Introduction
Treatment options remain very limited for patients
with advanced renal cell carcinoma (RCC). Metastasis and recurrence
occur in 20–30% of patients who have received radical resection.
This disease is resistant to chemotherapy and/or radiotherapy, and
the median survival is only 7–10 months (1). In recent years, small-molecule
targeted therapy, including tyrosine kinase inhibitors (TKIs), have
become the first-line treatment for advanced RCC, though prognosis
remains poor (2). However, RCC
appears to be sensitive to immunotherapy (3,4).
The chimeric antigen receptor (CAR) modified T-cell
(CAR-T) therapy is a newly developed adoptive treatment of cancers.
A CAR is a recombinant receptor construct composed of an
extracellular single-chain variable fragment (scFv) derived from an
antibody, a hinge and transmembrane domain, a costimulation
signaling domain (CD28 or CD137) and a CD3ζ signaling domain to
provide T-cell activation signals (5). The CAR thereby redirects T-cell
specificity to the tumor in a major histocompatibility complex
(MHC)-independent manner (6).
Currently, CAR-T therapy has shown potential for curative
therapeutic efficacy in patients with hematological malignancies,
particularly for treating B-cell malignancies with CD19-specific
CAR-T cells (7,8). Associated CAR-T products, CTL019
(Novartis, Basel, Switzerland) and Yescarta (KTE-C19; Gilead,
Foster City, CA, USA), have been approved in the past few months to
treat patients with B-cell malignancies. However, the therapeutic
efficacy of CAR-T cells in most solid tumors, including RCC,
remains less impressive (9).
Weijtens et al designed a first-generation CAR (scFv-FcRγ)
directed against carbonic anhydrase IX (CAIX), an RCC antigen, and
used the CAR-modified T cells to treat patients with
CAIX-expressing metastatic RCC (10,11).
Although the blood cytokine profiles mirrored CAR-T cell presence
and in vivo activity, no clinically objective responses were
observed in any of the 12 patients.
Natural killer (NK) cells are effector cells of the
innate immune system that are capable of killing tumor cells, as
well as producing cytokines without previous stimulation (12). The efficacy of autologous NK cells
in the immunotherapy of solid tumors was evaluated in various
clinical settings (13–15). Since autologous NK cells may be
functionally inhibited by the interaction between self-HLA class I
molecules on tumor cells and inhibitory NK receptors, and because
allogeneic NK cells do not attack non-hematopoietic tissues in the
recipient, the adoptive transfer of allogeneic NK cells may
represent a better approach (12).
Over the past decade, adoptive transfer of ex vivo-activated
or ex vivo-expanded allogeneic NK cells has emerged as a
promising immunotherapeutic strategy for cancers (16–18).
Given the potent cytolytic function of these cells and the shorter
lifespan, mature NK cells are considered as attractive candidate
effector cells to express CARs for the therapy of patients with
cancers. Primary human NK cells have been successfully engineered
to express CARs against a number of targets, including CD244
(19), GD2 (20) and EGFR (21).
NK92, a human NK-like cell line, was derived from
the peripheral blood of a female patient with non-Hodgkin's
lymphoma and is an IL-2 dependent immortalized cell line (22,23).
The general safety of infused NK92 cells has been established in
phase I clinical trials with clinical responses observed in 11
treated renal cancer patients (24). The adoptive transfer of CAR-NK92
cells has several theoretical advantages over the use of patient-
or donor-derived T cells or NK cells. The advantages are primarily
related to the lack of expression of inhibitory killer Ig-like
receptors (iKIRs), presumed lack of immunogenicity and
graft-vs.-host disease (GVHD), ease of expansion and availability
as an ‘off-the-shelf’ product, which can greatly reduce the
treatment cycle and cost of treatment.
However, NK92 also has obvious disadvantages, such
as tumorigenicity and the potential susceptibility to the EB virus.
Therefore, as a safety consideration, NK92 must be irradiated
before its clinical use. Researchers, such as Tam et al and
Tonn et al, have studied the safety of NK92 cells for
adoptive immunotherapy (25,26).
Their studies indicated that after the proper amount of γ-ray
irradiation, NK92 cells could not only be safe for clinical
application but could also maintain their cytotoxicity for a
certain period. Currently, NK92 cells have been successfully
engineered to express CARs against a number of targets, including
CD19 (27), CD20 (28), Her2 (29,30),
GD2 (31), EpCAM (32), CS1 (33), CD138 (34), EGFR (35–37),
CD3 (38) and CD5 (39). CAR-transduced NK (CAR-NK) cells
exhibit efficient in vitro and in vivo tumor cell
killing ability, although no clinical data from CAR-NK cell therapy
have been reported to date (40).
Results of current clinical trials have demonstrated
that mono-therapy with CAR-T cells is not an effective strategy for
solid tumors. The combination of CAR-T cells with chemotherapeutic
drugs may be a promising approach to enhance therapeutic efficacy
(41). Bortezomib (Velcade;
PS-341), which binds the catalytic site of the 26S proteasome, was
the first proteasome inhibitor brought into clinical use,
inhibiting the chymotrypsin-like and, to a lesser extent, the
trypsin-like and postglutamyl peptide-hydrolyzing activities
(42). The proteasome pathway plays
an important role in cellular homeostasis by degrading misfolded or
deleterious proteins to maintain normal cellular physiology
(43–45). The proteasome system of malignant
cells is usually overloaded by the accumulation of defective
proteins (44,45). Bortezomib was the first FDA-approved
proteasome inhibitor for the treatment of relapsed/refractory
multiple myeloma disease due to its impressive clinical activity
(44–46). However, the therapeutic efficacy in
solid tumors, including RCC, was limited when bortezomib was used
as a single agent (47,48). New data have revealed that
bortezomib could be an attractive candidate for combination with
antitumor natural killer cell and/or T-cell adoptive therapy due to
its intrinsic ability to increase the levels of immunostimulatory
cytokines and components of the Notch and NFκB signaling pathways
in these lymphocytes (49,50).
In the present study, we constructed a
third-generation CAR against CAIX, which consisted of an anti-CAIX
scFv, CD8 hinge and transmembrane regions, and intracellular signal
domains of CD28, CD137 and CD3ζ. The efficacy of combination
therapy with the CAIX-specific CAR-modified NK-92 (CAIX-CAR-NK92)
cells and bortezomib against RCC were investigated in vitro
and in a mouse model of human RCC. Our results indicated that the
combination of CAIX-CAR-NK92 cells with bortezomib is a promising
strategy to treat RCC.
Materials and methods
Cell culture
NK92 cells were purchased from the American Type
Culture Collection (ATCC; Manassas, VA, USA). NK92 cells and
transduced NK92 cells were incubated in α-modification of Eagle's
minimum essential medium (α-MEM; Gibco; Life Technologies; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 2 mM
L-glutamine, 0.2 mM Myo-inositol, 0.02 mM folic acid, 0.1 mM
2-mercaptoethanol, 400 IU/ml IL-2 (PeproTech, Inc., Rocky Hill, NJ,
USA), 12.5% fetal bovine serum (FBS) and 12.5% horse serum (Gibco,
Life Technologies; Thermo Fisher Scientific, Inc.), 1/100
penicillin/streptomycin (Gibco, Life Technologies; Thermo Fisher
Scientific, Inc.). The human renal cancer cell line OSRC-2 was
cultured in RPMI-1640 medium (Gibco, Life Technologies), and the
human renal cancer cell lines ACHN, Ketr-3 and human embryonic
kidney cells 293 were cultured in Dulbecco's modified Eagle's
medium (DMEM; Gibco; Life Technologies; Thermo Fisher Scientific,
Inc.). RPMI-1640 and DMEM medium were supplemented with 10% FBS and
1/100 penicillin/streptomycin when used. All of the cell lines were
cultured at 37°C in a humidified atmosphere with 5% CO2.
Cells in the logarithmic growth phase were used for all
experiments.
Construction of CAIX-specific CAR and
lentivirus preparation
The CAIX-CAR construct
(LV5-scFv-CD8-CD28-CD137-CD3ζ) was composed of anti-CAIX single
chain variable fragment (scFv, G250), CD8 (138–208 aa), CD28
(180–220 aa), 4-1BB (214–255 aa) and CD3ζ (52–164 aa) domains. The
encoding sequence of the CAR was inserted into a lentiviral vector
designated LV5-EF1a-Puro (Shanghai GenePharma Co., Ltd., Shanghai,
China). The methods for lentivirus preparation were the same as in
our previous study (51).
Transduction of NK92 cells
For lentivirus infection, wells of 24-well plates
were coated with RetroNectin (Takara Bio, Inc., Shiga, Otsu) (1 ml
of 25 µg/ml in PBS) overnight at 4°C. RetroNectin was later
removed, and the wells were blocked (30 min, room temperature) with
sterile-filtered PBS containing 2% BSA and washed with PBS. NK92
cells were adjusted to 2×105 cells/ml using α-MEM with
IL-2 and 2-mercaptoethanol. Next, the cells were loaded in the
RetroNectin-precoated 24-well plates followed by the addition of
lentivirus at an MOI of 50 in the presence of 5 µg/ml Polybrene
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), and they were
centrifuged (2 h, 380 × g, at 32°C) and incubated for 24 h. Next,
the cells were transferred to uncoated wells with fresh complete
medium and cultured for 12 h. Following this step, the infection
protocol was repeated. Starting from day 3 after the second
infection, transfected NK92 cells were repeatedly selected by
puromycin (Sigma-Aldrich; Merck KGaA) with a final concentration of
1 µg/ml every two weeks in order to establish a stable transfected
NK92 cell line. Control lentivirus (empty vector with a puromycin
select gene) transfected NK92 cells (Ctrl-NK92) were also selected
with puromycin.
FACS analysis
For CAR detection, 0.5×106 Ctrl-NK92 and
CAR-NK92 cells were washed once with FACS buffer (2% FBS in PBS)
and were blocked with 1:100 diluted goat serum in PBS at 4°C for 1
h. The blocked cells were resuspended in 50 µl FACS buffer
containing 2.5 µl Alexa Fluor 647-conjugated AffiniPure
F(ab')2 Fragment Goat Anti-mouse IgG, F(ab')2
Fragment-Specific antibody (dilution 1:200; cat. no. 115-606-072;
Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) and
were incubated at 4°C for 1 h. The cells were then washed,
suspended and evaluated with a FACS machine (FACSCanto II; BD
Biosciences, Franklin Lakes, NJ, USA). Unstained NK92 cells were
used as the control. The data were analyzed with FlowJo software
(7.6.1 version; FlowJo LLC, Ashland, OR, USA).
For the membrane surface expression of CAIX in renal
cancer cell lines, 0.5×106 cells were suspended in 50 µl
FACS buffer containing 2 µl FITC-conjugated mouse anti-human CAIX
antibody (dilution 1:10; cat. no. FAB2188F; R&D Systems, Inc.,
Minneapolis, MN, USA) and incubated at 4°C for 30 min. A
FITC-conjugated mouse IgG2A isotype antibody (dilution 1:10; cat.
no. IC003F R&D Systems, Inc.) was used as the control. FACS
machine detection and data analysis were aforementioned.
Western blot analysis
In total, 1×106 cells were lysed with
RIPA lysis buffer (Beyotime Institute of Biotechnology, Haimen,
China) with 0.1 mM phenylmethylsulfonyl fluoride (PMSF;
Sigma-Aldrich; Merck KGaA). The protein concentration was
determined with the BCA method. The mass of protein loaded per lane
was 60 µg. Total proteins were separated on a 10% SDS
polyacrylamide gel (SDS-PAGE). The proteins were subsequently
transferred to nitrocellulose membranes and incubated with a
primary rabbit anti-human CD3ζ antibody (dilution 1:1,000; cat. no.
ab40804; Abcam, Cambridge, MA, USA) or rabbit anti-human GAPDH
antibody (dilution 1:1,000; cat. no. GTX627408; GeneTex, Inc.,
Irvine, CA, USA) overnight at 4°C. The membranes were washed and
incubated with a goat anti-rabbit IgG (H+L) secondary antibody
(dilution 1:10,000; cat. no. VA001; Vicmed Life Sciences, Xuzhou,
China) at room temperature for 1 h. The blots were visualized using
an enhanced chemiluminescence (ECL) detection kit (Beyotime
Biotechnology).
Cytokine release assay
Effector cells were co-cultured with
1×104 target cells at an effector-to-target (E:T) ratio
of 1:1 in a final volume of 200 µl of α-MEM complete media in
triplicate sets of wells in round-bottom 96-well culture plates.
After 24 h, supernatants were assayed for the presence of IFN-γ,
perforin and granzyme B by ELISA using human IFN-γ ELISA kits and
human perforin ELISA kits (both from Dakewe Biotech Co., Ltd.,
Shenzhen, China) and human granzyme B ELISA kits (BioLegend, Inc.,
San Diego, CA, USA) according to the manufacturer's
instructions.
Cytotoxicity assay
The LDH release assays were performed with the
CytoTox 96® Non-Radioactive Cytotoxicity Assay Kit
(Promega Corp., Madison, WI, USA) according to the manufacturer's
instructions. Briefly, effector cells were incubated with
1×104 target cells at effector-to-target (E:T) ratios of
30:1, 10:1, 3:1, or 1:1 in a final volume of 100 µl of RPMI-1640
medium with 5% FBS in round-bottom 96-well culture plates at 37°C
for 4 h. The test was performed in three replicates for each E/T
ratio. The CAR-NK92 cells used in these assays were compared with
empty vector-transduced NK92 cells (Ctrl-NK92) in the assay. Target
cell cytotoxicity was calculated using the following formula: %
Cytotoxicity=100×[(experimental-effector spontaneous-target
spontaneous)/(target maximum-target spontaneous)].
In vivo efficacy studies
All protocols for the animal studies were reviewed
and approved by the Institutional Animal Care and Use Committee of
the Jiangsu Provincial Academy of Chinese Medicine (SCXK2012-005).
Six-week-old female NOD/SCID mice were purchased from Beijing
Huafukang Bioscience, Co., Inc. (Beijing, China). The housing
conditions of the mice included a constant temperature of 28°C and
ventilation was required 10–15 times per hour. Every day 10 h of
illumination and 14 h of darkness were maintained and food and
water were sterilized by high pressure steam. Each mouse was
inoculated subcutaneously with 3×106
Ketr-3luc+ cells (Ketr-3 cells expressing luciferase) in
the right flank on day 0. Next, the mice were randomly assigned to
six groups, including the untreated group, the bortezomib group,
the Ctrl-NK92 group, the CAR-NK92 group and the CAR-NK92+bortezomib
group. Bortezomib (5 µg/mouse in DMSO; Selleck Chemicals, Houston,
TX, USA) was injected 5 days after the tumor injection (day 5), and
24 h later, the mice in the associated groups were infused with
2.5×106 Ctrl-NK92 cells or CAR-NK92 cells. From the day
of the NK92 cell infusion, all mice were administered 2000 IU of
IL-2 daily by intraperitoneal (i.p.) injection for 60 days. The
bortezomib/NK92 cell treatment cycle was repeated once a week until
the experiment was terminated. During the treatment, the mice were
monitored frequently for subcutaneous xenograft progression. The
tumors were assessed with a caliper at their greatest length and
width twice a week to estimate the tumor volume after the tumors
had grown to a palpable size. The tumor volume was calculated using
the following formula: Tumor volume = length ×
(width)2/2.
On day 72, the experiment was terminated. Before the
mice were humanely sacrificed by cervical dislocations, the mice
bearing Ketr-3luc+ tumors were infused intraperitoneally
with D-luciferin (150 mg/kg; Berthod Technologies GmbH & Co.
KG, Bad Wildbad, Germany), anesthetized with 1% pentobarbital (50
mg/kg; Beyotime Institute of Biotechnology) by intraperitoneal
(i.p.) injection, and imaged using an in vivo imaging system
(Berthold Technologies GmbH & Co. KG).
Immunohistochemical analysis
The tumor tissues were fixed in 4% paraformaldehyde,
decalcified in saturated EDTA, and embedded in paraffin. The
embedded tumor tissues were cut into 3–5-µm thick sections. The
sections were stained with rabbit anti-human CD3ζ monoclonal
antibody (dilution 1:500; cat. no. ab40804; Abcam), and
HRP-conjugated anti-rabbit IgG (cat. no. SPN9001; ZSGB-BIO; OriGene
Technologies, Inc. Rockville, MD, USA) was used as a secondary
antibody followed by a peroxidase enzymatic reaction. Tissues were
visualized with diaminobenzidine (DAB; ZSGB-BIO; OriGene
Technologies, Inc.) and counterstained with hematoxylin. For the
quantification of infiltrated NK92 cells into the tumors, NK92
cells were counted in 10 randomly selected intratumoral fields of
each slide at an ×200 magnification.
Statistical analysis
All statistical analyses were performed with Prism
software version 6.0 (GraphPad Software, Inc., La Jolla, CA, USA).
Statistical analysis for the comparison of two groups was performed
using unpaired t-tests. For comparison of more than two groups,
one-way analysis of variance (ANOVA) was used, and multiple
comparisons between the groups were performed using the S-N-K
tests. For the bioluminescence results, the signal intensity was
log-transformed and compared by two-tailed Student's t-test.
P-values <0.05 were considered indicate a statistically
significant difference.
Results
Construction and expression of
CAIX-specific CAR
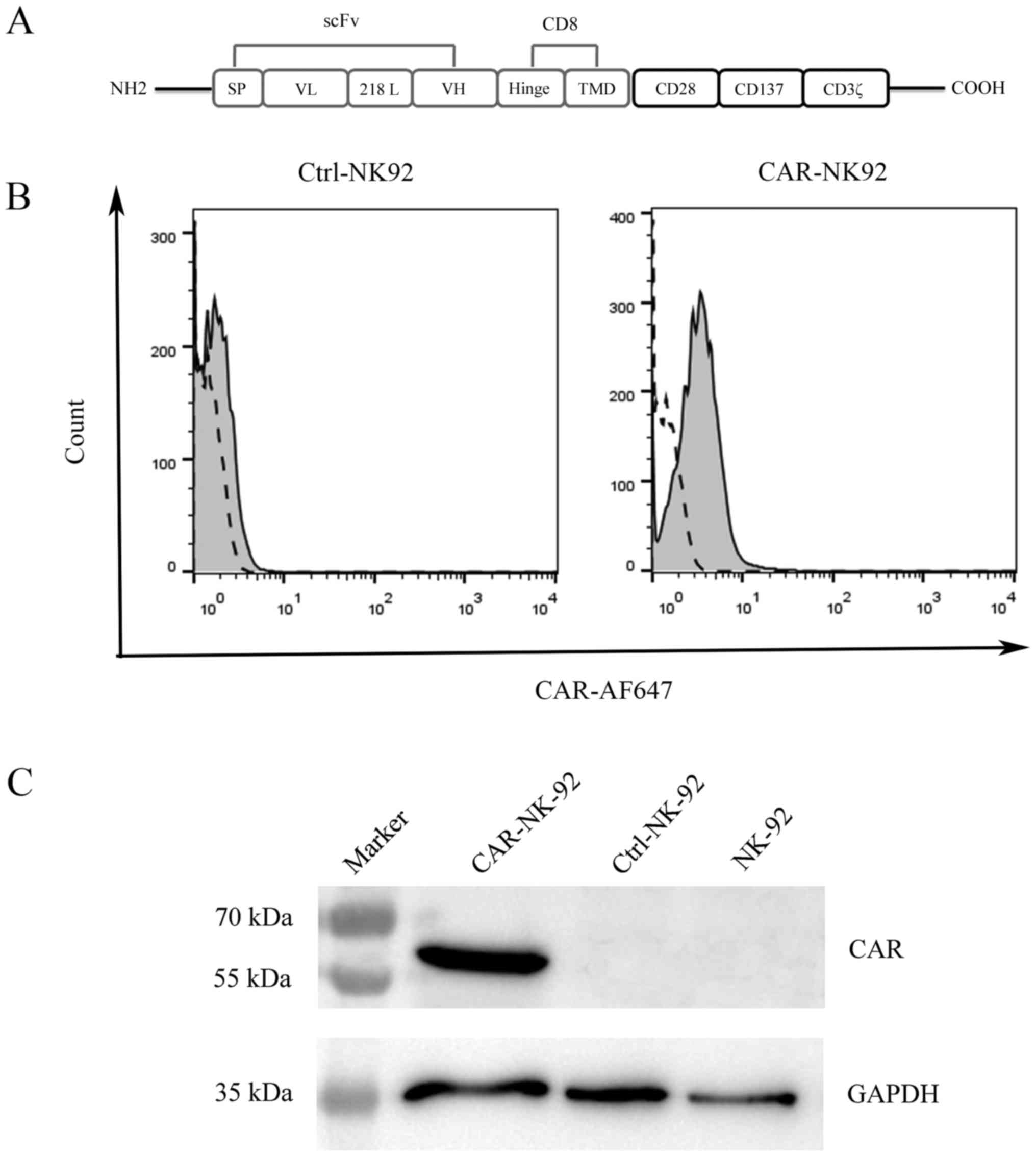
To generate a third-generation CAR specific to CAIX,
a single chain variable fragment (scFv) from an anti-CAIX
monoclonal antibody (G250) was constructed by linking the heavy
chain variable region and light chain variable region with a 218
linker (GSTSGSGKPGSGEGSTKG). This anti-CAIX scFv was fused with the
CD8 hinge and transmembrane domains, and CD28, 4-1BB costimulatory
signaling domains, followed by a CD3ζ activation domain to create a
CAIX-specific CAR (CAIX-CAR, Fig.
1A). We prepared the CAIX-CAR-modified NK92 cells
(CAIX-CAR-NK92) and control NK92 cells (Ctrl-NK92) by infecting
human NK92 cells with a lentivirus containing the encoding sequence
of CAIX-CAR and a control lentivirus (empty vector),
respectively.
Since the lentivirus vector contains a puromycin
selection marker, we generated stable CAIX-CAR-NK92 and Ctrl-NK92
cell lines through repeated selection of the infected NK92 cells
with puromycin. After selection for two weeks, the surface
expression of CAIX-CAR of the infected NK92 cells was detected by
FACS analysis with an Alexa Fluor 647-conjugated goat
F(ab')2 antibody reacting with the
F(ab')2/Fab portion of mouse IgG. Uninfected NK92 cells
were used as the negative control (Fig.
1B). To further examine CAIX-CAR expression in NK92 cells, we
performed western blot analysis using a rabbit anti-human CD3ζ
monoclonal antibody that recognized the ζ chain portion of human
CD3. As expected, CAIX-CAR was only detected in CAIX-CAR-NK92 cells
(Fig. 1C).
Cytokine release of CAIX-CAR-NK92
cells in vitro
To identify the potential target cell lines of
CAIX-CAR-NK92 cells for in vitro and in vivo
experiments, CAIX expression on the surface of renal cancer cells
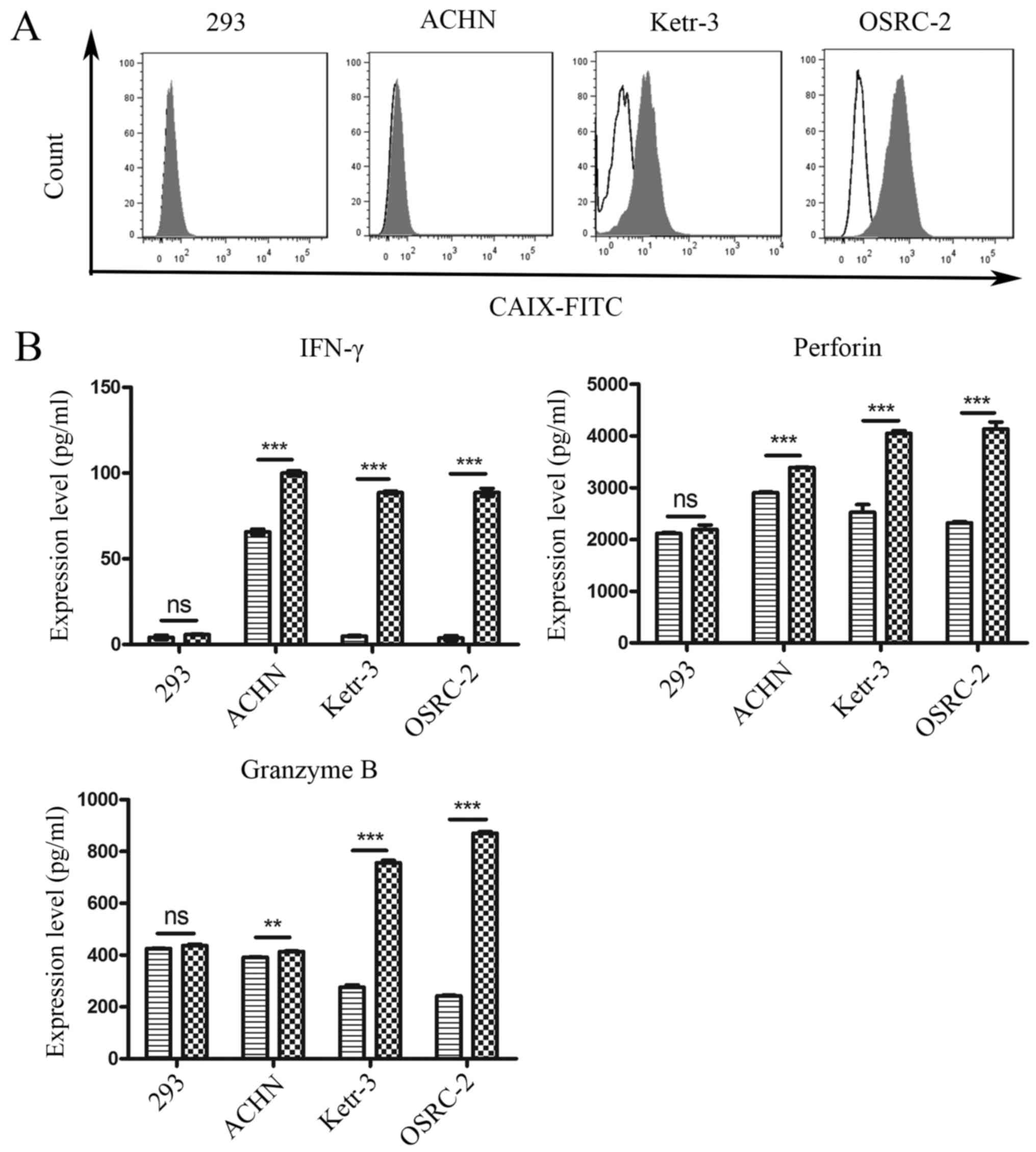
was determined. As shown in Fig.
2A, CAIX was highly expressed on the surface of human renal
cancer Ketr-3 and OSRC-2 cell lines, whereas its expression on the
surface of the human renal cancer cell line ACHN was low, and there
was no expression on the surface of 293 cells.
To examine whether CAR-NK92 cells specifically
recognized target cells and elicited specific effector cell
functions, Ctrl-NK92 and CAR-NK92 cells were co-cultured with
Ketr-3, OSRC-2, ACHN or 293 cell lines for 24 h. Next, released
cytokines, such as INF-γ, perforin, and granzyme B, which play an
important role in mediating adoptive immunotherapy efficacy, were
detected by ELISA in the cell culture supernatant. As revealed in
Fig. 2B, compared with Ctrl-NK92
cells, CAR-NK92 cells mediated robust cytokine secretion when
co-cultured with CAIX-positive tumor cell lines. However, the
cytokine release of CAR-NK92 cells and Ctrl-NK92 cells were not
significantly different for CAIX-negative 293 cells. These results
indicated that CAR-NK92 cells specifically recognized CAIX-positive
target cells and elicited effector cell functions.
Cytotoxicity of CAIX-CAR-NK92 cells
against CAIX-positive renal cancer cells in vitro
To further confirm whether CAIX-CAR-NK92 cells
specifically recognized CAIX-positive renal cancer cells and
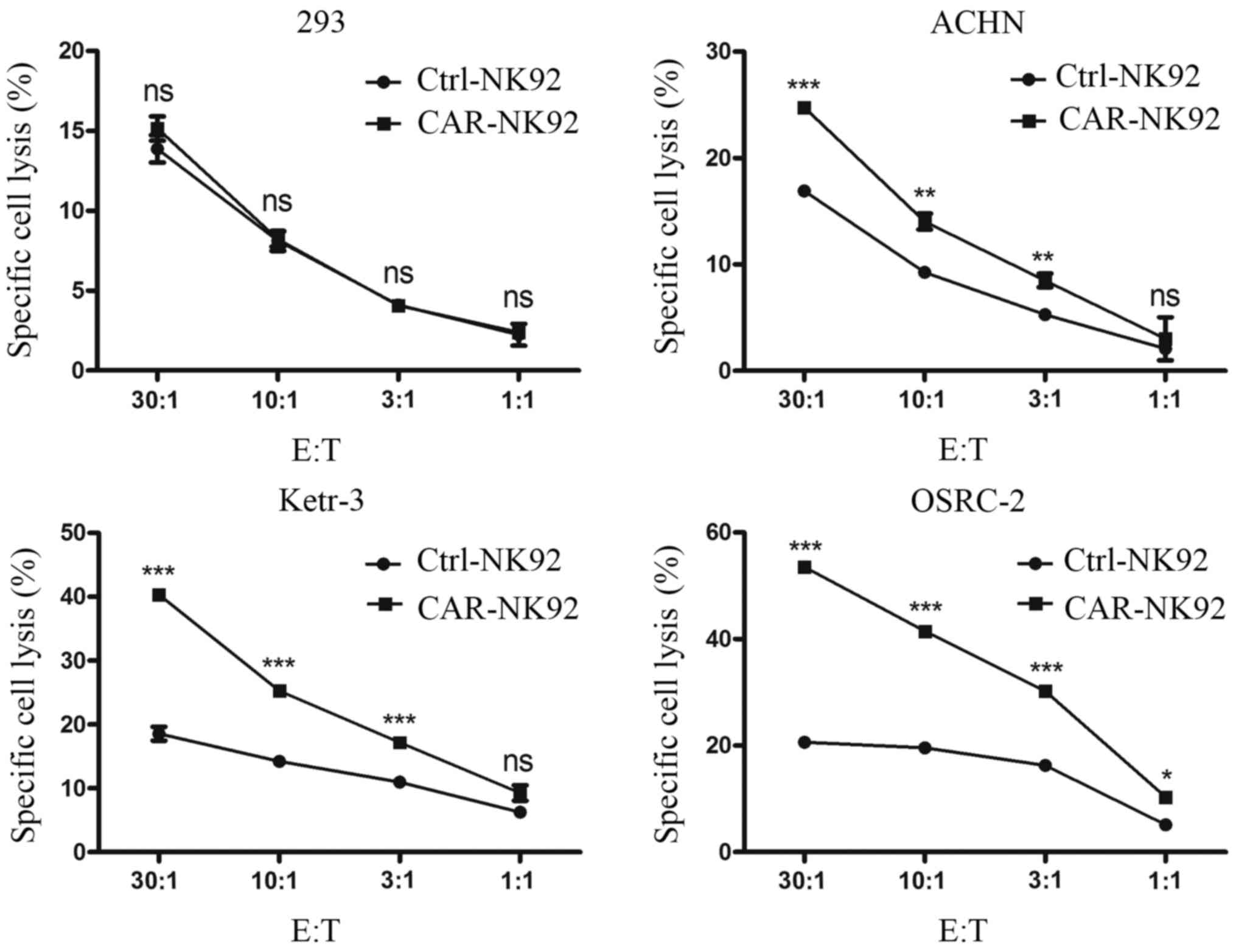
mediated specific cytotoxicity, dose-dependent lactate
dehydrogenase (LDH) release assays were performed. CAIX-CAR-NK92
cells and Ctrl-NK92 cells were co-cultured with Ketr-3, OSRC-2,
ACHN or 293 cells for 4 h. LDH released into the cell culture
supernatant was detected. As revealed in Fig. 3, compared with the Ctrl-NK92 cells,
CAIX-CAR-NK92 cells exhibited stronger killing activity against
CAIX-positive cancer cells Ketr-3, OSRC-2 and ACHN. However, the
difference in cytotoxicity between the Ctrl-NK92 and the
CAIX-CAR-NK92 cells against 293 cells was not significant.
Additionally, the cytotoxicity of CAIX-CAR-NK92 cells against
CAIX-positive tumor cells was positively correlated with the E:T
ratios (Fig. 3). These data
revealed that CAIX-CAR NK92 cells mediated specific killing against
CAIX-positive renal cancer cells.
Pretreatment with bortezomib increases
the sensitivity of renal cancer cells to CAIX-CAR-NK92 and NK92
cells in vitro
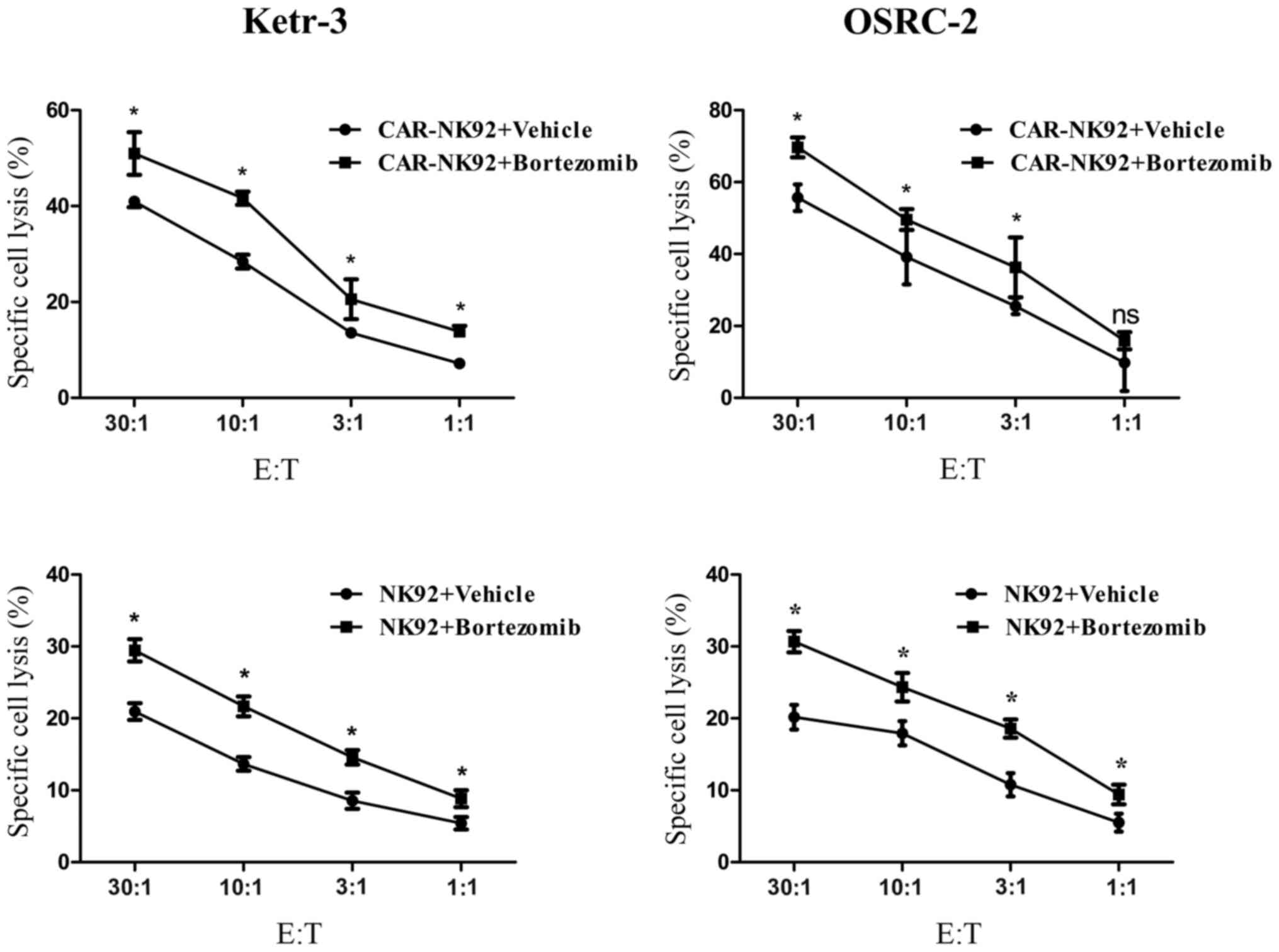
Previous studies revealed that low-dose bortezomib
sensitized human renal cancer cells to NK cells via the
upregulation of DNAM-1 and NKG2D ligands (52,53),
upregulation of DR4, and downregulation of HLA class I (54). To determine whether bortezomib
treatment could increase the sensitivity of renal cancer cells to
CAIX-CAR-NK92 or NK92 cell-mediated lysis, we treated Ketr-3 and
OSRC-2 cells with 10 nM bortezomib for 24 h (55). Next, we detected the killing
activity of the CAR-NK92 or NK92 cell lines respectively to the
bortezomib-treated Ketr-3 and OSRC-2 cells. The results revealed
that bortezomib treatment significantly increased the sensitivity
of Ketr-3 and OSRC-2 cells to the CAIX-specific CAR-NK92 cells and
NK92 cells and the synergistic efficacy was antigen independent
(Fig. 4).
Combination with bortezomib improves
the antitumor activity of CAIX-specific CAR-NK92 cells against RCC
xenografts
To examine the efficacy of combination therapy with
CAIX-CAR-NK92 cells and bortezomib against CAIX-positive RCC, we
established a subcutaneous xenograft model in NOD/SCID mice using
the human renal cancer line Ketr-3 cells expressing luciferase
(Ketr-3luc+). Next, the tumor-bearing mice were treated
with CAR-NK92 cells by tail vein injection and bortezomib by
peritoneal cavity injection. The treatment program is displayed in
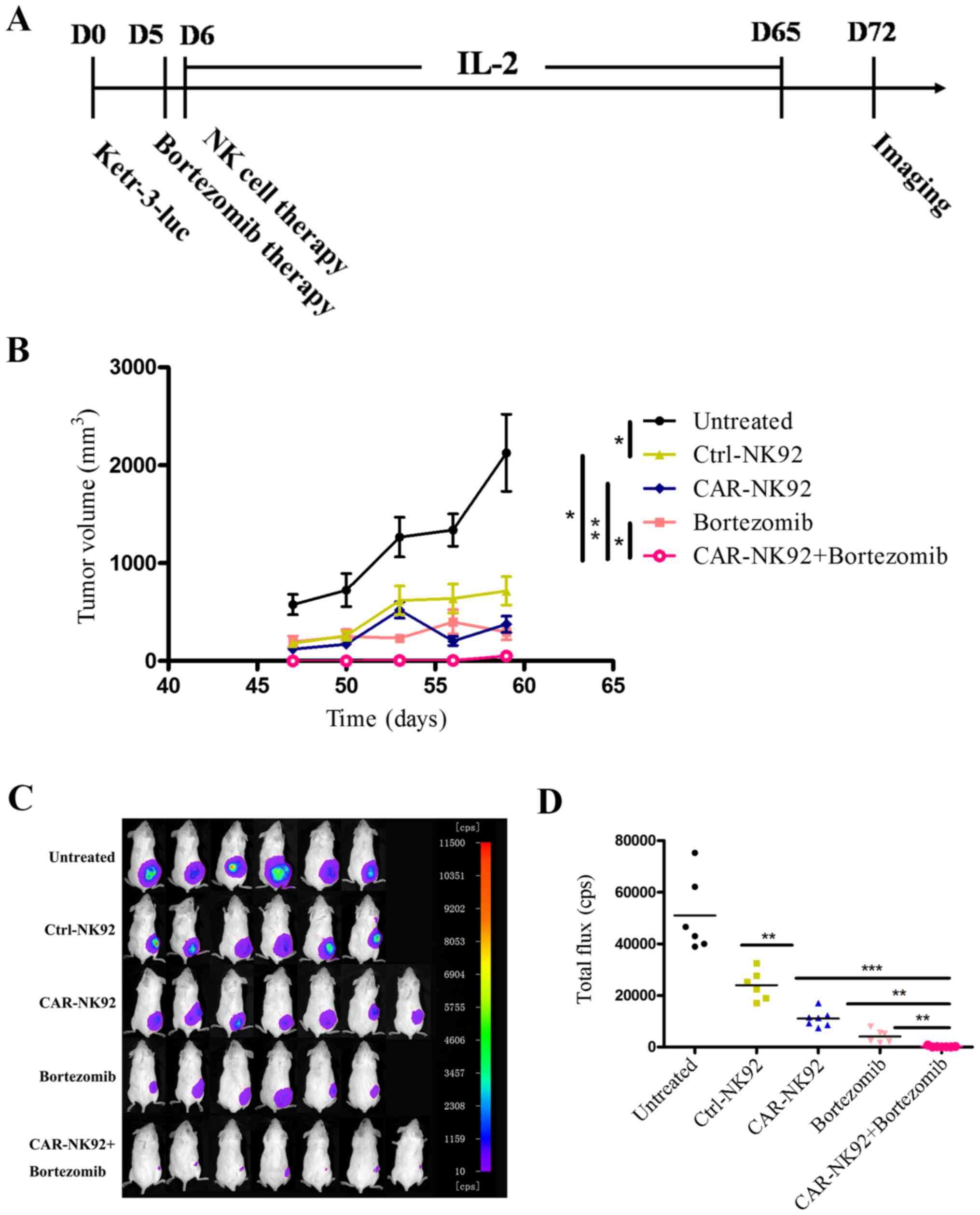
Fig. 5A.
As shown in Fig. 5,
both the results of the tumor growth curve (Fig. 5B) and in vivo imaging
(Fig. 5C and D) revealed that
Ctrl-NK92 cells, CAR-NK92 cells or bortezomib treatment alone
significantly reduced the growth of Ketr-3luc+ tumors
compared to the untreated group. In addition, treatment with
CAIX-CAR-NK92 cells significantly suppressed tumor growth compared
with the Ctrl-NK92 cells. These results demonstrated that
CAIX-CAR-NK92 cells can specifically kill CAIX-positive renal
cancer cells in vivo. Furthermore, the combination of
CAIX-CAR-NK92 cells and bortezomib significantly reduced the growth
of Ketr-3luc+ tumors compared to bortezomib or
CAIX-CAR-NK92 cells alone. The tumors of the mice that received
CAIX-CAR-NK92 cells plus bortezomib treatment were almost
eradicated. The values of the tumor volumes were concordant with
those of the in vivo imaging.
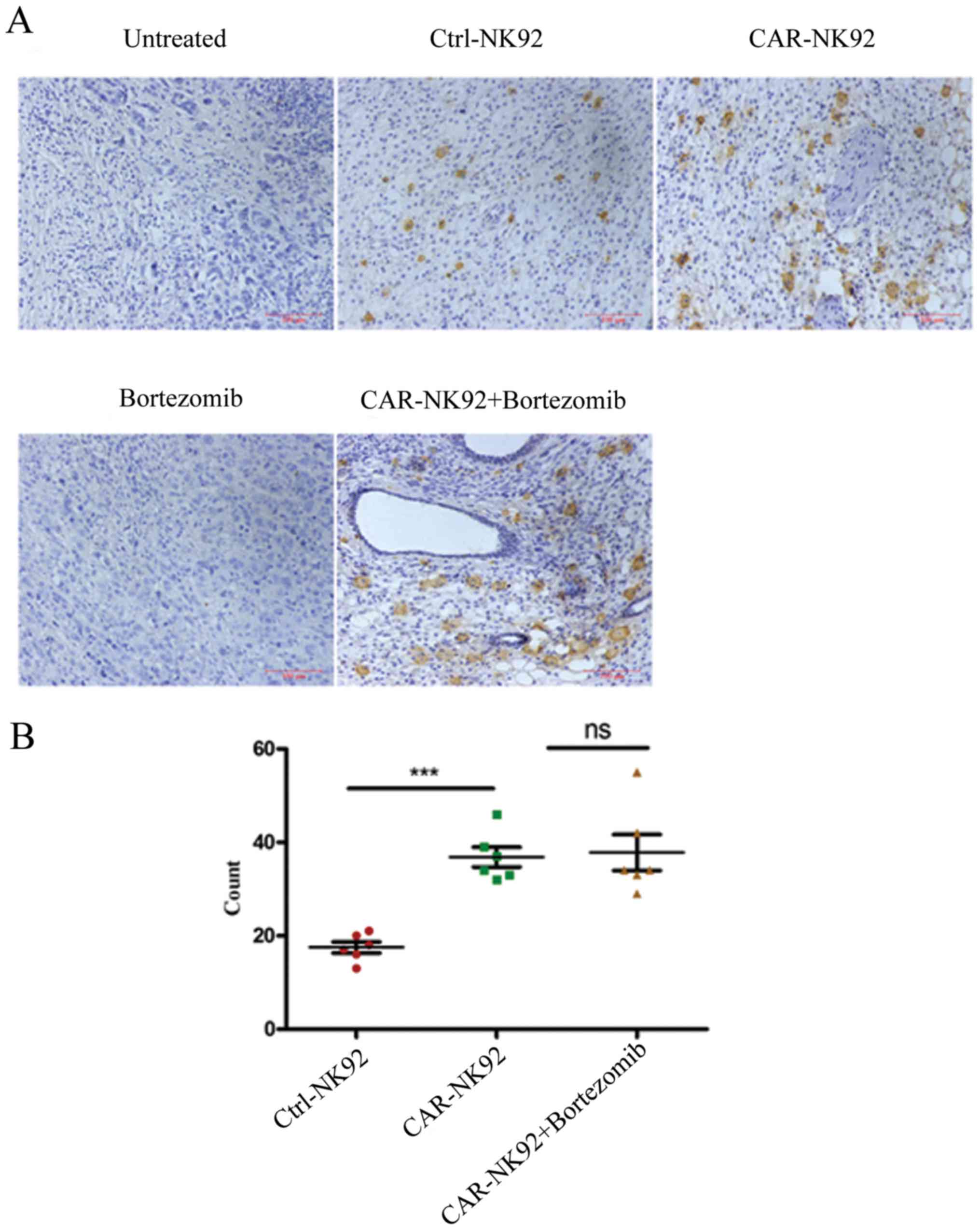
Detection of NK92 cell infiltration in
tumor tissues
Both our western blotting data (not shown) and a
previous study revealed that wild-type NK92 cells expressed CD3ζ
(28). Therefore, we used an
anti-human CD3ζ antibody to detect infiltrated NK92 cells in tumor
tissues by immunohistochemical (IHC) staining. As shown in Fig. 6A, CD3ζ-positive NK92 cells in the
tumor tissue of Ctrl-NK92, CAR-NK92 and CAR-NK92+bortezomib groups
were stained brown. However, no positive cells were observed in the
tumor tissues of the untreated and bortezomib groups. The
statistical results revealed that the number of NK92 cells in the
tumors of the CAR-NK92 group was significantly greater than in the
tumors of the Ctrl-NK92 group (P<0.001; Fig. 6B). The number of NK92 cells in the
tumor tissues of the CAR-NK92+bortezomib group was larger than in
the tumor tissues of the CAR-NK92 group. However, the difference
was comparable (Fig. 6B). These
findings indicated that CAIX-specific CAR-NK92 cells can traffic to
tumor sites.
Discussion
NK cells are a type of immune effector cells that
play important roles in immune surveillance. Numerous NK cell-based
anticancer therapies, including CAR-NK92 cells, are currently under
investigation. In the present study, for the first time to the best
of our knowledge, we constructed a third-generation CAR against
CAIX and developed the CAR-modified NK92 cell line. The CAR-NK92
cells exhibited robust CAIX-specific killing effects against
CAIX-positive renal cancer cells. In addition, we reported, for the
first time, that CAIX-CAR-NK92 cells exhibited synergistic
therapeutic efficacy with bortezomib against RCC in a mouse
model.
Early in 1996, Weijtens et al constructed a
first-generation CAR against CAIX, which was composed of a mouse
anti-human CAIX scFv and the Fc(epsilon)RI signaling receptor
gamma-chain (FcRγ) of mast cells, and they confirmed that
introduction of this CAR into cytotoxic T lymphocytes (CTLs)
rendered these lymphocytes specific for RCC (11). No clinical objective responses have
been observed in the clinical trials of this CAR (10). The CAR that we constructed is a
third-generation CAR with two costimulatory molecules, CD28 and
CD137. According to previous studies, incorporation of the CD28
signaling domain can enhance the proliferation and cytotoxicity of
CAR-T cells (56), and the CD137
signaling domain can enhance the persistence of CAR-T cells
compared with first generation CARs containing only one
costimulatory molecule (57).
In the in vivo studies, we confirmed that
bortezomib and CAIX-CAR-NK92 cells exhibited synergistic effects by
suppressing the growth of human RCC xenografts in NOD/SCID mice
(Fig. 5). We then investigated the
potential mechanisms. We revealed that the NK cells infiltrated
into tumor tissue, and that NK cells in the tumors of mice that
received treatment with CAR-NK92 cells and bortezomib were
comparable with those in the tumors of mice that received treatment
with CAR-NK92 alone (Fig. 6).
Therefore, bortezomib promoted the antitumor effects of CAR-NK92
but not by enhancing the infiltrating ability of CAR-NK92 cells
into tumors.
Bortezomib was reported to downregulate the
cell-surface expression of human lymphocyte antigen (HLA) class I
and to enhance natural killer cell-mediated lysis of myeloma
(54). Bortezomib was also reported
to enhance NK cell-mediated antitumor effects by upregulating the
expression of MICA/B, the ligands of NKG2D, and DR5, the receptor
of TRAIL in cancer cells (52,53).
Bortezomib can also enhance NK cell-mediated antitumor effects by
upregulating the expression of NK-cell activating receptors NKG2D,
TRAIL, and DNAM-1 in NK cells treated with bortezomib (52,58,59).
To investigate whether bortezomib enhanced the antitumor efficacy
of CAIX-CAR-NK92 cells by changing the expression of these
molecules in cancer cells or NK92 cells, we detected the expression
changes of CA9, Fas, MicA/B, HLA-A2, and DR4 in the RCC cell lines
Ketr-3, OSRC-2, ACHN, DNAM-1, NKG2D and NKp46 TRAIL in NK92 cells
after treatment with bortezomib at concentrations of 0, 5, 10 and
20 nM for 24 h. However, in general, these molecules did not
significantly change (data not shown). Therefore, the mechanism by
which bortezomib enhanced the therapeutic efficacy of CAIX-CAR-NK92
cells may not be by changing the expression of these molecules in
cancer cells or NK92 cells.
New data has revealed that besides the established
role of bortezomib in sensitizing tumors to cell death, it may act
as a multifaceted immunomodulatory drug and can sustain antitumor
immune effector functions and address tumor-associated
immunosuppression (49). Growing
evidence indicates that bortezomib administered at 15–20 nM doses
exhibited an intrinsic ability to increase the levels of
immunostimulatory cytokines and components of the Notch and NFκB
signaling pathways in lymphocytes, thereby amplifying their
effector functions either directly or indirectly (50,60,61).
Bortezomib also affected cytokine production in the tumor
microenvironment, thereby polarizing the cytokine milieu in favor
of antitumor immunity. Therefore, it is possible that bortezomib
enhances the antitumor immunity of immune effector cells by
overcoming tumor-induced suppression of numerous immune regulatory
networks (62). However, in the
present study, whether bortezomib enhanced the antitumor efficacy
of CAIX-CAR-NK92 cells by these mechanisms requires further
evaluation.
In conclusion, we constructed a third-generation CAR
against the RCC antigen CAIX. Our results revealed that
CAIX-specific CAR-NK92 cells have great potential to kill RCC
cells, and combination with bortezomib enhanced the effects of
CAR-NK92 cells against RCC in vitro and in vivo. The
present study is based on the NK92 cell line. Future studies of
this regimen can also be expanded to autologous or allogeneic
primary NK or T cells. This study provides an experimental basis
for the novel clinical regimen of CAIX-specific CAR-modified NK or
T cells for the treatment of RCC.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (nos. 81773253, 81502193 and
81571512); the Social Development Key Project of Jiangsu Province
(no. BE2016643); the Natural Science Foundation of Jiangsu Province
(BK20151167); and the Natural Science Key Project of Jiangsu
Provincial Education Department (17KJA320011).
Availability of data and materials
The datasets used or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
JZ and QF conceived and designed the study. QZ, JX,
JD, HoL, HuL, HaL, ML, YM and ZW performed the experiments. QZ and
JX wrote the paper. QZ, JX, JD, HoL and HuL reviewed and edited the
manuscript. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
All protocols for the animal studies were reviewed
and approved by the Institutional Animal Care and Use Committee of
the Jiangsu Provincial Academy of Chinese Medicine
(SCXK2012-005).
Patient consent for publication
Not applicable.
Competing interests
The authors have no commercial, proprietary, or
financial interest in the products or companies described in this
article and declare that they have no competing interests.
References
|
1
|
Kuusk T, Grivas N, de Bruijn R and Bex A:
The current management of renal cell carcinoma. Minerva Med.
108:357–369. 2017.PubMed/NCBI
|
|
2
|
Linehan WM and Ricketts CJ: Kidney cancer
in 2016: RCC-advances in targeted therapeutics and genomics. Nat
Rev Urol. 14:76–78. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fisher RI, Rosenberg SA and Fyfe G:
Long-term survival update for high-dose recombinant interleukin-2
in patients with renal cell carcinoma. Cancer J Sci Am. 6 Suppl
1:S55–S57. 2000.PubMed/NCBI
|
|
4
|
Rosenberg SA, Yang JC, Topalian SL,
Schwartzentruber DJ, Weber JS, Parkinson DR, Seipp CA, Einhorn JH
and White DE: Treatment of 283 consecutive patients with metastatic
melanoma or renal cell cancer using high-dose bolus interleukin 2.
JAMA. 271:907–913. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang Q, Li H, Yang J, Li L, Zhang B, Li J
and Zheng J: Strategies to improve the clinical performance of
chimeric antigen receptor-modified T cells for cancer. Curr Gene
Ther. 13:65–70. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Geyer MB and Brentjens RJ: Review: Current
clinical applications of chimeric antigen receptor (CAR) modified T
cells. Cytotherapy. 18:1393–1409. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kochenderfer JN, Dudley ME, Feldman SA,
Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes
MS, Sherry RM, et al: B-cell depletion and remissions of malignancy
along with cytokine-associated toxicity in a clinical trial of
anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood.
119:2709–2720. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cruz CR, Micklethwaite KP, Savoldo B,
Ramos CA, Lam S, Ku S, Diouf O, Liu E, Barrett AJ, Ito S, et al:
Infusion of donor-derived CD19-redirected virus-specific T cells
for B-cell malignancies relapsed after allogeneic stem cell
transplant: A phase 1 study. Blood. 122:2965–2973. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lim WA and June CH: The principles of
engineering immune cells to treat cancer. Cell. 168:724–740. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lamers CH, Klaver Y, Gratama JW, Sleijfer
S and Debets R: Treatment of metastatic renal cell carcinoma (mRCC)
with CAIX CAR-engineered T-cells-a completed study overview.
Biochem Soc Trans. 44:951–959. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weijtens ME, Willemsen RA, Valerio D, Stam
K and Bolhuis RL: Single chain Ig/gamma gene-redirected human T
lymphocytes produce cytokines, specifically lyse tumor cells, and
recycle lytic capacity. J Immunol. 157:836–843. 1996.PubMed/NCBI
|
|
12
|
Pietra G, Vitale C, Pende D, Bertaina A,
Moretta F, Falco M, Vacca P, Montaldo E, Cantoni C, Mingari MC, et
al: Human natural killer cells: News in the therapy of solid tumors
and high-risk leukemias. Cancer Immunol Immunother. 65:465–476.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rosenberg SA, Lotze MT, Muul LM, Leitman
S, Chang AE, Ettinghausen SE, Matory YL, Skibber JM, Shiloni E,
Vetto JT, et al: Observations on the systemic administration of
autologous lymphokine-activated killer cells and recombinant
interleukin-2 to patients with metastatic cancer. N Engl J Med.
313:1485–1492. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Law TM, Motzer RJ, Mazumdar M, Sell KW,
Walther PJ, O'Connell M, Khan A, Vlamis V, Vogelzang NJ and Bajorin
DF: Phase III randomized trial of interleukin-2 with or without
lymphokine-activated killer cells in the treatment of patients with
advanced renal cell carcinoma. Cancer. 76:824–832. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Parkhurst MR, Riley JP, Dudley ME and
Rosenberg SA: Adoptive transfer of autologous natural killer cells
leads to high levels of circulating natural killer cells but does
not mediate tumor regression. Clin Cancer Res. 17:6287–6297. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Miller JS, Soignier Y,
Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, McKenna
D, Le C, Defor TE, Burns LJ, et al: Successful adoptive transfer
and in vivo expansion of human haploidentical NK cells in patients
with cancer. Blood. 105:3051–3057. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rubnitz JE, Inaba H, Ribeiro RC, Pounds S,
Rooney B, Bell T, Pui CH and Leung W: NKAML: A pilot study to
determine the safety and feasibility of haploidentical natural
killer cell transplantation in childhood acute myeloid leukemia. J
Clin Oncol. 28:955–959. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Curti A, Ruggeri L, D'Addio A, Bontadini
A, Dan E, Motta MR, Trabanelli S, Giudice V, Urbani E, Martinelli
G, et al: Successful transfer of alloreactive haploidentical KIR
ligand-mismatched natural killer cells after infusion in elderly
high risk acute myeloid leukemia patients. Blood. 118:3273–3279.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Altvater B, Landmeier S, Pscherer S, Temme
J, Schweer K, Kailayangiri S, Campana D, Juergens H, Pule M and
Rossig C: 2B4 (CD244) signaling by recombinant antigen-specific
chimeric receptors costimulates natural killer cell activation to
leukemia and neuroblastoma cells. Clin Cancer Res. 15:4857–4866.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kailayangiri S, Altvater B, Spurny C,
Jamitzky S, Schelhaas S, Jacobs AH, Wiek C, Roellecke K, Hanenberg
H, Hartmann W, et al: Targeting Ewing sarcoma with activated and
GD2-specific chimeric antigen receptor-engineered human NK cells
induces upregulation of immune-inhibitory HLA-G. Oncoimmunology.
6:e12500502017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Muller N, Michen S, Tietze S, Töpfer K,
Schulte A, Lamszus K, Schmitz M, Schackert G, Pastan I and Temme A:
Engineering NK cells modified with an EGFRvIII-specific chimeric
antigen receptor to overexpress CXCR4 improves immunotherapy of
CXCL12/SDF-1α-secreting glioblastoma. J Immunother. 38:197–210.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Geller MA, Cooley S, Judson PL, Ghebre R,
Carson LF, Argenta PA, Jonson AL, Panoskaltsis-Mortari A,
Curtsinger J, McKenna D, et al: A phase II study of allogeneic
natural killer cell therapy to treat patients with recurrent
ovarian and breast cancer. Cytotherapy. 13:98–107. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gong JH, Maki G and Klingemann HG:
Characterization of a human cell-line (Nk-92) with phenotypical and
functional-characteristics of activated Natural killer cells.
Leukemia. 8:652–658. 1994.PubMed/NCBI
|
|
24
|
Arai S, Meagher R, Swearingen M, Myint H,
Rich E, Martinson J and Klingemann H: Infusion of the allogeneic
cell line NK-92 in patients with advanced renal cell cancer or
melanoma: A phase I trial. Cytotherapy. 10:625–632. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tam YK, Martinson JA, Doligosa K and
Klingemann HG: Ex vivo expansion of the highly cytotoxic human
natural killer cell line NK-92 under current good manufacturing
practice conditions for clinical adoptive cellular immunotherapy.
Cytotherapy. 5:259–272. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tonn T, Becker S, Esser R, Schwabe D and
Seifried E: Cellular immunotherapy of malignancies using the clonal
natural killer cell line NK-92. J Hematoth Stem Cell. 10:535–544.
2001. View Article : Google Scholar
|
|
27
|
Boissel L, Betancur M, Wels WS, Tuncer H
and Klingemann H: Transfection with mRNA for CD19 specific chimeric
antigen receptor restores NK cell mediated killing of CLL cells.
Leuk Res. 33:1255–1259. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Muller T, Uherek C, Maki G, Chow KU,
Schimpf A, Klingemann HG, Tonn T and Wels WS: Expression of a
CD20-specific chimeric antigen receptor enhances cytotoxic activity
of NK cells and overcomes NK-resistance of lymphoma and leukemia
cells. Cancer Immunol Immunother. 57:411–423. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schonfeld K, Sahm C, Zhang C, Naundorf S,
Brendel C, Odendahl M, Nowakowska P, Bönig H, Köhl U, Kloess S, et
al: Selective inhibition of tumor growth by clonal NK cells
expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol
Ther. 23:330–338. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang C, Burger MC, Jennewein L, Genßler
S, Schönfeld K, Zeiner P, Hattingen E, Harter PN, Mittelbronn M,
Tonn T, et al: ErbB2/HER2-Specific NK cells for targeted therapy of
glioblastoma. J Natl Cancer Inst. 108:2016. View Article : Google Scholar
|
|
31
|
Esser R, Muller T, Stefes D, Kloess S,
Seidel D, Gillies SD, Aperlo-Iffland C, Huston JS, Uherek C,
Schönfeld K, et al: NK cells engineered to express a GD2-specific
antigen receptor display built-in ADCC-like activity against tumour
cells of neuroectodermal origin. J Cell Mol Med. 16:569–581. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sahm C, Schönfeld K and Wels WS:
Expression of IL-15 in NK cells results in rapid enrichment and
selective cytotoxicity of gene-modified effectors that carry a
tumor-specific antigen receptor. Cancer Immunol Immunother.
61:1451–1461. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chu J, Deng Y, Benson DM, He S, Hughes T,
Zhang J, Peng Y, Mao H, Yi L, Ghoshal K, et al: CS1-specific
chimeric antigen receptor (CAR)-engineered natural killer cells
enhance in vitro and in vivo antitumor activity against human
multiple myeloma. Leukemia. 28:917–927. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jiang H, Zhang W, Shang P, Zhang H, Fu W,
Ye F, Zeng T, Huang H, Zhang X, Sun W, et al: Transfection of
chimeric anti-CD138 gene enhances natural killer cell activation
and killing of multiple myeloma cells. Mol Oncol. 8:297–310. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Genssler S, Burger MC, Zhang C, Oelsner S,
Mildenberger I, Wagner M, Steinbach JP and Wels WS: Dual targeting
of glioblastoma with chimeric antigen receptor-engineered natural
killer cells overcomes heterogeneity of target antigen expression
and enhances antitumor activity and survival. Oncoimmunology.
5:e11193542016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen X, Han J, Chu J, Zhang L, Zhang J,
Chen C, Chen L, Wang Y, Wang H, Yi L, et al: A combinational
therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1
for breast cancer brain metastases. Oncotarget. 7:27764–27777.
2016.PubMed/NCBI
|
|
37
|
Han J, Chu J, Chan Keung W, Zhang J, Wang
Y, Cohen JB, Victor A, Meisen WH, Kim SH, Grandi P, et al:
CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII
enhance killing of glioblastoma and patient-derived glioblastoma
stem cells. Sci Rep. 5:114832015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen KH, Wada M, Firor AE, Pinz KG, Jares
A, Liu H, Salman H, Golightly M, Lan F, Jiang X and Ma Y: Novel
anti-CD3 chimeric antigen receptor targeting of aggressive T cell
malignancies. Oncotarget. 7:56219–56232. 2016.PubMed/NCBI
|
|
39
|
Chen KH, Wada M, Pinz KG, Liu H, Lin KW,
Jares A, Firor AE, Shuai X, Salman H, Golightly M, et al:
Preclinical targeting of aggressive T-cell malignancies using
anti-CD5 chimeric antigen receptor. Leukemia. 31:2151–2160. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rezvani K and Rouce RH: The application of
natural killer cell immunotherapy for the treatment of cancer.
Front Immunol. 6:5782015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xu J, Tian K, Zhang H, Li L, Liu H, Liu J,
Zhang Q and Zheng J: Chimeric antigen receptor-T cell therapy for
solid tumors require new clinical regimens. Expert Rev Anticancer
Ther. 17:1099–1106. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Orlowski M and Wilk S: Catalytic
activities of the 20 S proteasome, a multicatalytic proteinase
complex. Arch Biochem Biophys. 383:1–16. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lilienbaum A: Relationship between the
proteasomal system and autophagy. Int J Biochem Mol Biol. 4:1–26.
2013.PubMed/NCBI
|
|
44
|
Chen D, Frezza M, Schmitt S, Kanwar J and
Dou QP: Bortezomib as the first proteasome inhibitor anticancer
drug: Current status and future perspectives. Curr Cancer Drug
Targets. 11:239–253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Manasanch EE and Orlowski RZ: Proteasome
inhibitors in cancer therapy. Nat Rev Clin Oncol. 14:417–433. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Richardson PG, Hideshima T and Anderson
KC: Bortezomib (PS-341): A novel, first-in-class proteasome
inhibitor for the treatment of multiple myeloma and other cancers.
Cancer Control. 10:361–369. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Huang Z, Wu Y, Zhou X, Xu J, Zhu W, Shu Y
and Liu P: Efficacy of therapy with bortezomib in solid tumors: A
review based on 32 clinical trials. Future Oncol. 10:1795–1807.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kondagunta GV, Drucker B, Schwartz L,
Bacik J, Marion S, Russo P, Mazumdar M and Motzer RJ: Phase II
trial of bortezomib for patients with advanced renal cell
carcinoma. J Clin Oncol. 22:3720–3725. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Pellom ST Jr, Singhal A and Shanker A:
Prospects of combining adoptive cell immunotherapy with bortezomib.
Immunotherapy. 9:305–308. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ames E, Hallett WH and Murphy WJ:
Sensitization of human breast cancer cells to natural killer
cell-mediated cytotoxicity by proteasome inhibition. Clin Exp
Immunol. 155:504–513. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang Q, Wang H, Li H, Xu J, Tian K, Yang
J, Lu Z and Zheng J: Chimeric antigen receptor-modified T cells
inhibit the growth and metastases of established tissue
factor-positive tumors in NOG mice. Oncotarget. 8:9488–9499.
2017.PubMed/NCBI
|
|
52
|
Armeanu S, Krusch M, Baltz KM, Weiss TS,
Smirnow I, Steinle A, Lauer UM, Bitzer M and Salih HR: Direct and
natural killer cell-mediated antitumor effects of low-dose
bortezomib in hepatocellular carcinoma. Clin Cancer Res.
14:3520–3528. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lundqvist A, Abrams SI, Schrump DS,
Alvarez G, Suffredini D, Berg M and Childs R: Bortezomib and
depsipeptide sensitize tumors to tumor necrosis factor-related
apoptosis-inducing ligand: A novel method to potentiate natural
killer cell tumor cytotoxicity. Cancer Res. 66:7317–7325. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Shi J, Tricot GJ, Garg TK, Malaviarachchi
PA, Szmania SM, Kellum RE, Storrie B, Mulder A, Shaughnessy JD Jr,
Barlogie B and van Rhee F: Bortezomib down-regulates the
cell-surface expression of HLA class I and enhances natural killer
cell-mediated lysis of myeloma. Blood. 111:1309–1317. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hallett WH, Ames E, Motarjemi M, Barao I,
Shanker A, Tamang DL, Sayers TJ, Hudig D and Murphy WJ:
Sensitization of tumor cells to NK cell-mediated killing by
proteasome inhibition. J Immunol. 180:163–170. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Finney HM, Lawson AD, Bebbington CR and
Weir AN: Chimeric receptors providing both primary and
costimulatory signaling in T cells from a single gene product. J
Immunol. 161:2791–2797. 1998.PubMed/NCBI
|
|
57
|
Carpenito C, Milone MC, Hassan R, Simonet
JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF,
Albelda SM, et al: Control of large, established tumor xenografts
with genetically retargeted human T cells containing CD28 and CD137
domains. Proc Natl Acad Sci USA. 106:3360–3365. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lundqvist A, Yokoyama H, Smith A, Berg M
and Childs R: Bortezomib treatment and regulatory T-cell depletion
enhance the antitumor effects of adoptively infused NK cells.
Blood. 113:6120–6127. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Niu C, Jin H, Li M, Zhu S, Zhou L, Jin F,
Zhou Y, Xu D, Xu J, Zhao L, et al: Low-dose bortezomib increases
the expression of NKG2D and DNAM-1 ligands and enhances induced NK
and γδ T cell-mediated lysis in multiple myeloma. Oncotarget.
8:5954–5964. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Seeger JM, Schmidt P, Brinkmann K, Hombach
AA, Coutelle O, Zigrino P, Wagner-Stippich D, Mauch C, Abken H,
Krönke M and Kashkar H: The proteasome inhibitor bortezomib
sensitizes melanoma cells toward adoptive CTL attack. Cancer Res.
70:1825–1834. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Thounaojam MC, Dudimah DF, Pellom ST Jr,
Uzhachenko RV, Carbone DP, Dikov MM and Shanker A: Bortezomib
enhances expression of effector molecules in anti-tumor
CD8+ T lymphocytes by promoting Notch-nuclear factor-κB
crosstalk. Oncotarget. 6:32439–32455. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Pellom ST Jr, Dudimah DF, Thounaojam MC,
Uzhachenko RV, Singhal A, Richmond A and Shanker A: Bortezomib
augments lymphocyte stimulatory cytokine signaling in the tumor
microenvironment to sustain CD8+ T cell antitumor
function. Oncotarget. 8:8604–8621. 2017. View Article : Google Scholar : PubMed/NCBI
|