Introduction
Breast cancer is the most common type of carcinoma
and the second most common cause of cancer-associated mortality in
females (1). MicroRNAs (miRNAs) are
a group of small, single-stranded, non-coding RNAs that regulate
gene expression by partial base pairing with the 3′-untranslated
region (3′-UTR) or enhancer region of targeted genes (2–5). In
addition to regulating particular mRNAs directly, miRNAs can also
affect the association between effectors and target mRNAs (6). Each miRNA may regulate a variety
proteins and serve an important role in more than one key cellular
progress, including cell proliferation, cell survival and cell fate
determination (7). Evidence has
indicated that deregulation of miRNA expression may contribute
toward the development of various types of human diseases,
including cancer. The miRNAs are therefore becoming increasingly
appreciated as potential biomarkers for the diagnosis, treatment
and prognosis of diseases.
Recent studies have identified microRNA-424-5p
(miR-424) as a crucial regulator in the development of several
types of cancer, including bladder (8) and cervical cancer (9). In bladder cancer, the decreased
expression of miR-424 was associated with invasive tumor growth,
advanced clinical stage and a poor prognosis. Increased miR-424
levels inhibited tumor growth and invasive ability. In cervical
cancer, miR-424 may act as an anti-oncogene by suppressing cell
growth. However, the role of miR-424 in breast cancer remains
poorly defined.
The aim of the present study was to define the
function of miR-424 in breast cancer cells. The experiments
performed indicated that miR-424 may suppress cell proliferation
and arrest cells in the G2/M cell phase by negatively regulating
cyclin-dependent kinase 1 (CDK1) mRNA in human breast cancer, and
that this may occur through the Hippo pathway and the extracellular
signal-regulated kinase (ERK) pathway. The results of the present
study provided novel evidence for the role of miR-424 in breast
cancer.
Methods and materials
Specimens, cell lines and culture
conditions
The present study collected 17 pairs of breast
cancer and matched adjacent normal control samples from the
Department of Breast and Thyroid Surgery of Shanghai Tenth People's
Hospital (Shanghai, China) between February and April 2017, which
were histologically confirmed to be invasive ductal breast cancer.
The patients were females aged between 34 and 74 years, with a mean
age of 55 years. All these tissues were immediately snap-frozen to
−196°C in liquid nitrogen. None of these patients had received any
radiotherapy or chemotherapy prior to surgery.
Human triple-negative breast cancer MDA-MB-231 and
HCC1937 cell lines, and the non-malignant breast epithelial MCF-10A
cell line, were used. The cells were purchased from the Chinese
Academy of Sciences. The HEK-293T cell line was a gift from the
laboratory department of Shanghai Tenth People's Hospital. All
cells were cultured in Dulbecco's modified Eagle's medium (DMEM;
cat. no. C11995500BT; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), supplemented with 10% fetal bovine serum (FBS;
cat. no. 900-108; Gemini Bio Products, West Sacramento, CA, USA),
penicillin (100 U/ml) and streptomycin (100 µg/ml; PS; cat. no.
15140-163; Gibco; Thermo Fisher Scientific, Inc.). Cells were
incubated at 37°C in a humidified atmosphere containing 5%
CO2.
Transfection assays
The cells (1×106) were cultured in 6-well
plates (Corning Incorporated, Corning, NY, USA). The cell density
reached 30–50% confluency after <24 h. Next, the miR-424 mimics
or miR-424 inhibitor or negative control (NC) or CDK1 small
interfering RNA (siR-CDK1) or siR-NC were transfected separately
into cells at a final concentration of 100 nmol/l using
Lipofectamine® 2000 (cat. no. 11668-019; Invitrogen;
Thermo Fisher Scientific, Inc.) in DMEM. The medium was replaced
with complete DMEM after 4–6 h of incubation. The cells were
incubated as described earlier and used for future analyses after
24 h.
miR-424 mimics (5′-CAGCAGCAAUUCAUGUUUUGAA-3′),
miR-424 inhibitor (5′-GUCGUCGUUAAGUACAAAACUU-3′) and NC mimics
(5′-UUUGUACUACACAAAAGUACUG-3′) were chemosynthesized by Guangzhou
RiboBio Co., Ltd. (Guangzhou, China). siR-CDK1 (sense,
5′-GGCACUGAAUCAUCCAUAUTT-3′ and antisense,
5′-AUAUGGAUGAUUCAGUGCCTT-3′) and siR-NC (sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′) were synthesized by Sangon Biotech
Co., Ltd. (Shanghai, China).
MTT assays
Cell proliferation ability was estimated by the MTT
assay (cat. no. A100793-0001; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) every 24 h for a period of 120 h. Cells were plated into
96-well plates (Corning Incorporated) at a density of 500
cells/well. In brief, the MTT solution was added to each well and
continuously incubated for 4 h. Subsequently, the medium was
replaced by dimethylsulfoxide (DMSO; cat. no. A503039-0500;
Sigma-Aldrich; Merck KGaA) to dissolve the purple formazan. The
absorbance at optical density (OD) of 492 nm was measured by a
microplate spectrophotometer (BioTek Instruments, Inc., Winooski,
VT, USA).
Plate colony formation assays
Cells were plated in 6-well plates at a density of
500 cells/well and incubated for 7–14 days or until the colonies
were visible to the eye. The colonies were fixed in 95% ethanol for
10 min, dried and stained with 0.1% crystal violet solution for 15
min at room temperature. Next, the plates were gently washed three
times with water. Colonies with diameters of >1.5 mm were
counted as live cells.
Protein extraction and western blot
analyses
Following transfection for 48–72 h, the cells were
resuspended in 80 µl/well radioimmunoprecipitation assay lysis
buffer (cat. no. P0013C; Beyotime Institute of Biotechnology,
Haimen, China) for 20 min. The protein concentrations were
quantified using a bicinchoninic acid protein assay kit (P0010;
Beyotime Institute of Biotechnology). Protein samples were
denatured with 6× SDS sample loading buffer (P0015F; Beyotime
Institute of Biotechnology) at 100°C for 10 min. Equal amounts of
protein (30 µg) from each sample were separated by 10% SDS-PAGE and
transferred onto 0.45-µm nitrocellulose membranes (Beyotime
Institute of Biotechnology). The membranes were blocked with 5%
skimmed milk for 60 min at room temperature and then probed with
antibodies against CDK1 (dilution, 1:1,000; cat. no. BS6467;
Bioworld Technology, Inc., Freemont, CA, USA), proliferating cell
nuclear antigen [PCNA; dilution, 1:1,000; cat. no. 13110; Cell
Signaling Technology, Inc., Danvers, MA, USA (CST)], Yes-associated
protein (YAP; dilution, 1:1,000; cat. no. 14074; CST), ERK
(dilution, 1:10,000; cat. no. ab184699; Abcam, Cambridge, UK),
phosphorylated ERK (p-ERK; dilution, 1:10,000; cat. no. ab201015;
Abcam) and β-actin (dilution, 1:1,000; cat. no. BS6498; Bioworld
Technology, Inc.) overnight at 4°C. The membranes were incubated
with IRDye® 800CW-conjugated anti-mouse (dilution,
1:1,000; cat. no. 926-32210; LI-COR Biosciences, Lincoln, NE, USA)
or IRDye® 800CW-conjugated anti-rabbit secondary
antibodies (dilution, 1:1,000; cat. no. 926-32211; LI-COR
Biosciences) for 60 min. Finally, the bands were detected using an
Odyssey Scanning system (LI-COR Biosciences).
Cell cycle assays
The cells were trypsinized and centrifuged at 1,000
rpm for 5 min at room temperature, and washed in cold PBS twice.
Then cells were fixed in cold 70% ethanol at 4°C overnight. Each
sample were resuspended in 300 µl 0.05 g/l propidium iodide
(PI/RNase Staining Buffer Solution, 550825, BD Pharmingen) for 30
min in the dark at room temperature. Next, cell cycles were
analyzed by flow cytometry (FACSCanto™ II; BD Biosciences, Franklin
Lakes, NJ, USA) and the software used for analysis was ModFit LT
3.2 (Flow Cytometry DNA Modeling Software, Verity Software House,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the cells (following
transfection for 48 h) or tissues using TRIzol® reagent
(cat. no. 15596-026; Invitrogen; Thermo Fisher Scientific, Inc.).
RNA was reverse transcribed using a PrimeScript™ RT-PCR kit (cat.
no. RR037A; Takara Biotechnology Co., Ltd., Dalian, China)
according to the manufacturer's protocol. The SYBRGreen PCR master
mix (cat. no. KK4601; Kapa Biosystems, Inc., Wilmington, MA, USA)
was used for RT-qPCR, which was followed by detection using a
7900HT fast RT-PCR instrument (Applied Biosystems; Thermo Fisher
Scientific, Inc.).
miR-424 (miRQ0001341-1-1) and U6 (MQP-0201) primers,
purchased from Guangzhou RiboBio Co., Ltd., were used for the
detection of miR-424. The primer sequences were as follows: miR-424
forward, 5′-CAGCAGCAAUUCAUGUUUUGAA-3′ and reverse,
5′-CAGTGCGTGTCGTGGAGT-3′; and U6 (internal standard) forward,
5′-CTCGCTTCGGCAGCACA-3′ and reverse, 5′-AACGCTTCACGAATTTGCGT-3′.
For CDK1 mRNA analyses, β-actin was used as an internal standard.
The primer sequences (Sangon Biotech Co., Ltd.) were as follows:
CDK1 forward, 5′-GGATGTGCTTATGCAGGATTCC-3′ and reverse,
5′-CATGTACTGACCAGGAGGGATAG-3′; and β-actin forward,
5′-CAGAGCCTCGCCTTTGCC-3′ and reverse, 5′-GTCGCCCACATAGGAATC-3′. The
amplification procedures were as follows: 3 min at 95°C, followed
by 40 cycles of 3 sec at 95°C and 30 sec at 60°C.
The relative expression of miRNA or mRNA was
assessed using the 2−ΔΔCq method (10).
Dual-luciferase reporter assay
The psiCHECK-2/CDK1 3′-UTR wild-type and mutant
reporter plasmids (G0001) were purchased from Shanghai Integrated
Biotech Solutions, China. HEK-293T cells were transiently
co-transfected with 40 ng wild-type or mutant reporter plasmids
with 100 nmol/l miR-424 or NC using Lipofectamine® 2000
reagent. Following transfection for 5 h, the complete medium was
changed. Following transfection for 24 h, the cells were detected
using a Dual Luciferase Assay (cat. no. E1910; Promega Corporation,
Madison, WI, USA). The Renilla luciferase (RL) activity was
normalized to firefly luciferase (FL) activity and the ratio of
RL/FL was recorded. Each sample was tested with three
replicates.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism version 6.0 (GraphPad Software, Inc., La Jolla, CA,
USA). Data from >3 independent experiments are presented as the
mean ± standard deviation. The differences between two groups were
compared using Student's t-test. Differences between >2 groups
were compared using one-way analysis of variance followed by
Tukey's post hoc test. Kaplan-Meier, followed by the log-rank test,
was used for survival analyses. P<0.05 were considered to
indicate a statistically significant difference.
Results
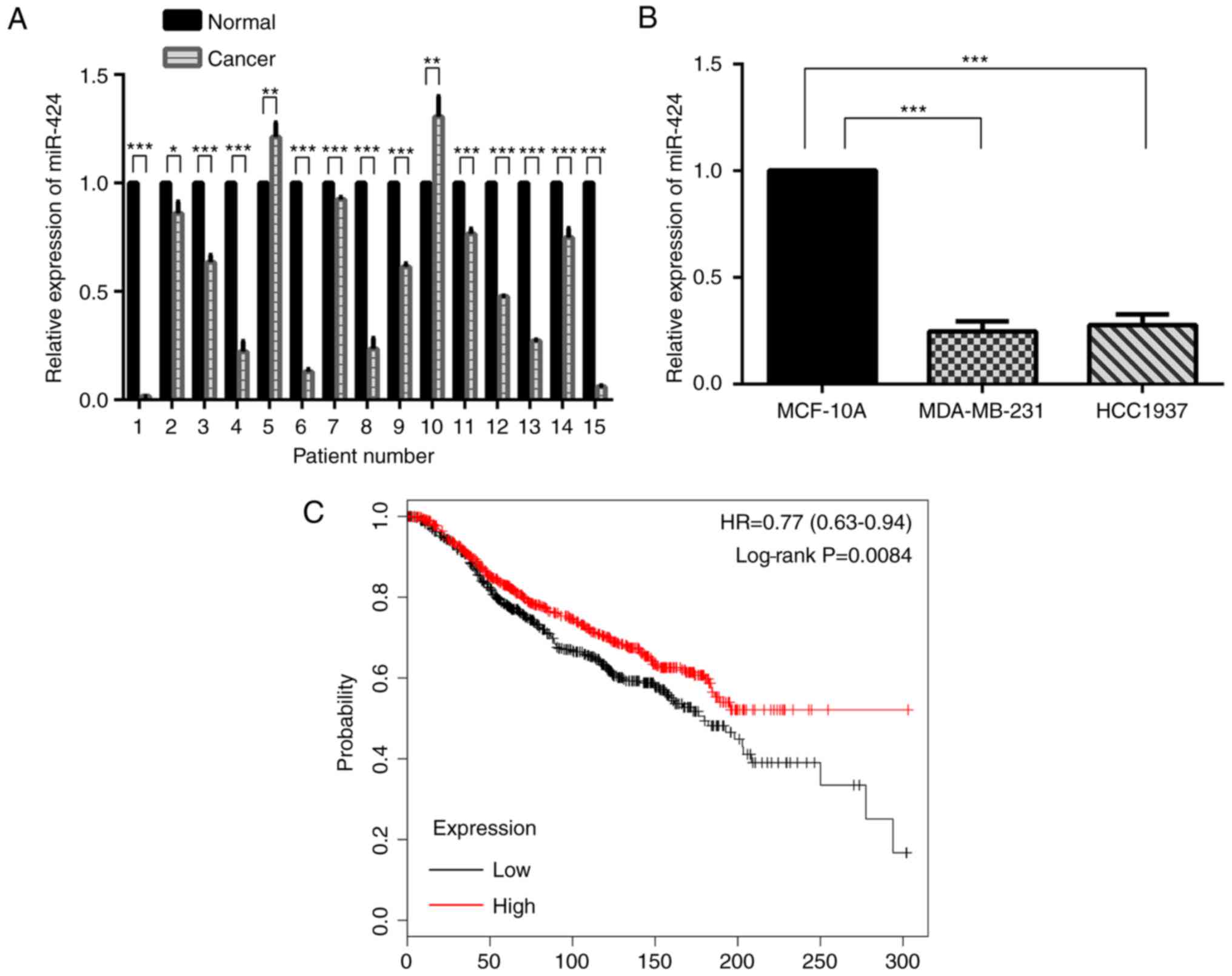
miR-424 expression is decreased in
breast cancer
The expression of miR-424 was decreased in human
breast cancer samples compared with that in matched normal breast
tissue samples (Fig. 1A). In
MDA-MB-231 and HCC-1937 cells, miR-424 expression was also
decreased compared with that in MCF-10A cells (Fig. 1B). Kaplan-Meier survival analyses
suggested that miR-424 acted as an anti-oncogene (Fig. 1C).
miR-424 suppresses cell proliferation
in breast cancer
To begin with, RT-qPCR was conducted to examine the
expression levels of miR-424 in MDA-MB-231 and HCC-1937 cells. As
demonstrated in Fig. 2A, cells were
successfully transfected with miR-424 mimics or inhibitors, leading
to increased or decreased miR-424 expression, respectively. The
MDA-MB-231 and HCC-1937 cell lines were used in the subsequent
experiments. In order to determine the effects of miR-424 in breast
cancer cells, the effect of miR-424 on cell proliferation was
verified. The expression of miR-424 was upregulated and
downregulated when compared with that in NC. The cells were
respectively transfected with miR-424 mimics, inhibitor or NC,
prior to MTT assays and colony formation assays being performed
following transfection for 24 h. The MTT assay results demonstrated
that cell proliferation was decreased in cells transfected with
miR-424 mimics, compared with the NC cell group, while the cells
transfected with inhibitors exhibited the opposite results
(Fig. 2B and C). The colony
formation assays also indicated that the number of colonies in
cells with upregulated miR-424 was less, and in cells with
downregulated miR-424, the colony number was greater than that in
the NC group (Fig. 2D and E). In
addition, western blot analysis was used to detect PCNA protein
levels to represent the level of cell proliferation, which was also
decreased by upregulation of miR-424 (Fig. 2F and G). All these data revealed
that miR-424 suppresses cell proliferation in breast cancer.
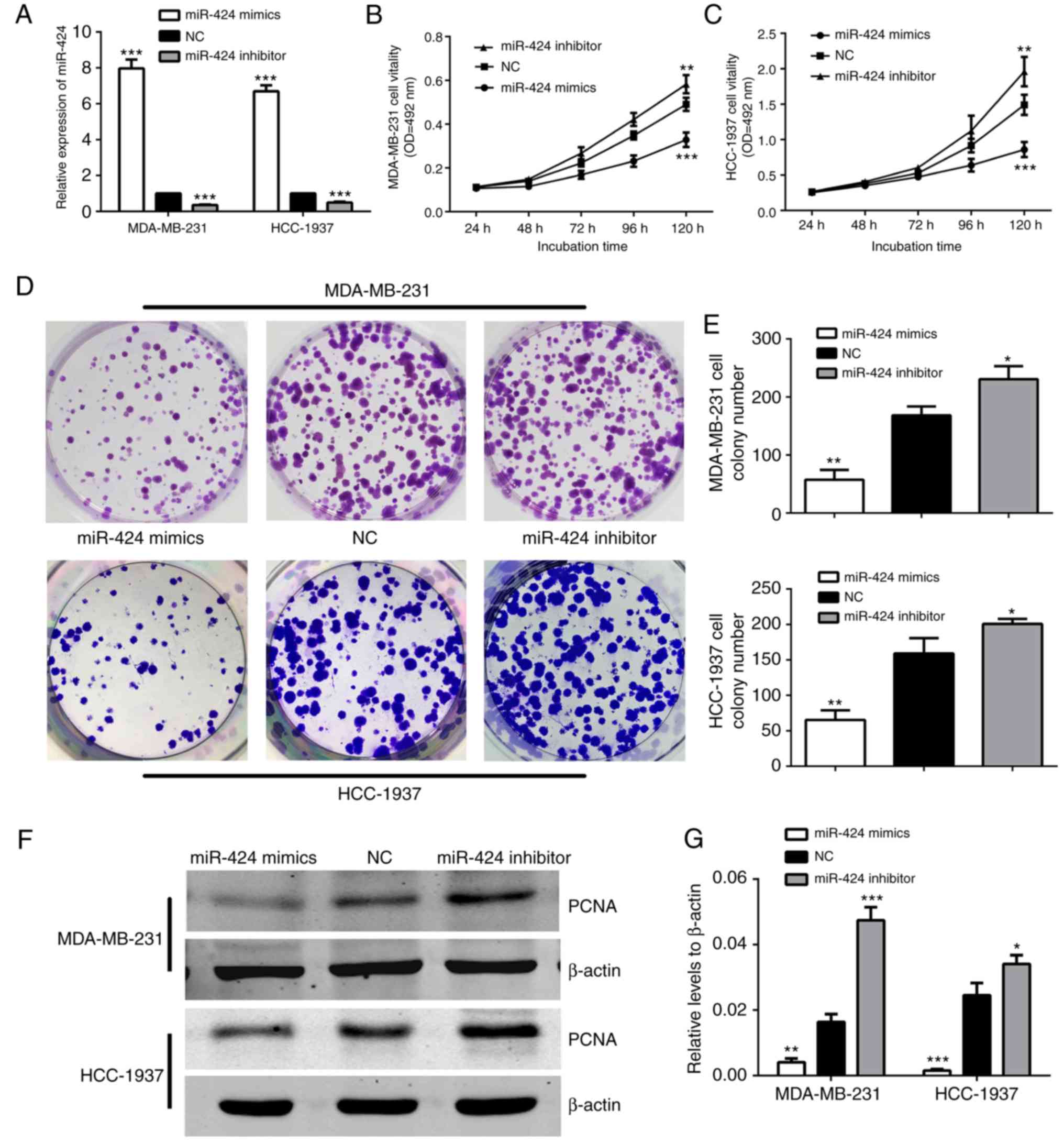 | Figure 2.miR-424 inhibited MDA-MB-231 and
HCC-1937 cell proliferation. (A) The relative expression of miR-424
was detected by reverse transcription-quantitative polymerase chain
reaction in cells following transfection with miR-424 mimics,
inhibitors or NC for 48 h. ***P<0.001. (B) The MTT assay was
used to assess the proliferation level of MDA-MB-231 cells. The
data represent the OD at 492 nm ± SD. **P<0.01, ***P<0.001.
(C) The MTT assay was used to assess the proliferation level of
HCC-1937 cells. The data represent the OD at 492 nm ± SD.
**P<0.01, ***P<0.001. (D) The colony formation assays
demonstrated the colony forming ability of the cells. MDA-MB-231
and HCC-1937 cells transfected with miR-424 mimics, inhibitors or
NC are shown. (E) Cells transfected with miR-424 mimics exhibited
fewer colonies than the NC group. *P<0.05, **P<0.01. (F)
Western blots demonstrated the relative expression of PCNA protein.
(G) The graph shows the mean ± SD of PCNA protein levels relative
to β-actin, which was used as a control. All protein levels were
quantified by measuring the IDV of each protein band. *P<0.05,
**P<0.01, ***P<0.001. miR, microRNA; NC, negative control;
SD, standard deviation; PCNA, proliferating cell nuclear antigen;
IDV, integrated density value; OD, optical density. |
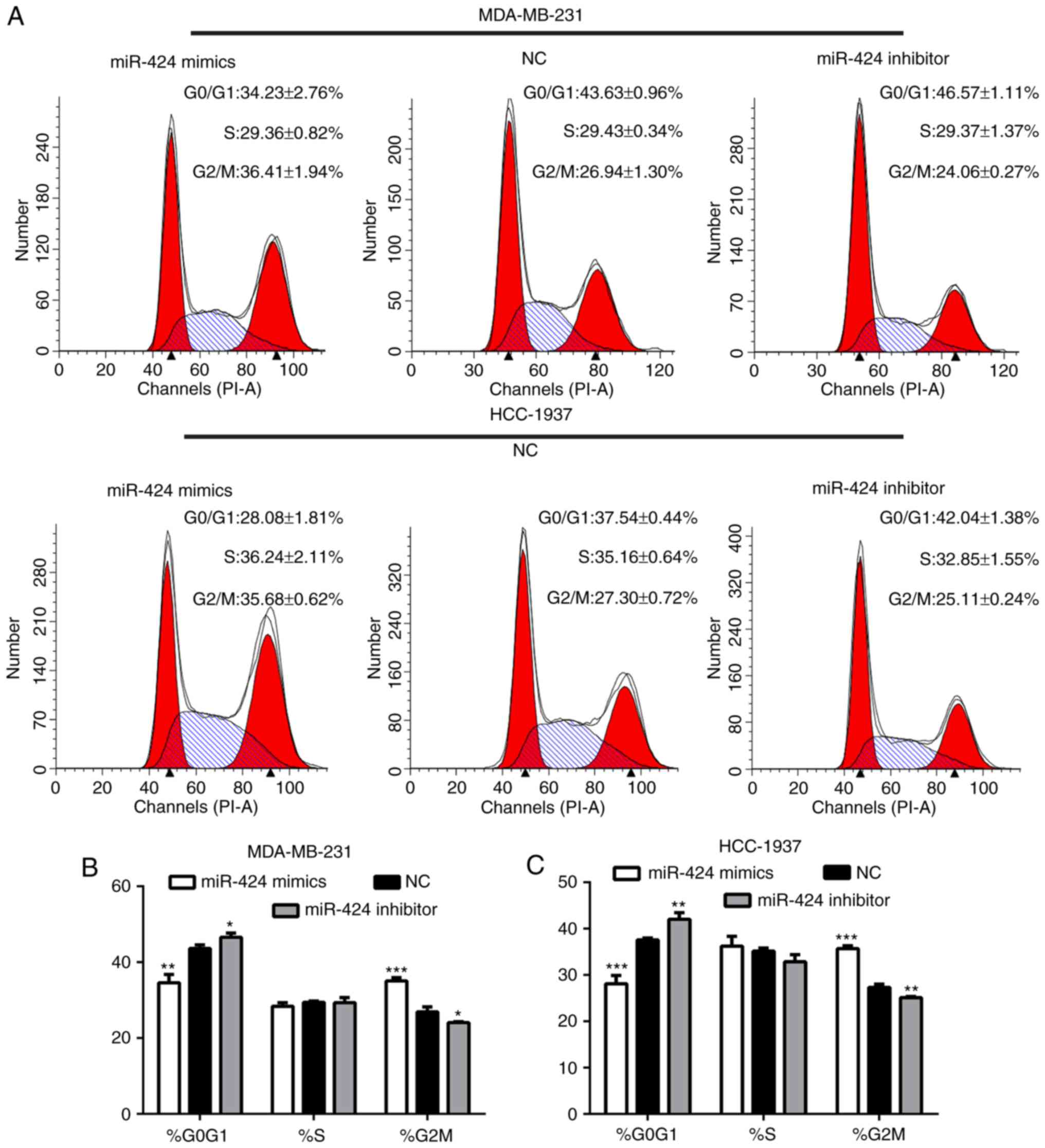
miR-424 regulates the cell cycle of
breast cancer cells
Following transfection for 48 h, the cells were
collected for flow cytometry analyses. This demonstrated that the
proportion of G2/M phase cells in the miR-424 mimic group was
significantly increased compared with that of the inhibitor and NC
groups, and that the proportion of G0/G1 phase cells was decreased
(Fig. 3). This result indicated
that miR-424 regulated the cell cycle by arresting cells in the
G2/M cell phase.
miR-424 suppresses CDK1 expression and
certain key genes in the Hippo and ERK pathways
To further investigate the mechanism of action of
miR-424 in breast cancer, TargetScan (http://www.targetscan.org/) was searched to identify
target genes of miR-424. It suggested that CDK1 may be a potential
target of miR-424. To determine whether miR-424 regulated
endogenous CDK1, miR-424 mimics, inhibitors and NC were transfected
into MDA-MB-231 and HCC-1937 cells, prior to the levels of CDK1
protein being detected by western blot analysis, and mRNA was
monitored by RT-qPCR 48 h after transfection. It revealed that
miR-424 mimics significantly suppressed the expression of
endogenous CDK1 protein and mRNA compared with that of the NC
group. By contrast, the miR-424 inhibitor enhanced the expression
of CDK1 protein and mRNA (Fig.
4).
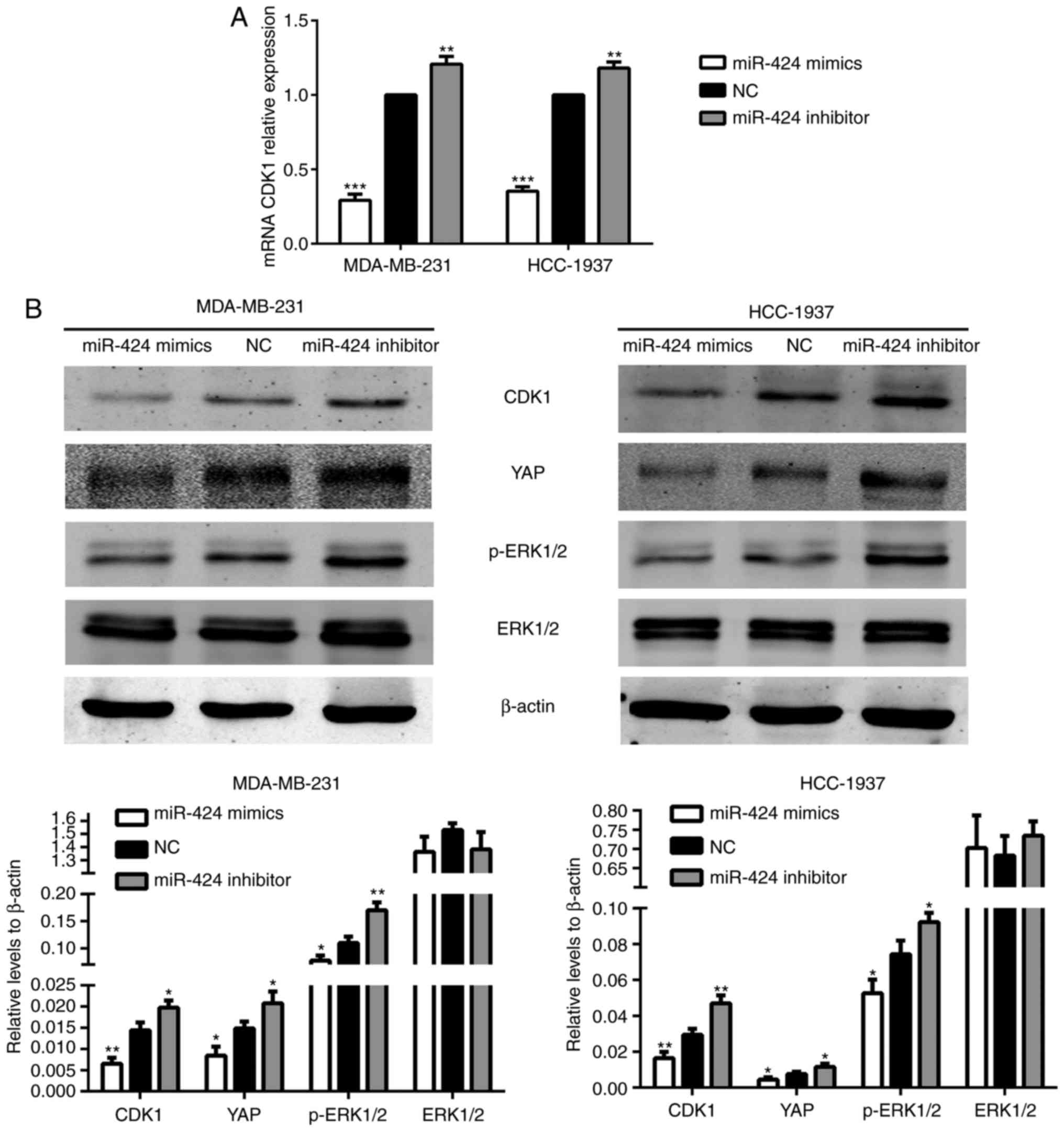 | Figure 4.miR-424 increased the levels of CDK1
and YAP, and the phosphorylation of ERK1/2. (A) The relative
expression of CDK1 mRNA in transfected cells. **P<0.01,
***P<0.001. (B) The relative expression of CDK1, YAP, ERK1/2 and
p-ERK1/2 proteins. *P<0.05, **P<0.01. miR, microRNA; CDK,
cyclin-dependent kinase; YAP, yes-associated protein; ERK,
extracellular signal-regulated kinase; p-ERK, phosphorylated
ERK. |
The Hippo and ERK pathways have been extensively
studied in breast cancer in recent years. Therefore, the present
study attempted to identify an association between these two
pathways and miR-424. In the Hippo pathway, it was revealed that
the protein expression level of YAP was decreased in the miR-424
mimic group and was increased in the miR-424 inhibitor group,
compared with the NC group. In the ERK pathway, the expression of
ERK1/2 did not differ between each group, while the protein
expression level of p-ERK1/2 was decreased in the miR-424 mimic
group (Fig. 4B). We hypothesized
that these results may be due to variations in CDK1, and subsequent
results verified this hypothesis.
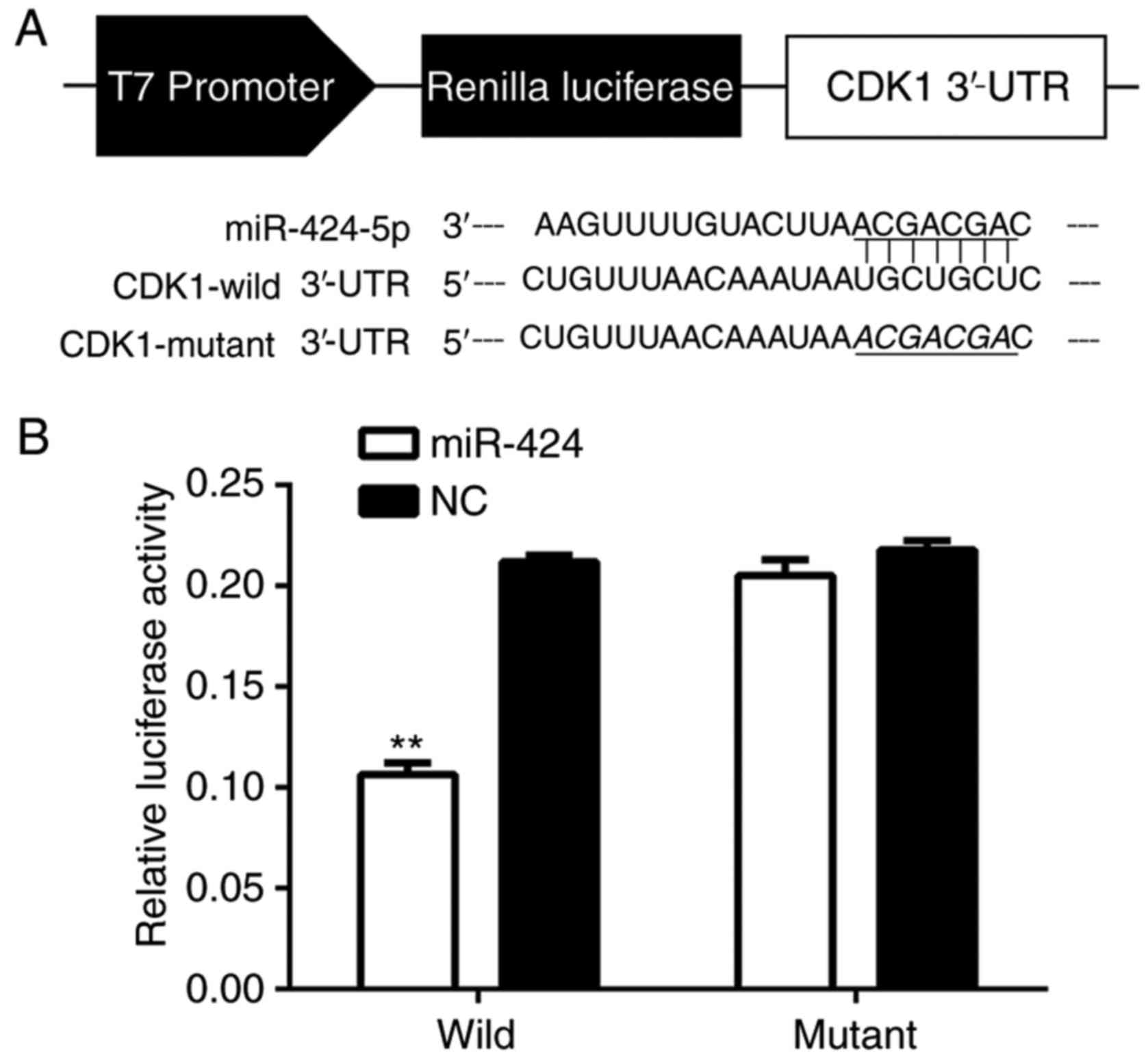
CDK1 is a direct target of
miR-424
To determine if miR-424 binded to CDK1, the miR-424
and CDK1 mRNA sequences were analyzed and seven complementary
nucleotides were identified (Fig.
5A). The luciferase reporter assay was used in the HEK-293T
cell line, and the luciferase activity was suppressed in cells
co-transfected with psi-CHECK-2/CDK1 3′-UTR and miR-424 mimics
compared with NC, indicating that CDK1 was a direct target of
miR-424 (Fig. 5B).
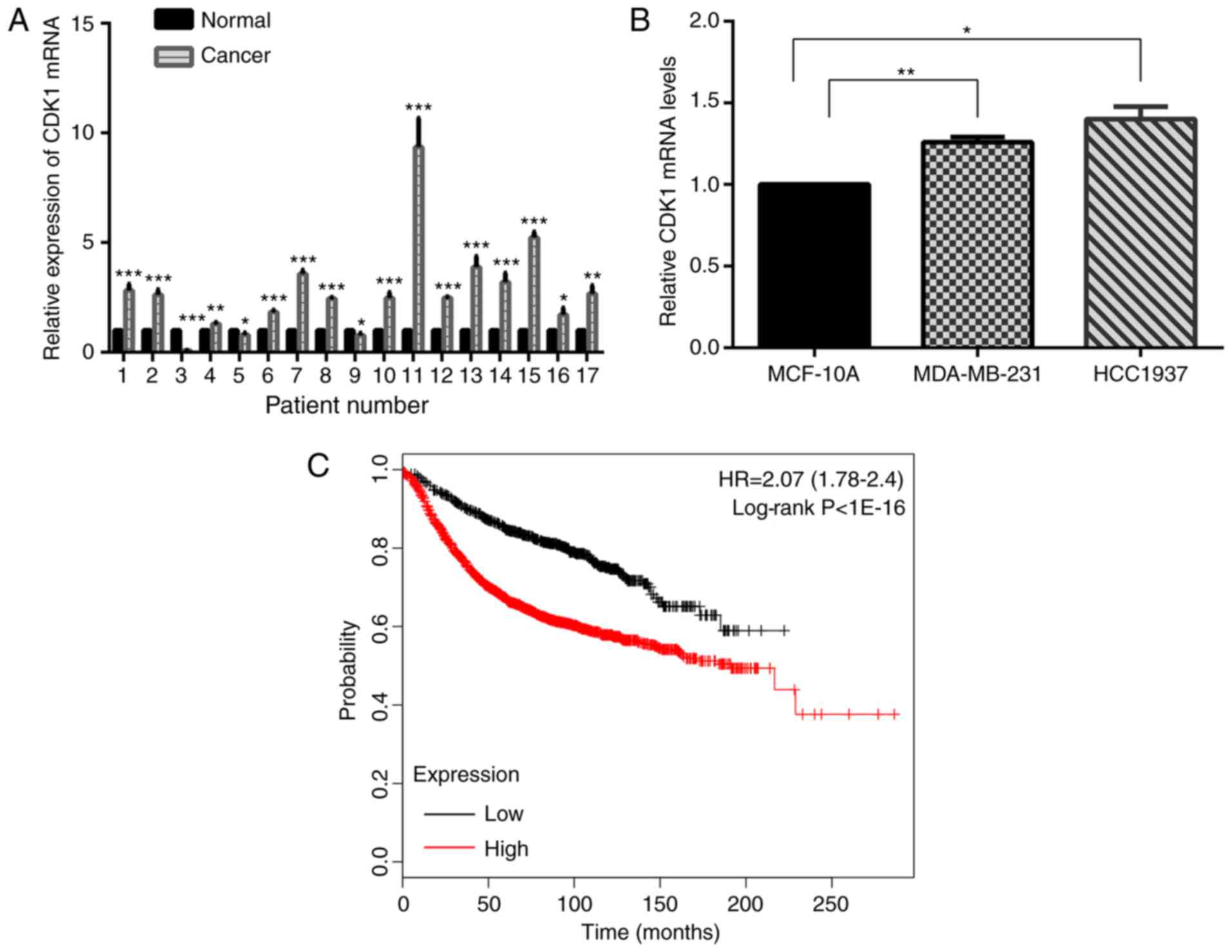
CDK1 promotes cell proliferation and
regulates the cell cycle in breast cancer
The expression level of CDK1 mRNA was tested in 17
pairs of breast cancer and matched normal specimens. It
demonstrated that CDK1 was increased in human breast cancer
specimens compared with normal breast specimens (Fig. 6A). Further study yielded the same
results in MDA-MB-231 and HCC-1937 cells, compared with MCF-10A
cells (Fig. 6B). Kaplan-Meier
survival analyses suggested that CDK1 acted as an oncogene
(Fig. 6C).
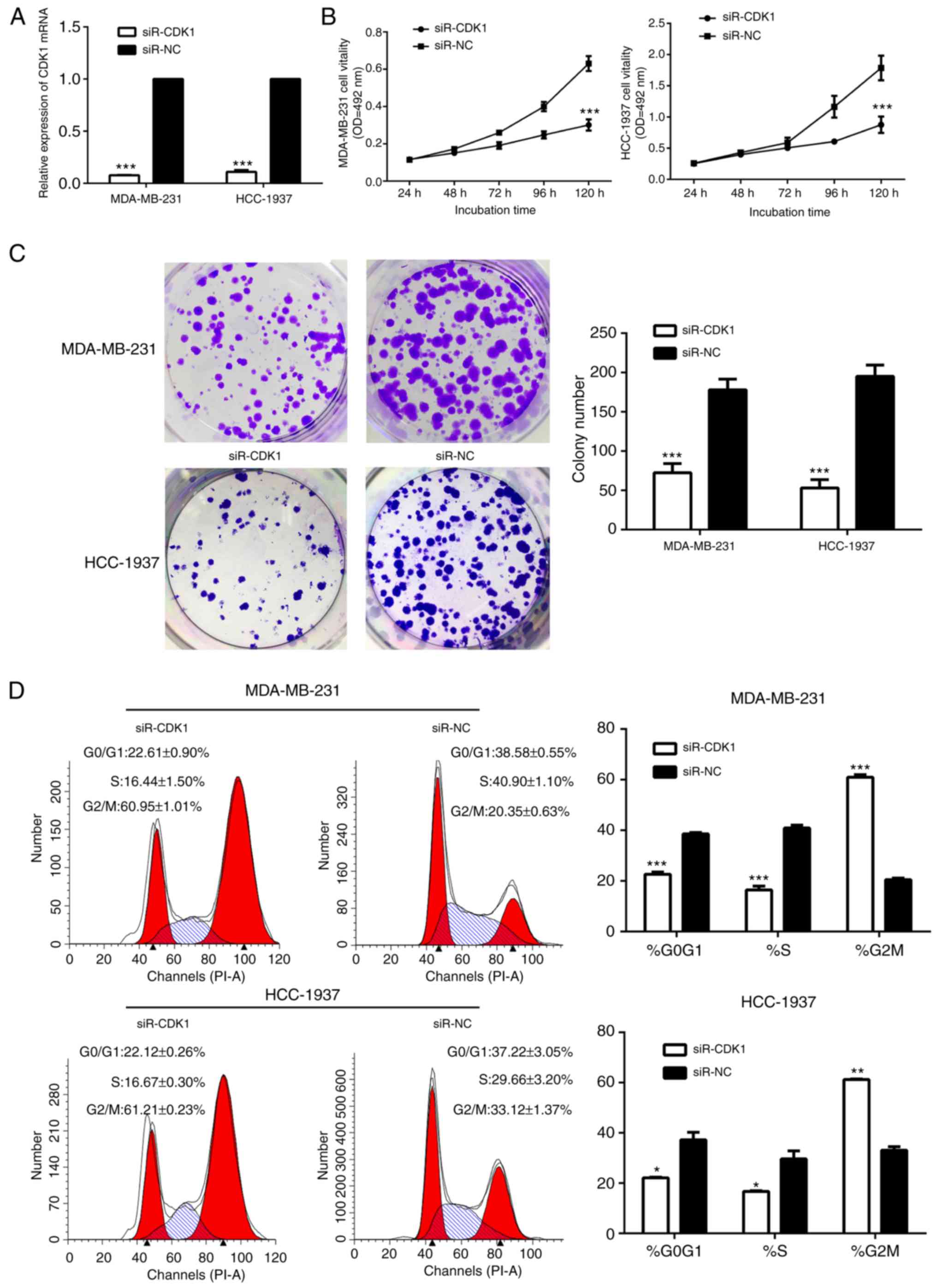
Therefore, the present study investigated the role
of CDK1 in breast cancer. The cells in the siR-CDK1-treated group
exhibited notably decreased CDK1 protein and mRNA expression,
compared with the siR-NC-treated group, suggesting that CDK1 was
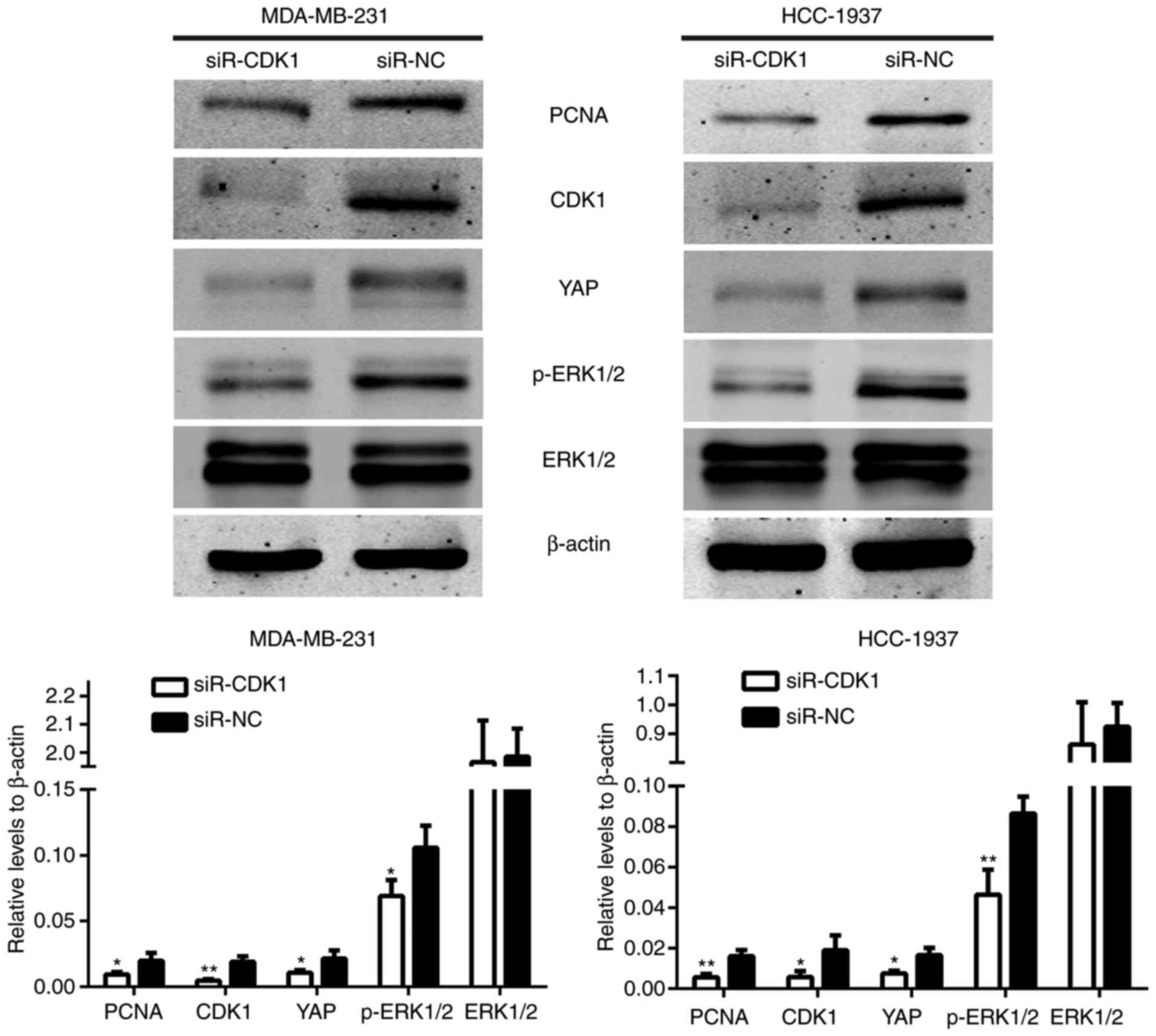
successfully silenced by siR-CDK1 (Figs. 7A and 8). Silencing of CDK1 inhibited cell
proliferation (Fig. 7B and C), and
arrested the cells in the G2/M phase (Fig. 7D). In addition, the levels of
associated proteins were decreased by silencing of CDK1, which
further confirmed the results of the present study (Fig. 8).
As demonstrated in Fig.
4B, the protein expression levels were regulated by miR-424.
The protein expression levels of these genes in the
siR-CDK1-treated group and the siR-NC-treated group were also
assessed. The results demonstrated that the protein expression
levels of YAP and p-ERK1/2 were also induced in the
siR-CDK1-treated group compared with the siR-NC-treated group
(Fig. 8). These data demonstrated
that the effect of miR-424 on regulating the Hippo and ERK pathways
were achieved through regulating CDK1.
Discussion
It has been reported that there are an estimated 1.7
million females expected to be diagnosed with breast cancer by
2020, and there has been a 26% increase from current recorded
levels (11,12). Although the mortality rate in this
decade has decreased, breast cancer remains a major life
threatening disease with 1.3 million newly diagnosed cases each
year among females worldwide (13).
Therefore, it is crucial to investigate and develop more effective
ways to diagnose and treat breast cancer at an early stage.
As a type of potential regulator, miRNAs serve a key
role in tumorigenesis and the progression of breast cancer
(14). They participate in numerous
different biological processes, including cell proliferation,
differentiation, invasion, migration and transcription. miRNAs may
act as oncogenes or anti-oncogenes, such that their dysregulation
is associated with the initiation and progression of breast cancer
(15–17). miRNA-based therapies have already
been used as vital strategies for breast cancer treatment (18). Additionally, it has been reported
that several miRNAs participated in cell tumorigenesis and
metastasis (19). The
overexpression of anti-oncogenic miRNAs could therefore be a novel
therapeutic approach for breast cancer treatment (20–22).
The present study emphasized the role of miR-424 and its possible
mechanism in breast cancer.
In recent years, several studies reported that
miR-424 may act as an anti-oncogene in certain types of cancer.
Therefore, the expression level of miR-424 was measured, and it was
revealed to be decreased in human breast cancer tissues and cell
lines, compared with normal tissues and cells, respectively.
Similar observations have been reported in several different tumor
types, including minimal deviation adenocarcinoma (23), cervical cancer (9) and bladder cancer (8). Although the number of clinical
specimens was not large enough to confirm this conclusion, these
results still have utility. Kaplan-Meier survival analyses
indicated that overexpression of miR-424 resulted in longer overall
survival times. We hypothesized that miR-424 served an
anti-oncogene role in breast cancer. Therefore, miR-424 mimics and
inhibitors were transfected into MDA-MB-231 and HCC-1937 cells to
upregulate or downregulate the expression of miR-424, respectively,
and miR-424 expression was determined by RT-qPCR. Next, MTT and
colony formation assay results demonstrated that high miR-424
expression remarkably repressed the proliferation and colony
formation ability of breast cancer cells. Flow cytometric analysis
indicated that miR-424 significantly arrested cells in the G2/M
phase. Furthermore, the expression levels of PCNA, which are
associated with cell proliferation, were also inhibited by miR-424.
To improve understanding regarding the functions of miR-424, it was
critical to identify its target genes. TargetScan revealed that
CDK1 may be a direct target of miR-424.
CDKs are a family of serine/threonine kinases that
are critical regulatory enzymes for cell cycle transitions
(24). Aberrant activation of CDKs
may enhance tumor proliferation and chromosomal instability;
therefore, they were focuses for the development of anticancer drug
(25). In fact, numerous effective
and selective inhibitors of CDKs have been identified in recent
years, including inhibitors of CDK4/6 that have recently been
approved by the FDA (26–29). CDK1 is the only CDK that is able to
initiate mitosis. Its association with cyclin B is the primary
impetus for entry into mitosis. CDK1 is required for mammalian cell
proliferation, as confirmed by mouse knockout experiments (30). Additionally, there have been related
reports regarding cell cycle and CDKs in recent years. CDK8 is able
to promote cell proliferation in breast cancer, and is directly
regulated by miRNA-26b and miRNA-107 (31–33).
As an upstream component of CDK1, cyclin-dependent kinase
regulatory subunit 2 (CKS2) is involved in the progression of
thyroid papillary cancer cells, while CDK1 and cyclin B are
regulated by the miR-7-CKS2 axis (34). Western blot analysis demonstrated
that upregulation of miR-424 significantly inhibited CDK1 protein
expression, while RT-qPCR confirmed that miR-424-overexpression
significantly suppressed the level of CDK1 mRNA. Next, miR-424
directly binds to CDK1 as confirmed by luciferase reporter assays.
Subsequently, it was revealed that the effect of CDK1 in the
survival rate and relative expression levels in cancer tissues or
cells was opposite to the effects of miR-424 in human breast
cancer. Following decreasing the CDK1 levels by transfection with
siR-CDK1, it was revealed that the effects on cell proliferation
and the cell cycle were similar to those identified following
upregulation of miR-424. Therefore, we hypothesized that the effect
of miR-424 on these biological processes was exerted through
targeting CDK1.
Furthermore, the present study measured the
expression levels of certain Hippo and ERK pathway proteins in
cells with miR-424 upregulation or downregulation. The expression
alterations of miR-424 in MDA-MB-231 and HCC-1937 cells changed the
protein expression levels of YAP and p-ERK1/2, but not those of
ERK1/2. Similar results were also observed in CDK1-silenced cells.
YAP is a core downstream molecule of the Hippo signaling pathway in
mammals, which acts as an oncogene in the initiation and
development of breast cancer. A previous study demonstrated that
numerous proteins can promote the progression of breast cancer
through the Hippo signaling pathway (35). The results of the present study
suggested that miR-424 affected the Hippo pathway through targeting
CDK1 and then regulating the expression level of YAP. The ERK
pathway is a classic mitogen-activated protein kinase (MAPK)
signaling cascade that regulates cell proliferation, malignant
transformation, autophagy and differentiation, with core members,
including Ras, Raf, MEK1/2 and ERK1/2 (36,37).
ERK1/2 are serine-threonine kinases that are activated through
phosphorylation by MEK1/2 (38).
They served important roles in malignant cell transformation. Based
on these results, we hypothesized that the miR-424-CDK1 axis
altered the activation levels of ERK1/2 rather than their protein
expression levels.
In conclusion, the present study demonstrated that
miR-424 acted as a tumor suppressor in breast cancer cells.
miR-424-overexpression suppressed cell growth and disrupted the
cell cycle, likely by targeting CDK1 and further regulating the
Hippo and ERK pathways; therefore, miR-424 may be a potential novel
therapeutic target for human breast cancer.
Acknowledgements
The authors would like to thank all the teachers at
the Central Laboratory of Shanghai Tenth People's Hospital
(Shanghai, China) for providing support.
Funding
The present study was supported by the National
Natural Sciences Foundation of China (grant no. 81272240), the
Shanghai Municipal Health Bureau of Shanghai, China (grant no.
201640097) and the Shanghai Municipal Science and Technology
Commission of Shanghai, China (grant no. 17411967200).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DX and LF conceived and designed the study. DX, HS
and TW performed the experiments. HX, BZ and CW analyzed the data.
DX, KH and DL wrote the manuscript. KH and DL were also involved in
the conception of this study. JH, CJ and YD acquired the reagents,
the materials and the analysis tools. All authors read and approved
the manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
All procedures involving human participants in the
present study were performed in accordance with the Ethical
Standards of the Institutional and/or National Research Committee,
and with the 1964 Declaration of Helsinki and its later amendments
or comparable ethical standards.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bombonati A and Sgroi DC: The molecular
pathology of breast cancer progression. J PATHOL. 223:307–31. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yates LA, Norbury CJ and Gilbert RJ: The
long and short of microRNA. Cell. 153:516–519. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Esteller M: Non-coding RNAs in human
disease. Nat Rev Genet. 12:861–874. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xiao M, Li J, Li W, Wang Y, Wu F, Xi Y,
Zhang L, Ding C, Luo H, Li Y, et al: MicroRNAs activate gene
transcription epigenetically as an enhancer trigger. RNA Biol.
14:1326–1334. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Friedman RC, Farh KK, Burge CB and Bartel
DP: Most mammalian mRNAs are conserved targets of microRNAs. Genome
Res. 19:92–105. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dvinge H, Git A, Graf S, Salmon-Divon M,
Curtis C, Sottoriva A, Zhao Y, Hirst M, Armisen J, Miska EA, et al:
The shaping and functional consequences of the microRNA landscape
in breast cancer. Nature. 497:378–382. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cekaite L, Eide PW, Lind GE, Skotheim RI
and Lothe RA: MicroRNAs as growth regulators, their function and
biomarker status in colorectal cancer. Oncotarget. 7:6476–6505.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu CT, Lin WY, Chang YH, Lin PY, Chen WC
and Chen MF: DNMT1-dependent suppression of microRNA424 regulates
tumor progression in human bladder cancer. Oncotarget.
6:24119–24131. 2015.PubMed/NCBI
|
|
9
|
Zhou Y, An Q, Guo RX, Qiao YH, Li LX,
Zhang XY and Zhao XL: miR424-5p functions as an anti-oncogene in
cesrvical cancer cell growth by targeting KDM5B via the Notch
signaling pathway. Life Sci. 171:9–15. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang J, Kong X, Li J, Luo Q, Li X, Shen
L, Chen L and Fang L: miR-96 promotes tumor proliferation and
invasion by targeting RECK in breast cancer. Oncol Rep.
31:1357–1363. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gottesman MM: Mechanisms of cancer drug
resistance. Annu Rev Med. 53:615–627. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rahmani-Nezhad S, Safavi M, Pordeli M,
Ardestani SK, Khosravani L, Pourshojaei Y, Mahdavi M, Emami S,
Foroumadi A and Shafiee A: Synthesis, in vitro cytotoxicity and
apoptosis inducing study of 2-aryl-3-nitro-2H-chromene derivatives
as potent anti-breast cancer agents. Eur J Med Chem. 86:562–569.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tirona MT, Sehgal R and Ballester O:
Prevention of breast cancer (part I): Epidemiology, risk factors,
and risk assessment tools. Cancer Invest. 28:743–750. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jahagirdar D, Purohit S, Jain A and Sharma
NK: Export of microRNAs: A bridge between breast carcinoma and
their neighboring cells. Front Oncol. 6:1472016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Luo Q, Li X, Li J, Kong X, Zhang J, Chen
L, Huang Y and Fang L: MiR-15a is underexpressed and inhibits the
cell cycle by targeting CCNE1 in breast cancer. Int J Oncol.
43:1212–128. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li J, Kong X, Zhang J, Luo Q, Li X and
Fang L: Correction: MiRNA-26b inhibits proliferation by targeting
PTGS2 in breast cancer. Cancer Cell Int. 13:172013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Braza-Boils A, Mari-Alexandre J, Gilabert
J, Sanchez-Izquierdo D, Espana F, Estelles A and Gilabert-Estellés
J: MicroRNA expression profile in endometriosis: Its relation to
angiogenesis and fibrinolytic factors. Hum Reprod. 29:978–988.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gambari R, Brognara E, Spandidos DA and
Fabbri E: Targeting oncomiRNAs and mimicking tumor suppressor
miRNAs: New trends in the development of miRNA therapeutic
strategies in oncology (Review). Int J Oncol. 49:5–32. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jahagirdar D, Purohit S, Jain A and Sharma
NK: Export of microRNAs: A bridge between breast carcinoma and
their neighboring cells. Front Oncol. 6:1472016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bertoli G, Cava C and Castiglioni I: The
potential of miRNAs for diagnosis, treatment and monitoring of
breast cancer. Scand J Clin Lab Invest Suppl. 245:S34–S39. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bertoli G, Cava C and Castiglioni I:
MicroRNAs: New biomarkers for deiagnosis, prognosis, therapy
prediction and therapeutic tools for breast cancer. Theranostics.
5:1122–1143. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cava C, Bertoli G and Castiglioni I:
Integrating genetics and epigenetics in breast cancer: Biological
insights, experimental, computational methods and therapeutic
potential. BMC Syst Biol. 9:622015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee H, Kim KR, Cho NH, Hong SR, Jeong H,
Kwon SY, Park KH, An HJ, Kim TH, Kim I, et al: MicroRNA expression
profiling and Notch1 and Notch2 expression in minimal deviation
adenocarcinoma of uterine cervix. World J Surg Oncol. 12:3342014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nurse P, Masui Y and Hartwell L:
Understanding the cell cycle. Nat Med. 4:1103–1116. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–1566.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lapenna S and Giordano A: Cell cycle
kinases as therapeutic targets for cancer. Nat Rev Drug Discov.
8:547–566. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Asghar U, Witkiewicz AK, Turner NC and
Knudsen ES: The history and future of targeting cyclin-dependent
kinases in cancer therapy. Nat Rev Drug Discov. 14:130–146. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Finn RS, Crown JP, Lang I, Boer K,
Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, et
al: The cyclin-dependent kinase 4/6 inhibitor palbociclib in
combination with letrozole vs letrozole alone as first-line
treatment of oestrogen receptor-positive, HER2-negative, advanced
breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study.
Lancet Oncol. 16:25–35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sherr CJ, Beach D and Shapiro GI:
Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov.
6:353–367. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Santamaria D, Barriere C, Cerqueira A,
Hunt S, Tardy C, Newton K, Cáceres JF, Dubus P, Malumbres M and
Barbacid M: Cdk1 is sufficient to drive the mammalian cell cycle.
Nature. 448:811–815. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li J, Li X, Kong X, Luo Q, Zhang J and
Fang L: MiRNA-26b inhibits cellular proliferation by targeting CDK8
in breast cancer. Int J Clin Exp Med. 7:558–565. 2014.PubMed/NCBI
|
|
32
|
Li XY, Luo QF, Wei CK, Li DF, Li J and
Fang L: MiRNA-107 inhibits proliferation and migration by targeting
CDK8 in breast cancer. Int J Clin Exp Med. 7:32–40. 2014.PubMed/NCBI
|
|
33
|
Li XY, Luo QF, Wei CK, Li DF and Fang L:
siRNA-mediated silencing of CDK8 inhibits proliferation and growth
in breast cancer cells. Int J Clin Exp Pathol. 7:92–100.
2013.PubMed/NCBI
|
|
34
|
Hua K, Jin J, Zhang H, Zhao B, Wu C, Xu H
and Fang L: MicroRNA-7 inhibits proliferation, migration and
invasion of thyroid papillary cancer cells via targeting CKS2. Int
J Oncol. 49:1531–1540. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shi P, Feng J and Chen C: Hippo pathway in
mammary gland development and breast cancer. Acta Biochim Biophys
Sin. 47:53–59. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cargnello M and Roux PP: Activation and
function of the MAPKs and their substrates, the MAPK-activated
protein kinases. Microbiol Mol Biol Rev. 75:50–83. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chang F, Steelman LS, Lee JT, Shelton JG,
Navolanic PM, Blalock WL, Franklin RA and McCubrey JA: Signal
transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine
receptors to transcription factors: Potential targeting for
therapeutic intervention. Leukemia. 17:1263–1293. 2003. View Article : Google Scholar : PubMed/NCBI
|