and 95.1 µM, respectively). In order to determine whether
olaparib toxicity could be increased when used in combination,
cells were treated with all three drugs together at various
concentrations. Notably, the combination indexes were <0.9
(Fig. 1D and E), thus indicating a
synergistic inhibitory effect that was stronger in MDA-MB-231 than
in CAL51 cells, which could limit the adverse effects of olaparib
in clinical use (16). As expected,
the IC50 values of each drug when used in combination
were much lower than those obtained for individual drugs (Table I) and, this effect was only observed
in TNBC cells; no synergistic effect was detected in MCF-7 cells
(CI values >1.1) (Fig. 1F).
 | Table I.Olaparib, CBP and BKM120 exert
synergistic inhibitory effects on triple-negative breast cancer
cell viability. |
Table I.
Olaparib, CBP and BKM120 exert
synergistic inhibitory effects on triple-negative breast cancer
cell viability.
|
| IC50
(µM) Drug alonea | IC50
(µM) Drug combinationb |
|---|
|
|
|
|
|---|
| Cell line | Olaparib | CBP | BKM120 | Olaparib | CBP | BKM120 |
|---|
| CAL51 | 44.1 | 1.2 | 4.8 | 3.3 | 0.8 | 0.6 |
| MDA-MB-231 | 95.1 | 16.5 | 11.1 | 12.5 | 3.1 | 2.5 |
To evaluate the efficacy of the three drug
combination on colony formation, cell cultures were treated for 10
days with drugs, either alone or in combination, and were then
stained with crystal violet (Fig.
2). Confirming the results of the aforementioned short-term
sensitivity assays, individual drugs inhibited proliferation of
TNBC cells to some extent and olaparib was the least toxic drug. A
combination of two drugs improved inhibition of MDA-MB-231 and
CAL51 cell proliferation; the inhibitory effect was strongest in
response to olaparib and CBP. Notably, the strongest inhibitory
effect was observed when the three drugs were used in
combination.
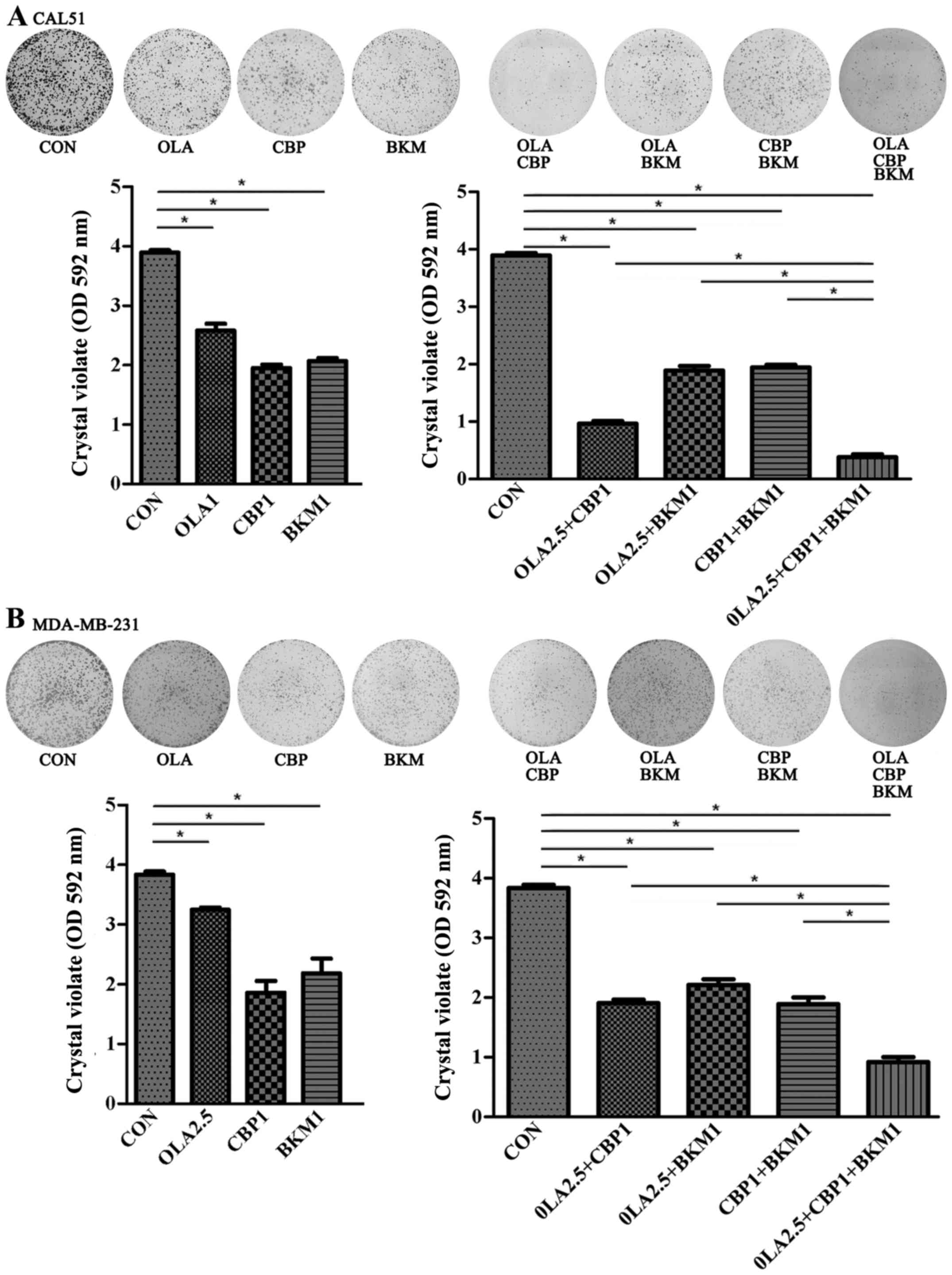 | Figure 2.OLA, CBP and BKM exert synergistic
inhibitory effects on triple-negative breast cancer cell
proliferation. (A) CAL51 and (B) MDA-MB-231 cells were seeded in
6-well plates and after 1 day were treated with the drugs, either
alone or in combination; cells were treated for 10 days before
being stained with crystal violet. Concentrations used were: OLA,
2.5 µM; CBP, 1 µM; BKM, 1 µM. The dye was solubilized and the
optical density was measured at 592 nm. Images are representative
of three experiments. Numerical data are presented as the means ±
standard deviation of three independent experiments. *P<0.05.
BKM, NVP-BKM120; CBP, carboplatin; CON, control (vehicle); OD,
optical density; OLA, olaparib. |
The results of these short- and long-term drug
assays indicated that olaparib, CBP and BKM120 may exert a strong
synergistic inhibitory effect on TNBC cells.
Olaparib, CBP and BKM120 affect cell
cycle progression in TNBC cells
In order to gain insight into the possible
mechanisms underlying the toxic effects of olaparib, CBP and BKM120
on TNBC cells, the present study assessed their effects on cell
cycle progression by flow cytometry (Fig. 3). As single drugs, olaparib and CBP
led to an increase in the proportion of CAL51 cells at the
G2/M phase of the cell cycle (4.6 and 9.2% increase
compared with the control group respectively), whereas in
MDA-MB-231 cells this was only apparent with olaparib (6.5%
increase compared with the control group), as CBP produced an
increase in the proportion of cells in S phase (10.1% increase
compared with the control group). In both cells, BKM120 had little
effect. Although there is no conclusive evidence regarding this
differential effect between cell lines, it may be hypothesized that
this is likely due to their different doubling times; CAL51 cells
divide faster than MDA-MB-231 cells. Therefore, the effects of some
drugs are seen later in CAL51 (G2/M) than in MDA-MB-231
cells (S phase). Combining olaparib and CBP led to an increase in
the proportion of cells in G2/M phase in CAL51 cells
(16.3% increase) and S phase in MDA-MB-231 cells (10.1% increase).
As expected, the strongest effect on cell cycle arrest was observed
when the three drugs were combined: CAL51 cells exhibited a 20.6%
increase in the number of cells at G2/M phase, whereas
MDA-MB-231 cells exhibited a 14.2% increase in the number of cells
at S phase, all compared with the control group.
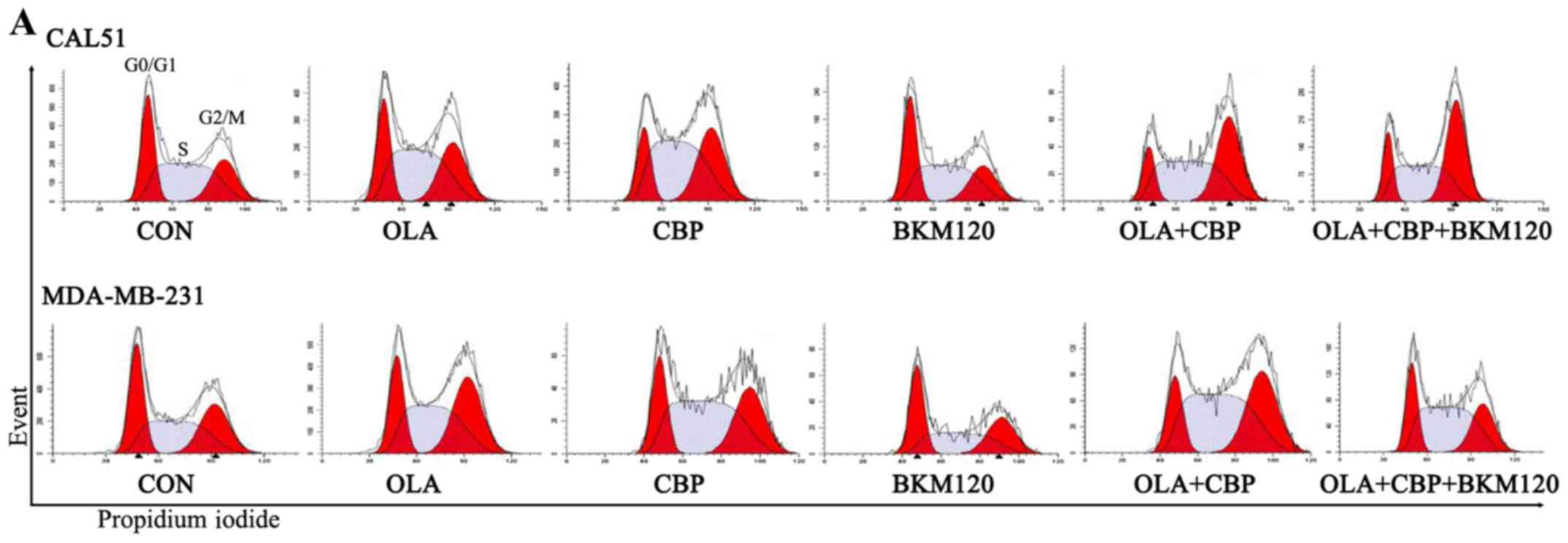 | Figure 3.OLA, CBP and BKM block cell cycle
progression in triple-negative breast cancer cells. Cells were
treated with OLA (1.25 µM), CBP (0.5 µM) and BKM (0.5 µM), alone or
in combination, for 48 h and were then stained with PI. (A) Flow
cytometry plots indicating cell cycle phases. The rugged line
represents cells detected by flow cytometry after PI staining; the
smooth line and red peaks are the best fits for cell cycle phases
obtained using the flow cytometry software (ModFit LT; Verity
Software House, Inc., Topsham, ME, USA). (B) Histogram data are
presented as the means of three independent experiments. For
clarity, error bars have been omitted in histograms showing bars
with three cell cycle phases (typical variation between
experiments: ±10%). *P<0.05. BKM, NVP-BKM120; CBP, carboplatin;
CON, control (vehicle); OD, optical density; OLA, olaparib; PI,
propidium iodide. |
Olaparib, CBP and BKM120 cause DNA
damage in TNBC cells
Due to their mode of action and toxicity in TNBC
cells, the present study aimed to determine the effects of the
three treatments on the expression of key molecules involved in DNA
damage (Fig. 4). Phosphorylation of
the histone variant H2AX serves a key role in the DNA damage
response and its expression is routinely used to detect genotoxic
effects. In addition, PARP inhibition leads to the accumulation of
DNA DSBs (19) The phosphorylated
levels of H2AX were increased in both MDA-MB-231 and CAL51 cells
following treatment with each of the three drugs, whereas the
levels of PADPR, which is synthesized by PARP, were decreased in
both cell lines; as expected, it was more markedly decreased
following the addition of BKM120 (Fig.
4A and B). Phosphorylation of the checkpoint kinases Chk1 and
Chk2, whose activation by ATM results in cell cycle arrest and cell
death (confirmed by PARP cleavage), was increased following
treatment with olaparib, CBP and BKM120 (Fig. 4A and B). Phosphorylation of Chk2 was
more evident in CAL51 cells than in MDA-MB-231 cells, indicating
that the latter may not be as sensitive to olaparib and CBP as the
former, as expression levels were only altered in the groups
treated with high concentrations. It is possible that BKM120
toxicity in MDA-MB-231 cells is associated with other mechanisms
than those affecting Chk2 phosphorylation.
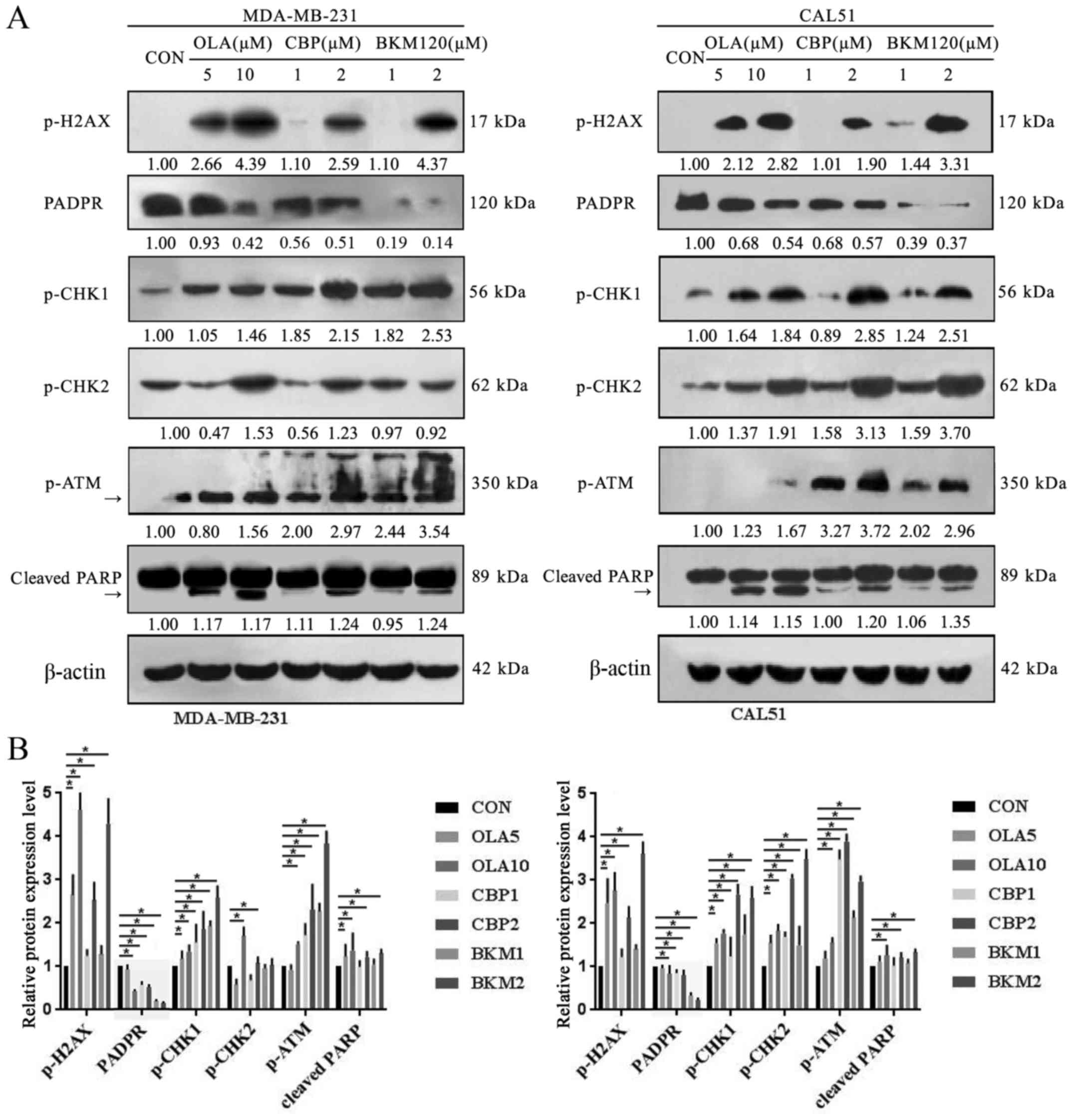 | Figure 4.OLA, CBP and BKM exert toxic effects
on triple-negative breast cancer cells by producing DNA damage. (A
and B) Cells were treated with vehicle (CON), OLA, CBP or BKM for
48 h, and protein expression was determined by western blotting.
(A) p-ATM antibody recognizes a non-specific band (heavier than 350
kDa) in MDA-MB-231 cells; the 350 kDa band is indicated by an
arrow. Cleaved PARP is indicated with an arrow. BKM increases NHEJ
DNA repair. (C and D) Cells were treated with CBP (0.5 µM) alone or
in combination with OLA (1.25 µM) and BKM (0.5 µM) for 24 h, and
protein expression was determined by western blotting. β-actin was
used as a loading control. Representative images of three
independent blots are shown. Semi-quantification of blots was
performed using ImageJ software (National Institutes of Health,
Bethesda, MD, USA) and values are normalized to those of β-actin
and expressed as relative to those of untreated cells. Band
intensity is indicated below each gel track and histogram data are
presented as the means ± standard deviation of three blots.
*P<0.05. ATM, ATM serine/threonine kinase; BKM, NVP-BKM120; CBP,
carboplatin; Chk, checkpoint kinase; CON, control (vehicle); OLA,
olaparib; p-, phosphorylated; PARP, poly(ADP-ribose)
polymerase. |
Since the formation of RAD51 foci is considered a
marker of HR DNA repair (19,20)
and γ-H2AX foci are considered to be a sensitive and selective
signal for the existence of DSBs (21), the present study monitored foci
formation in MDA-MB-231 and CAL51 cells after single, double and
triple drug combinations. CAL51 and MDA-MB-231 cells treated with a
combination of olaparib, CBP and BKM120 exhibited very low RAD51
foci (marker of HR DNA repair), which was reduced compared with in
response to other certain treatments, particularl CBP, when used
singly or in combination, i.e. olaparib and CBP (Fig. 5). γ-H2AX foci formation (a marker of
DSBs) was high when olaparib was combined with either CBP or CBP
and BKM120, although CBP alone produced a higher level of DSBs
(Fig. 5). Overall, these findings
suggested that cells receiving the triple drug combination may
exhibit less active HR repair leading to cell death.
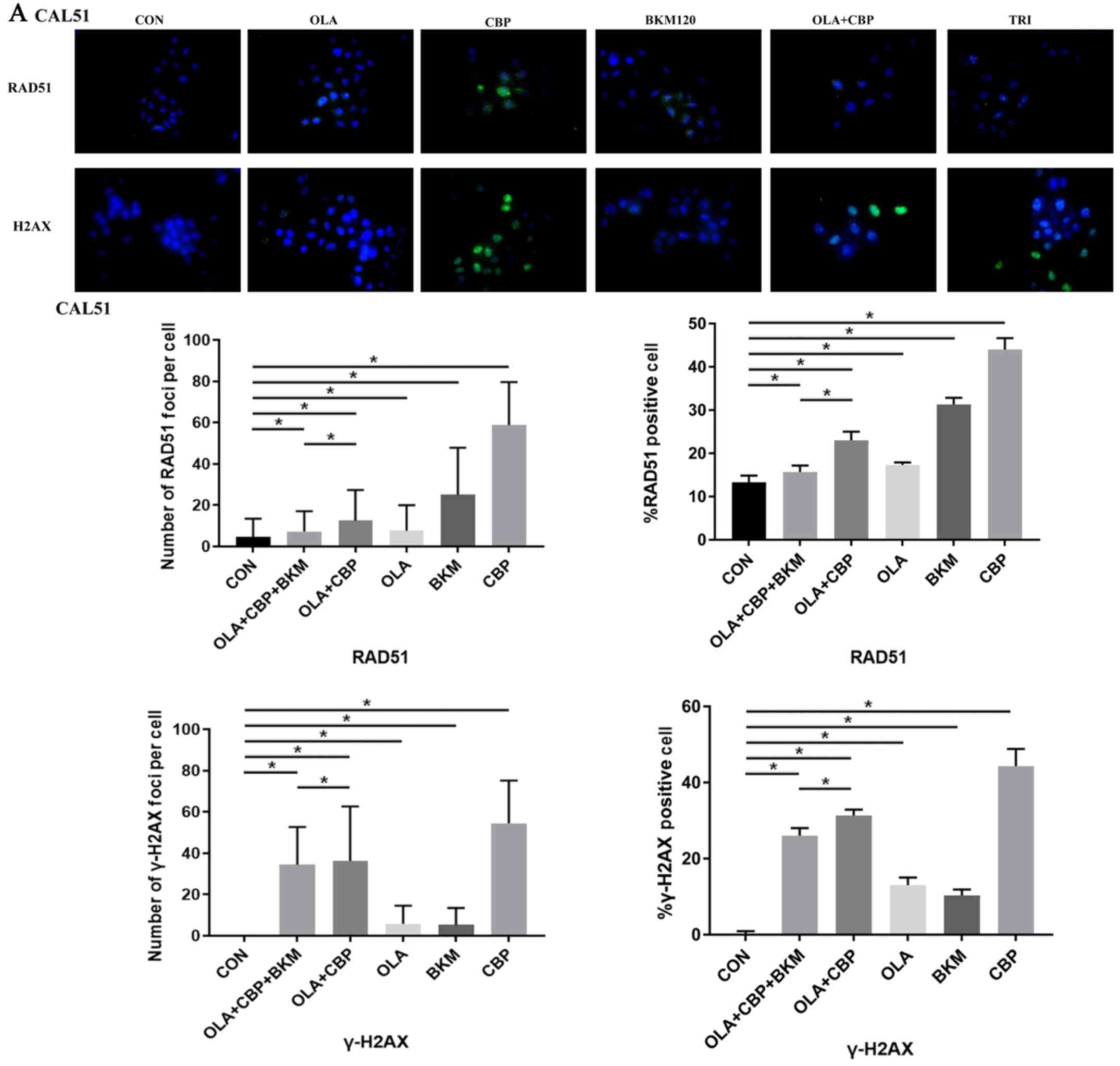 | Figure 5.Combination of OLA, CBP and BKM
promotes DNA damage in triple-negative breast cancer cells. (A)
Cells (CAL51) were treated with vehicle (CON), 1.2 µM OLA, 0.5 µM
CBP, 0.5 µM BKM or a combination, for 48 h. RAD51 and γ-H2AX
expression was visualized by immunofluorescence using primary
specific antibodies and Alexa Fluor 488-conjugated secondary
antibodies. Nuclei were stained with DAPI. The percentage of RAD51
and γ-H2AX positive cells (≥5 foci) and the number of RAD51 and
γ-H2AX foci/cell were determined by counting at least 100 cells
from each sample. Images were captured at ×400 magnification.. Data
are presented as the mean ± standard deviation of three independent
experiments. *P>0.05. BKM, NVP-BKM120; CBP, carboplatin; CON,
control (vehicle); OLA, olaparib; RAD51, DNA repair protein RAD51
homolog 1. Combination of OLA, CBP and BKM promotes DNA damage in
triple-negative breast cancer cells. (B) Cells (MDA-MB-231) were
treated with vehicle (CON), 1.2 µM OLA, 0.5 µM CBP, 0.5 µM BKM or a
combination, for 48 h. RAD51 and γ-H2AX expression was visualized
by immunofluorescence using primary specific antibodies and Alexa
Fluor 488-conjugated secondary antibodies. Nuclei were stained with
DAPI. The percentage of RAD51 and γ-H2AX positive cells (≥5 foci)
and the number of RAD51 and γ-H2AX foci/cell were determined by
counting at least 100 cells from each sample. Images were captured
at ×400 magnification.. Data are presented as the mean ± standard
deviation of three independent experiments. *P>0.05. BKM,
NVP-BKM120; CBP, carboplatin; CON, control (vehicle); OLA,
olaparib; RAD51, DNA repair protein RAD51 homolog 1. |
Overall, the three drugs used in this study,
olaparib, CBP, and BKM120, may exert their toxic effects on TNBC
cells by producing DNA damage.
BKM120 treatment increases NHEJ DNA
repair and growth inhibition of TNBC cells
Since BKM120 leads to an increase in NHEJ DNA repair
(22), the present study evaluated
the functionality of the NHEJ repair axis following treatment with
the combination of the three drugs. The aggregation of 53BP1 helps
to amplify the ATM signal and activation of cell cycle check
points. When DSBs occur, ATM initiates cell cycle check points by
phosphorylation of p53 and other cell cycle regulatory proteins
(14). As expected, the expression
of 53BP1, p27, p21 and p-ATM was increased in MDA-MB-231 and CAL51
cells treated with a combination of the three drugs (Fig. 4C and D). In addition, CAL51 cells
exhibited an increase in p53 expression, which was not observed in
MDA-MB-231 cells as they possess mutated p53. The increase in 53BP1
expression in the combination group compared with in the olaparib
and CBP group was also confirmed by immunofluorescence (Fig. 6). In addition, a decrease in BRCA1
expression was detected in the combination group compared with in
the olaparib and CBP group (Fig.
6), which may lead to a decrease in HR repair (23), and thus an increase in response to
the PARP inhibitor.
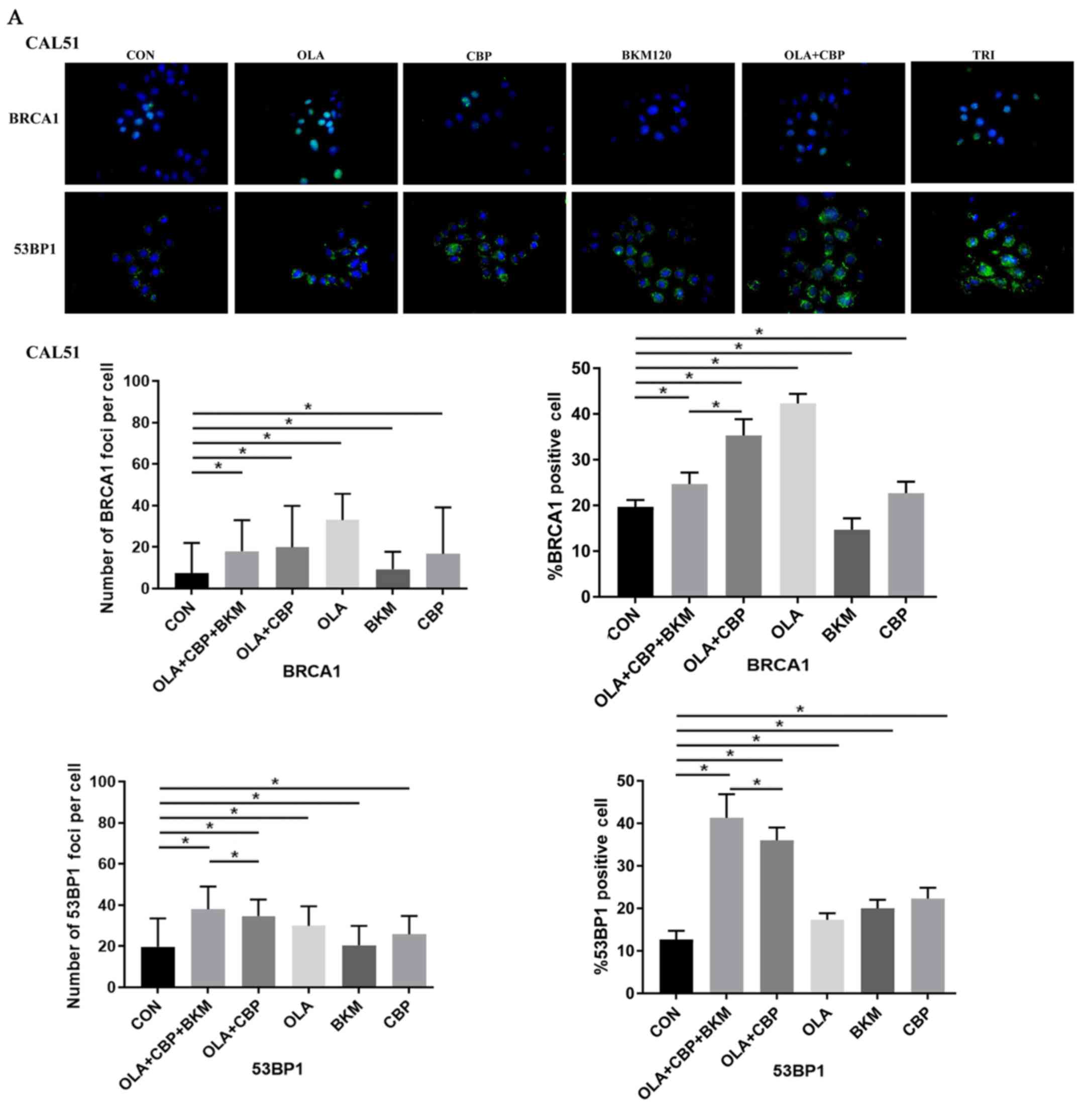 | Figure 6.Immunofluorescence of BRCA1 and
53BP1. (A) Cells (CAL51) were treated with vehicle (CON), 1.2 µM
OLA, 0.5 µM CBP, 0.5 µM BKM or a combination, for 48 h. BRCA1 and
53BP1 expression was visualized by immunofluorescence using primary
specific antibodies and Alexa Fluor 488-conjugated secondary
antibodies. Nuclei were stained with DAPI. The percentage of BRCA1
and 53BP1 positive cells (≥5 foci) and the number of BRCA1 and
53BP1 foci/cell were determined by counting ≥100 cells from each
sample. Images were captured at ×400 magnification.. Data are
presented as the mean ± standard deviation of three independent
experiments. *P>0.05. BKM, NVP-BKM120; BRCA1, BRCA1, DNA
repair-associated; CBP, carboplatin; CON, control (vehicle); OLA,
olaparib. Immunofluorescence of BRCA1 and 53BP1. (B) Cells
(MDA-MB-231) were treated with vehicle (CON), 1.2 µM OLA, 0.5 µM
CBP, 0.5 µM BKM or a combination, for 48 h. BRCA1 and 53BP1
expression was visualized by immunofluorescence using primary
specific antibodies and Alexa Fluor 488-conjugated secondary
antibodies. Nuclei were stained with DAPI. The percentage of BRCA1
and 53BP1 positive cells (≥5 foci) and the number of BRCA1 and
53BP1 foci/cell were determined by counting ≥100 cells from each
sample. Images were captured at ×400 magnification.. Data are
presented as the mean ± standard deviation of three independent
experiments. *P>0.05. BKM, NVP-BKM120; BRCA1, BRCA1, DNA
repair-associated; CBP, carboplatin; CON, control (vehicle); OLA,
olaparib. |
These findings indicated that a combination of
olaparib, CBP and BKM120 may lead to an increase in NHEJ DNA
repair, contributing to growth inhibition in TNBC cells.
Discussion
TNBC refers to a heterogeneous group of tumors that
possess aggressive clinical features (24) and lack targeted therapeutic options.
TNBC is currently treated with standard chemotherapeutics,
including CBP and other platinum drugs (25). In addition, ~15% of TNBC cases
harbor a germline mutation in BRCA1 or BRCA2; >80% of BRCA1
mutation-associated breast cancer cases and 35% of BRCA2
mutation-associated breast cancer cases possess the TNBC phenotype
(26,27). Therefore, therapeutic strategies
targeting these pathways are currently being studied. Olaparib is
an oral PARP inhibitor that, amongst other effects, blocks
base-excision repair by trapping PARP at the site of DNA damage,
and leads to the collapse of DNA replication forks and the
accumulation of DNA DSBs (28).
Furthermore, TNBC frequently presents copy number variations at
certain loci, such as those involved in DNA damage repair and PI3K
pathways (29). BKM120, which is a
pan-class I PI3K inhibitor currently in Phase I/II clinical trials,
has demonstrated antiproliferative, pro-apoptotic and antitumor
activity in various cell lines, as well as in xenograft models of
cancers with or without aberrant PI3K pathway activation (30). The combination of olaparib and CBP
is tolerable and has modest activity in patients with sporadic TNBC
(31). The present findings
indicated that the combination of CBP, olaparib and BKM120 may
exert synergistic inhibitory effects on MDA-MB-231 and CAL51 TNBC
cell lines.
DNA damage repair serves an important role in the
maintenance of genome stability; however, this same phenomenon can
lead to drug resistance in cancer cells. Therefore, blocking the
DNA repair pathway is an important method for the treatment of
tumors (32). DNA damage can be
divided into two types: DNA single-strand breaks (SSBs) and DSBs.
In the S phase of the cell cycle, several unrepaired SSBs can lead
to disintegration of the duplicating forks and can subsequently
result in DSBs. The repair of DSBs in DNA occurs through two
methods of repair, HR and NHEJ (33). HR repair uses high homologous sister
chromosome fragments as a template to repair damaged DNA, and is
considered an error-free repair mechanism (34). NHEJ is an error-prone repair
mechanism, which results in the accumulation of damaged DNA that
can cause cell death (33). NHEJ
begins in the G1 phase, in which the accuracy of repair
is low, whereas HR begins in the S/G2 phase, in which
the accuracy of repair is high. Furthermore, the repair of DSBs is
strictly regulated by the cell cycle. Deficient HR repair leads to
activation of alternate DNA repair pathways, including the base
excision repair and NHEJ pathways, which require PARP. HR repair
dysfunction sensitizes cells to PARP, which leads to further
chromosomal instability, cell cycle arrest and apoptosis (35,36);
TNBC normally expresses high levels of PARP-1 (37,38).
Therefore, olaparib can induce DSBs and inhibit HR repair, whereas
PI3K inhibitors can influence cell cycle progression, inhibit HR
repair and enhance NHEJ repair (39). Conversely, BKM120 can induce the
expression of BRCA1 (40);
therefore, BKM120 may promote HR repair and sensitize TNBC to PARP
inhibition.
In summary, the present study demonstrated that a
combination of three drugs, olaparib (a PARP inhibitor), CBP (a
drug which crosslinks with purine bases in the DNA, interfering
with DNA repair mechanisms and producing DNA damage) and BKM120 (a
pan-class I PI3K inhibitor) act synergistically on TNBC cells via
DNA damage, enhancing NHEJ and inhibiting HR. As TNBC lacks
targeted therapies, there is an urgent need to identify novel
effective treatments for this type of breast cancer. The present
study provided a strong rationale to explore the therapeutic use of
olaparib in combination with CBP and BKM120, first in animal
models, and later in clinical trials on patients with TNBC.
Acknowledgements
The authors would like to thank AstraZeneca for the
generous gift of olaparib, and Dr S. Raguz (Imperial College
London) for English language support. Part of this study was
presented at the ASCO 2018 Annual Meeting (Abstract no.
e12535).
Funding
The present study was supported by the Chinese
National Natural Sciences Foundation (grant no. 81672623 to ZJ) and
the National Science and Technology Support program (grant no.
2015BAI12B15 to ZJ).
Availability of data and materials
All data generated or analyzed during this study are
included in this publication article.
Authors' contributions
JZ designed the research; HZ and YH designed the
experiments; HZ and QY performed the research; QY performed the
cell culture and MTT assays; HZ performed western blotting and
immunofluorescence; YH performed the cell cycle analysis; HZ, YH
and JZ wrote the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dawson SJ, Provenzano E and Caldas C:
Triple negative breast cancers: Clinical and prognostic
implications. Eur J Cancer. 45:27–40. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Murai J, Huang SYN, Das BB, Renaud A,
Zhang Y, Doroshow JH, Ji J, Takeda S and Pommier Y: Trapping of
PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res.
72:5588–5599. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rouleau M, Patel A, Hendzel MJ, Kaufmann
SH and Poirier GG: PARP inhibition: PARP1 and beyond. Nat Rev
Cancer. 10:293–301. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu P, Cheng H, Roberts TM and Zhao JJ:
Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev
Drug Discov. 8:627–644. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fruman DA and Rommel C: PI3K and cancer:
Lessons, challenges and opportunities. Nat Rev Drug Discov.
13:140–156. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gopal AK, Kahl BS, de Vos S,
Wagner-Johnston ND, Schuster SJ, Jurczak WJ, Flinn IW, Flowers CR,
Martin P, Viardot A, et al: PI3Kδ inhibition by idelalisib in
patients with relapsed indolent lymphoma. N Engl J Med.
370:1008–1018. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Burger MT, Pecchi S, Wagman A, Ni ZJ,
Knapp M, Hendrickson T, Atallah G, Pfister K, Zhang Y, Bartulis S,
et al: Identification of NVP-BKM120 as a potent, selective, orally
bioavailable class I PI3 kinase inhibitor for treating cancer. ACS
Med Chem Lett. 2:774–779. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Maira SM, Pecchi S, Huang A, Burger M,
Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, et al:
Identification and characterization of NVP-BKM120, an orally
available pan-class I PI3-kinase inhibitor. Mol Cancer Ther.
11:317–328. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brachmann SM, Kleylein-Sohn J, Gaulis S,
Kauffmann A, Blommers MJ, Kazic-Legueux M, Laborde L, Hattenberger
M, Stauffer F, Vaxelaire J, et al: Characterization of the
mechanism of action of the pan class I PI3K inhibitor NVP-BKM120
across a broad range of concentrations. Mol Cancer Ther.
11:1747–1757. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Juvekar A, Burga LN, Hu H, Lunsford EP,
Ibrahim YH, Balmañà J, Rajendran A, Papa A, Spencer K, Lyssiotis
CA, et al: Combining a PI3K inhibitor with a PARP inhibitor
provides an effective therapy for BRCA1-related breast cancer.
Cancer Discov. 2:1048–1063. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Matulonis U, Wulf GM, Birrer MJ, Westin
SN, Farooq S, Bell-McGuin KM, Obermayer E, Whalen C, Spagnoletti T,
Luo W, et al: Phase I study of oral BKM120 and oral olaparib for
high-grade serous ovarian cancer (HGSC) or triple-negative breast
cancer (TNBC). Ann Oncol. 28:512–518. 2017.PubMed/NCBI
|
|
12
|
Isakoff SJ, Mayer EL, He L, Traina TA,
Carey LA, Krag KJ, Rugo HS, Liu MC, Stearns V, Come SE, et al:
TBCRC009: A multicenter phase II clinical trial of platinum
monotherapy with biomarker assessment in metastatic triple-negative
breast cancer. J Clin Oncol. 33:1902–1909. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Weichselbaum RR, Ishwaran H, Yoon T,
Nuyten DS, Baker SW, Khodarev N, Su AW, Shaikh AY, Roach P, Kreike
B, et al: An interferon-related gene signature for DNA damage
resistance is a predictive marker for chemotherapy and radiation
for breast cancer. Proc Natl Acad Sci USA. 105:18490–18495. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Daley JM and Sung P: 53BP1, BRCA1, and the
choice between recombination and end joining at DNA double-strand
breaks. Mol Cell Biol. 34:1380–1388. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
O'Donovan PJ and Livingston DM: BRCA1 and
BRCA2: Breast/ovarian cancer susceptibility gene products and
participants in DNA double-strand break repair. Carcinogenesis.
31:961–967. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hu Y, Guo R, Wei J, Zhou Y, Ji W, Liu J,
Zhi X and Zhang J: Effects of PI3K inhibitor NVP-BKM120 on
overcoming drug resistance and eliminating cancer stem cells in
human breast cancer cells. Cell Death Dis. 6:e20202015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu Y, Qiu Y, Yague E, Ji W, Liu J and
Zhang J: miRNA-205 targets VEGFA and FGF2 and regulates resistance
to chemotherapeutics in breast cancer. Cell Death Dis. 7:e22912016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Modesti M and Kanaar R: DNA repair:
Spot(light)s on chromatin. Curr Biol. 11:R229–R232. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Baumann P and West SC: Role of the human
RAD51 protein in homologous recombination and double-stranded-break
repair. Trends Biochem Sci. 23:247–251. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lundin C, Schultz N, Arnaudeau C, Mohindra
A, Hansen LT and Helleday T: RAD51 is involved in repair of damage
associated with DNA replication in mammalian cells. J Mol Biol.
328:521–535. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang L, Graham PH, Hao J, Ni J, Bucci J,
Cozzi PJ, Kearsley JH and Li Y: PI3K/Akt/mTOR pathway inhibitors
enhance radiosensitivity in radioresistant prostate cancer cells
through inducing apoptosis, reducing autophagy, suppressing NHEJ
and HR repair pathways. Cell Death Dis. 5:e14372014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tung NM and Garber JE: BRCA1/2 testing:
Therapeutic implications for breast cancer management. Br J Cancer.
119:141–152. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Abramson VG, Lehmann BD, Ballinger TJ and
Pietenpol JA: Subtyping of triple-negative breast cancer:
Implications for therapy. Cancer. 121:8–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang L, Liu Q, Chen S and Shao Z:
Cisplatin versus carboplatin in combination with paclitaxel as
neoadjuvant regimen for triple negative breast cancer. OncoTargets
Ther. 10:5739–5744. 2017. View Article : Google Scholar
|
|
26
|
Foulkes WD, Smith IE and Reis-Filho JS:
Triple-negative breast cancer. N Engl J Med. 363:1938–1948. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Podo F, Buydens LMC, Degani H, Hilhorst R,
Klipp E, Gribbestad IS, Van Huffel S, van Laarhoven HW, Luts J,
Monleon D, et al: Triple-negative breast cancer: Present challenges
and new perspectives. Mol Oncol. 4:209–229. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee JM, Hays JL, Annunziata CM, Noonan AM,
Minasian L, Zujewski JA, Yu M, Gordon N, Ji J, Sissung TM, Figg WD,
et al: Phase I/Ib Study of olaparib and carboplatin in BRCA1 or
BRCA2 mutation-associated breast or ovarian cancer with biomarker
analyses. J Natl Cancer Inst. 106:dju0892014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Robertson L, Hanson H, Seal S,
Warren-Perry M, Hughes D, Howell I, Turnbull C, Houlston R, Shanley
S, Butler S, et al: BRCA1 testing should be offered to individuals
with triple-negative breast cancer diagnosed below 50 years. Br J
Cancer. 106:1234–1238. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Juvekar A, Hu H, Yadegarynia S, Lyssiotis
CA, Ullas S, Lien EC, Bellinger G, Son J, Hok RC, Seth P, et al:
Phosphoinositide 3-kinase inhibitors induce DNA damage through
nucleoside depletion. Proc Natl Acad Sci USA. 113:E4338–E4347.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee JM, Hays JL, Chiou VL, Annunziata CM,
Swisher EM, Harrell MI, Yu M, Gordon N, Sissung TM, Ji J, et al:
Phase I/Ib study of olaparib and carboplatin in women with triple
negative breast cancer. Oncotarget. 8:79175–79187. 2017.PubMed/NCBI
|
|
32
|
Zhang L, Reyes A and Wang X: The Role of
DNA repair in maintaining mitochondrial DNA stability. Adv Exp Med
Biol. 1038:85–105. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ma A and Dai X: The relationship between
DNA single-stranded damage response and double-stranded damage
response. Cell Cycle. 17:73–79. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee JM, Ledermann JA and Kohn EC: PARP
Inhibitors for BRCA1/2 mutation-associated and BRCA-like
malignancies. Ann Oncol. 25:32–40. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Farmer H, McCabe N, Lord CJ, Tutt AN,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bryant HE, Schultz N, Thomas HD, Parker
KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ and Helleday T:
Specific killing of BRCA2-deficient tumours with inhibitors of
poly(ADP-ribose) polymerase. Nature. 434:913–917. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Domagala P, Huzarski T, Lubinski J, Gugala
K and Domagala W: PARP-1 expression in breast cancer including
BRCA1-associated, triple negative and basal-like tumors: Possible
implications for PARP-1 inhibitor therapy. Breast Cancer Res Treat.
127:861–869. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ossovskaya V, Koo IC, Kaldjian EP, Alvares
C and Sherman BM: Upregulation of poly (ADP-ribose) polymerase-1
(PARP1) in triple-negative breast cancer and other primary human
tumor types. Genes Cancer. 1:812–821. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ayub A, Yip WK and Seow HF: Dual
treatments targeting IGF-1R, PI3K, mTORC or MEK synergize to
inhibit cell growth, induce apoptosis, and arrest cell cycle at G1
phase in MDA-MB-231 cell line. Biomed Pharmacother. 75:40–50. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ibrahim YH, García-García C, Serra V, He
L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzmán M, Grueso J,
et al: PI3K inhibition impairs BRCA1/2 expression and sensitizes
BRCA-proficient triple-negative breast cancer to PARP inhibition.
Cancer Discov. 2:1036–1047. 2012. View Article : Google Scholar : PubMed/NCBI
|














