Introduction
Liver cancer remains an important global public
health issue accounting for 9.1% of all cancer-related deaths
worldwide (1) due to its aggressive
nature and extremely poor survival rate (2). Primary liver cancer is rare during
childhood, representing 1% of all pediatric neoplasms (3,4). More
than 80% of all liver cancer in infant and children can be
identified as hepatoblastoma, which occurs at an annual incidence
of 0.05–0.15 patients per 100,000 individuals (3,5). The
vast majority of primary liver cancer in adults is hepatocellular
carcinoma (HCC), accounting for more than 85% of all primary liver
cancer cases (6). Although
considerable effort has been made in recent decades to improve the
diagnosis and treatment of HCC, it remains the second leading cause
of cancer-associated mortality worldwide (7). One of the most promising developments
in liver cancer treatment has been targeted delivery of cytotoxic
chemotherapy agents directly into tumors. Selective injection of
embolizing agents, in combination with doxorubicin, into the
arteries feeding tumors, or transarterial chemoembolization (TACE),
has been demonstrated to provide a survival benefit in patients
with unresectable liver cancer and has now become the standard
treatment for patients with intermediate stage HCC (8). Unfortunately, advanced liver cancer
often has a poor response to TACE and prognosis due to acquired
chemoresistance and tumor recurrence. Therefore, understanding the
precise molecular mechanisms underlying liver cancer
chemoresistance could potentially identify novel therapeutic
targets and improve the treatment of liver cancer.
Sirtuin (SIRT) proteins are a family of
evolutionarily conserved NAD+-dependent deacetylases. In
mammals, seven SIRT proteins have been identified, which share
NAD+ binding and catalytic domains, but target different
substrates (9). SIRT proteins
modulate numerous biological processes, including metabolism
(10,11), cell survival (12–14),
differentiation (15), DNA repair
(16) and cancer development
(17). These effects are primarily
achieved by deacetylating lysine residues on histones,
transcription factors or coactivators. At present, sterol
regulatory element-binding protein-1c (18), forkhead box O3 (FOXO3) (19), p53 (20) and p65 (21) are well-established SIRT targets.
SIRT1 deacetylates p53 and restrains DNA damage-induced p53
acetylation, thereby suppressing cell cycle arrest and apoptosis
(22). In addition, previous
reports indicate that multiple SIRT family members control
FOXO3-dependent apoptosis through deacetylation of FOXO3 (13,14,19,23).
Previous studies have also demonstrated that SIRT1 and SIRT7
proteins are involved in cancer transformation and clinical outcome
through both genetic and epigenetic alterations (15,24).
SIRT6, one of the SIRT proteins, plays critical
roles in controlling metabolism, genomic stability, inflammation,
aging and cancer progression (25–28).
SIRT6-deficient animals present with early lethality due to
profound abnormalities, including hypoglycemia and premature aging
(27). Conditional disruption of
SIRT6 in hepatocytes leads to increased glycolysis, triglyceride
synthesis, reduced beta oxidation and ultimately, fatty liver
formation (26). SIRT6 is reported
to be a tumor suppressor through both deacetylation-dependent and
-independent activity in multiple types of cancer, including
pancreatic, breast, colorectal and lung cancer (29–31).
However, the role and mechanistic function of SIRT6 in liver cancer
remain largely unexplored. Marquardt et al (17) reported that SIRT6 mRNA is
downregulated in HCC, but others observed that SIRT6 protein levels
in HCC cell lines and HCC patient tissues are upregulated (32). A recent study demonstrated that
SIRT6 was upregulated in patients with HCC and it serves as an
anti-apoptotic factor by suppressing Bax (33), suggesting that SIRT6 may play a role
in chemotherapy-induced cell death.
The aim of the present study was to investigate the
role of SIRT6 in doxorubicin-induced cell death in liver cancer
cell lines. It was identified that in response to doxorubicin,
SIRT6 was significantly downregulated. Restorative expression of
SIRT6, but not enzyme-inactivated SIRT6 mutant, abolished
doxorubicin-induced cell death. It was also revealed that
transcriptional factor FOXO3 serves as a target of SIRT6 in this
event. In response to doxorubicin treatment, FOXO3 was rapidly
activated and translocated into the nucleus, binding to its target
genes Bim and p27, which further induced cell death. Overexpression
of SIRT6 blocked nuclear translocation of FOXO3 and apoptosis. In
the absence of FOXO3, overexpression of SIRT6 no longer prevented
doxorubicin-induced cell death. The present findings present a
novel mechanism that controls FOXO3 activation and revealed that
SIRT6 is a pivotal regulatory factor in determining liver cancer
chemosensitivity. Therapeutic strategies that inhibit SIRT6 or
activate FOXO3 may offer novel options for the treatment of liver
cancer.
Materials and methods
Cell culture, plasmids and
transfection
HepG2, Huh7 and HeLa cells were purchased from the
American Type Culture Collection (ATCC; Manassas, VA, USA) and
routinely preserved in Dulbecco's modified Eagle's medium (DMEM;
Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (FBS; Invitrogen; Thermo
Fisher Scientific, Inc.), 50 U/ml penicillin and 50 mg/ml
streptomycin. Transfection of cells was performed in serum-free
medium (Opti-MEM, Invitrogen; Thermo Fisher Scientific, Inc.) using
X-tremeGENE™ HP DNA Transfection reagent (Roche Diagnostics,
Indianapolis, IN, USA) according to the manufacturer's protocol.
pECE-HA-FOXO3, SIRT6 Flag and pCDNA3.1 SIRT6_H133Y plasmids were
respectively provided by M. Greenberg, Eric Verdin and Katrin Chua
via Addgene, Inc. (Cambridge, MA, USA). Short hairpin (sh)RNA
targeting FOXO3 (MISSION shRNA plasmid DNA FOXO3; TRCN0000010335,
TRCN0000235487) was purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany).
Antibodies and chemicals
Anti-human influenza hemagglutinin (HA) antibody
(cat. no. ab9110) and anti-SIRT4 (cat. no. ab124521) were purchased
from Abcam (Cambridge, MA, USA). Anti-FOXO3 (cat. no. 75D8),
anti-acetylated-lysine (cat. no. 9441), anti-SIRT1 (cat. no. D1D7),
anti-SIRT6 (cat. no. D8D12), anti-ubiquitin (cat. no. P4D1),
anti-cleaved caspase-3 (cat. no. 9661), anti-Bim (cat. no. C34C5),
anti-p27 (cat. no. D69C12), anti-p-FOXO3 S253 (cat. no. 9466) and
anti-poly (ADP ribose) polymerase (PARP; cat. no. 9542) were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Anti-GAPDH (FL-335) was purchased from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA). Anti-Flag (M2) antibody, cycloheximide
(CHX) and doxorubicin hydrochloride were purchased from
Sigma-Aldrich (Merck KGaA).
Immunofluorescence
For indirect immunofluorescence, cells grown on
coverslips were fixed with 4% paraformaldehyde at room temperature
for 5 min and 0.2% Triton X-100 was used for cell permeation. The
coverslips were inverted and 40 µl droplets of blocking buffer (4%
goat serum) was incubated at room temperature for 45 min to prevent
non-specific binding. Subsequently, cells were incubated with
rabbit anti-HA (dilution 1:100) or rabbit anti-SIRT6 (dilution
1:100) and mouse anti-Flag (dilution 1:1,000) for 1 h at room
temperature. Coverslips were washed with phosphate-buffered saline
(PBS), followed by incubation for 1 h at room temperature in the
dark with Alexa Fluor 488-conjugated goat anti-rabbit IgG (dilution
1:5,000; cat. no. A27.34; Molecular Probes; Thermo Fisher
Scientific, Inc.). Additionally, DAPI was added for 5 min at room
temperature to stain nuclear DNA. Images were acquired using a
Nikon Eclipse Ti microscope (Nikon Corp., Tokyo, Japan).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated cells using the TRIzol
reagent (Thermo Fisher Scientific, Inc.) followed by cDNA
generation with an RNA reverse transcription kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Subsequently, a CFX96
real-time system (Bio-Rad Laboratories, Hercules, CA, USA) was used
to perform qPCR. Reaction volumes of 25 µl were used, containing
12.5 µl SYBR Green PCR Master Mix (Applied Biosystems; Thermo
Fisher Scientific, Inc.), 10.5 µl 1 µmol/l primer stock and 2 µl
cDNA (1:10 diluted). Primer sequences are presented in Table I.
 | Table I.Primer sequences used for reverse
transcription-quantitative polymerase chain reaction and ChIP
assays. |
Table I.
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction and ChIP
assays.
| Primer | Sequence
(5′-3′) |
|---|
| SIRT1 forward |
TAGCCTTGTCAGATAAGGAAGGA |
| SIRT1 reverse |
ACAGCTTCACAGTCAACTTTGT |
| SIRT4 forward |
GCTTTGCGTTGACTTTCAGGT |
| SIRT4 reverse |
CCAATGGAGGCTTTCGAGCA |
| SIRT6 forward |
CCCACGGAGTCTGGACCAT |
| SIRT6 reverse |
CTCTGCCAGTTTGTCCCTG |
| GAPDH forward |
GAAGGTGAAGGTCGGAGTC |
| GAPDH reverse |
GAAGATGGTGATGGGATTTC |
| p27 ChIP
forward |
TGCGCGCTCCTAGAGCTC |
| p27 ChIP
reverse |
TTTCTCCCGGGTCTGCAC |
| Bim ChIP
forward |
AGGCTAGGGTACACTTCG |
| Bim ChIP
reverse |
AGGCTCGGACAGGTAAAG |
Western blot analysis
Total cell lysates prepared from cells were used for
detection. Briefly, cells were washed twice with ice cold PBS and
then cell lysis were made by using RIPA buffer (50 mM Tris, pH 7.5,
150 mM sodium chloride, 1% NP-40, 0.2% SDS, 0.5% sodium
deoxycholate, 0.1 mM EDTA and 1% protease and phosphatase
inhibitors) (Sigma-Aldrich; Merck KGaA). After centrifugation at
16,000 × g for 15 min, supernatants were collected. Cell lysates
(25 µg) were separated by 10% SDS-PAGE and transferred to
polyvinylidene difluoride membranes (Immobilon-P membranes; EMD
Millipore, Billerica, MA, USA). Membranes were blocked with
blocking buffer (5% skim milk, 0.1% Tween-20 in PBS) for 1 h at
room temperature. Following incubation with primary antibodies
(1:1,000) overnight at 4°C, membranes were then incubated with
horseradish peroxidase-conjugated secondary antibodies (cat. nos.
31460 and 31430). Thermo Fisher Scientific, Inc.). Signals were
detected using the ECL Plus Western Blotting Detection system (GE
Healthcare, Chicago, IL, USA) with the ODYSSEY Fc, Dual-Mode
Imaging system (LI-COR Biosciences, Lincoln, NE, USA).
Chromatin immunoprecipitation (ChIP)
assay
ChIP assays were performed as previously described
(34). Cells were fixed, washed and
harvested followed by shearing of genomic DNA using sonication.
Sonicated DNA (20 µl) was purified and used as input DNA control.
Sheared DNA was cleared and chromatin-bound DNAs were
immunoprecipitated using FOXO3a and 50 µl anti-HA magnetic beads
(Dynabeads M-280 Sheep anti-rabbit IgG; Invitrogen; Thermo Fisher
Scientific, Inc.). The eluted DNA from the beads was precipitated
and analyzed by PCR using multiple primer sets, as listed in
Table I.
Immunoprecipitation
HeLa cells were seeded at 4×106
cells/10-cm plate and transiently transfected with 4 µg Flag-SIRT6.
At 1 day after transfection, cells were lysed with RIPA buffer as
described above. Cell extracts (400 µg) were subjected to
immunoprecipitation with 50 µl anti-Flag M2 magnetic beads
(Sigma-Aldrich; Merck KGaA) in each experiment. Western blotting
was used to analyze immune complexes.
Caspase-3/-7 activity and TUNEL
assay
Caspase-3/-7 activity was measured using the
Caspase-Glo 3/7 Assay System (Promega Corp., Madison, WI, USA),
according to the manufacturer's protocol. For TUNEL assays, at room
temperature, cells were fixed with 4% paraformaldehyde for 10 min
and then rinsed with PBS. TUNEL staining was performed using the
DeadEND Fluorometric TUNEL system (Promega Corp.) according to the
manufacturer's protocol. Five or more randomly selected fields were
examined for quantification of TUNEL staining.
Statistical analysis
Three biological replicates and two technical
repetitions were performed for each assay unless indicated
otherwise. Representative results are presented. Differences
between two groups were calculated using a two-tailed unpaired
Student's t-test. Statistical significance among multiple groups
was calculated using one-way ANOVA followed by Turkey's test.
Variance between groups met the assumptions or the appropriate
test. Unless otherwise stated, a P-value of <0.05 was considered
statistically significant.
Results
Downregulation of SIRT6 following
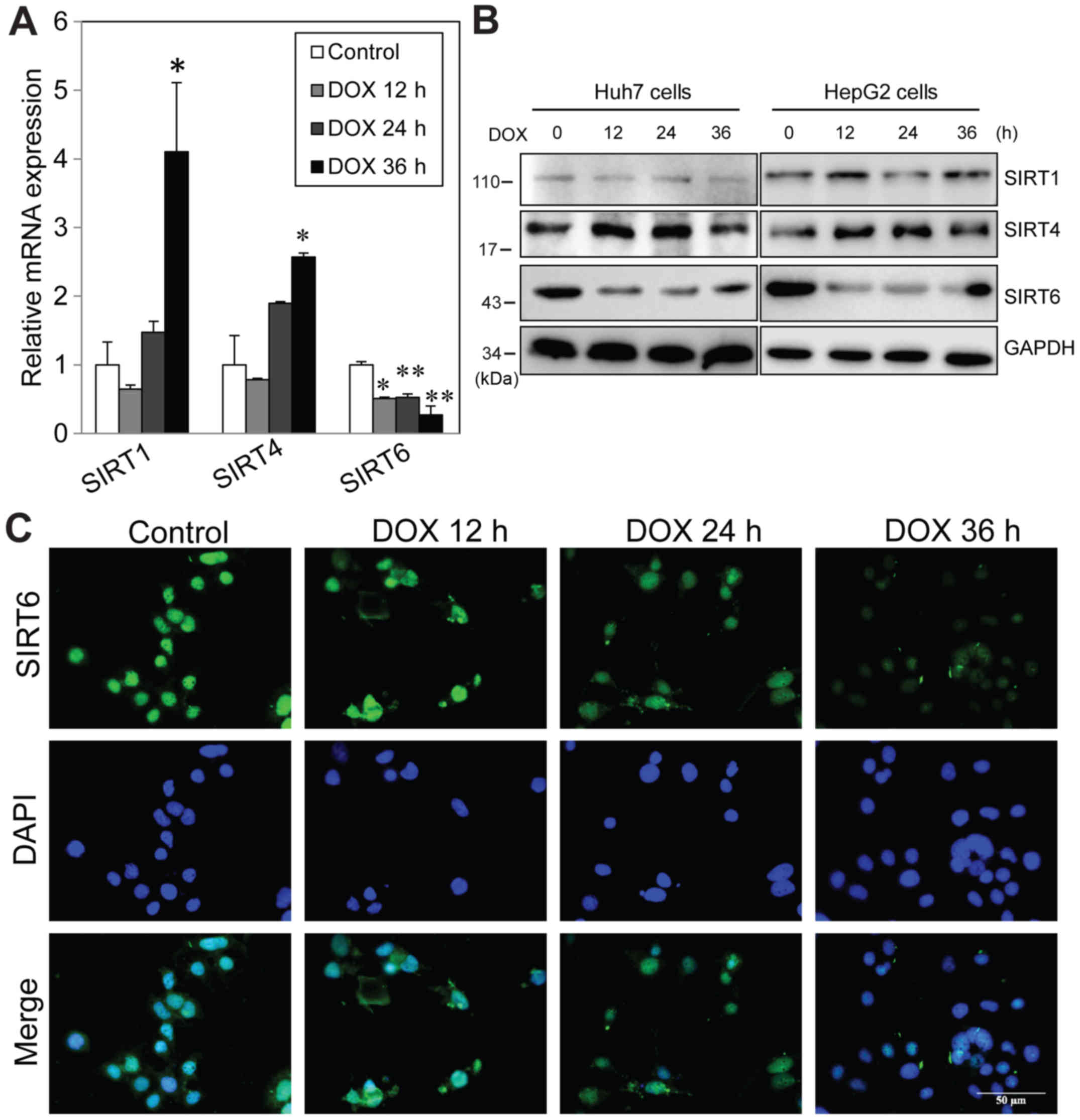
doxorubicin treatment of liver cancer cells
In order to examine the effects of doxorubicin
treatment on SIRT family member proteins, HepG2 cells were treated
with 1 µM doxorubicin, and then mRNA and protein expression was
measured at various time-points. Doxorubicin treatment resulted in
significant increases in SIRT1 and SIRT4 mRNA expression and
downregulation of SIRT6 mRNA level by 36 h (Fig. 1A). Western blot analysis indicated
that there was no change in SIRT1 protein level following treatment
and SIRT level was increased from 12 h post-treatment. Consistent
with the mRNA results, SIRT6 protein was also significantly reduced
12 h after treatment and this effect lasted at least 36 h (Fig. 1B). Similarly, it was also observed
that doxorubicin induced a significant decrease in SIRT6 protein
expression in Huh7 cells (Fig. 1B).
Immunofluorescence was performed to evaluate the cellular
localization of SIRT6 in untreated and treated cells. The results
indicated that SIRT6 protein localized to both the cytosol and
nucleus in untreated cells. Consistent with the western blotting
results, it was identified that doxorubicin decreased global SIRT6
intensity from 24 h post-treatment (Fig. 1C). These data indicated that
doxorubicin treatment downregulates SIRT6 mRNA and protein levels
in liver cancer cells. The inconsistencies between SIRT1 mRNA and
protein levels also suggested that there may be
post-transcriptional regulation following doxorubicin
treatment.
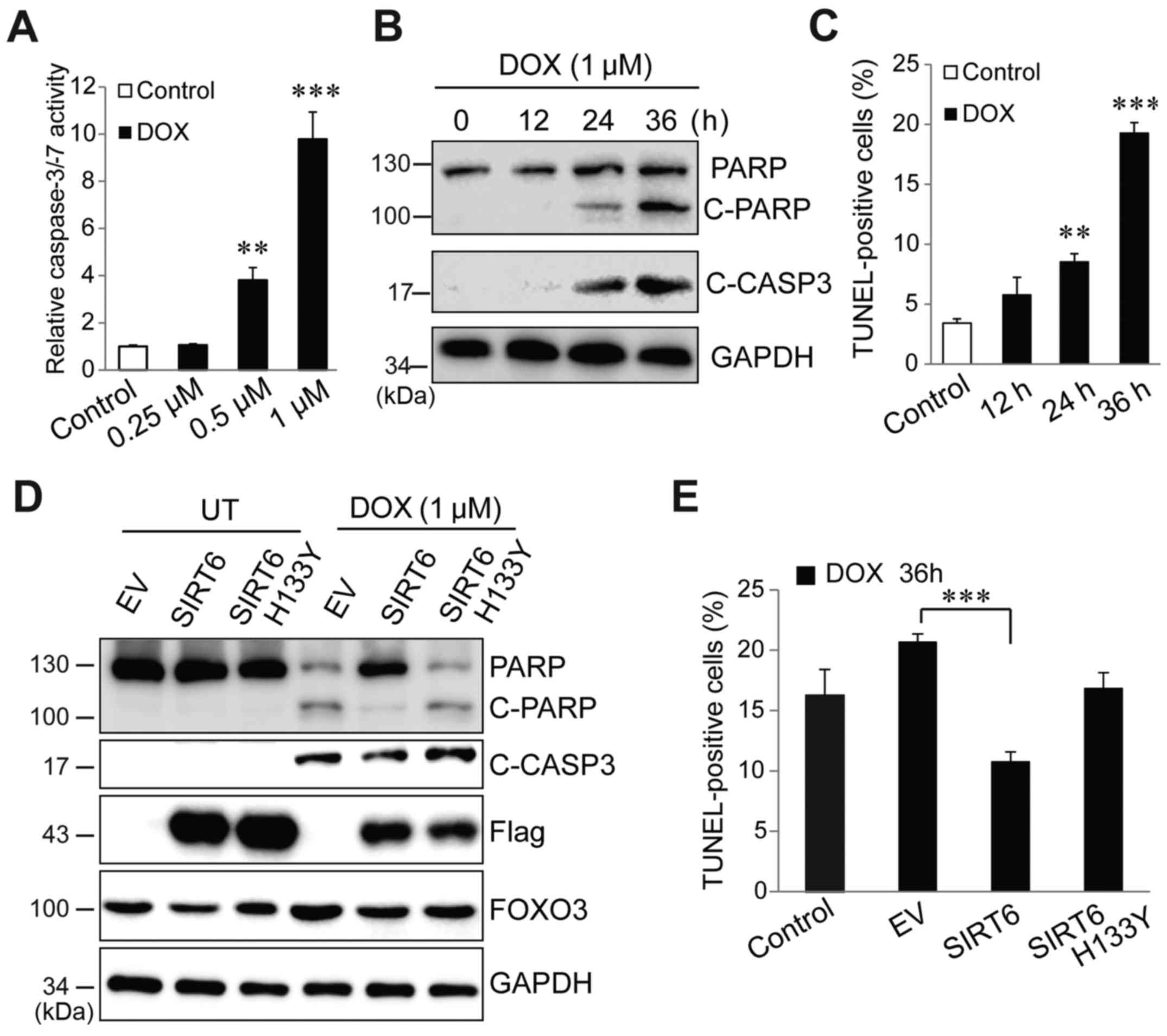
To determine whether downregulation of SIRT6 is
required for doxorubicin to cause liver cancer cell apoptosis,
HepG2 cells were first treated with different concentrations of
doxorubicin. A dose-dependent activation of caspase-3/-7 was
observed after 36 h of doxorubicin treatment (Fig. 2A). Western blot analysis was
performed to evaluate apoptotic signals by measuring PARP and
caspase-3 cleavage, and it was identified found that 1 µM
doxorubicin caused a significant increase in cleaved PARP and
caspase-3 (Fig. 2B). Therefore,
this concentration was used for subsequent experiments (Fig. 2B). A TUNEL assay indicated that 1 µM
doxorubicin induced a significant increase in TUNEL-positive cells
from 24 h post-treatment (Fig. 2C).
Then, Flag-tagged SIRT6 and inactive SIRT6 H133Y mutant were
overexpressed in Huh7 cells followed by treatment with 1 µM
doxorubicin. In the absence of doxorubicin, overexpression of SIRT6
and H133Y mutant exerted no effects on PARP cleavage and caspase-3
activation. However, in the presence of doxorubicin, SIRT6
overexpression almost completely abolished doxorubicin-induced PARP
and caspase-3 cleavage (Fig. 2D). A
TUNEL assay indicated that restorative expression of SIRT6, but not
inactive SIRT6, blocked doxorubicin-induced cell death (Fig. 2E). These data indicated that SIRT6
plays a critical role in determining cell fate in response to
doxorubicin but this effect requires SIRT6 deacetylase enzyme
activity.
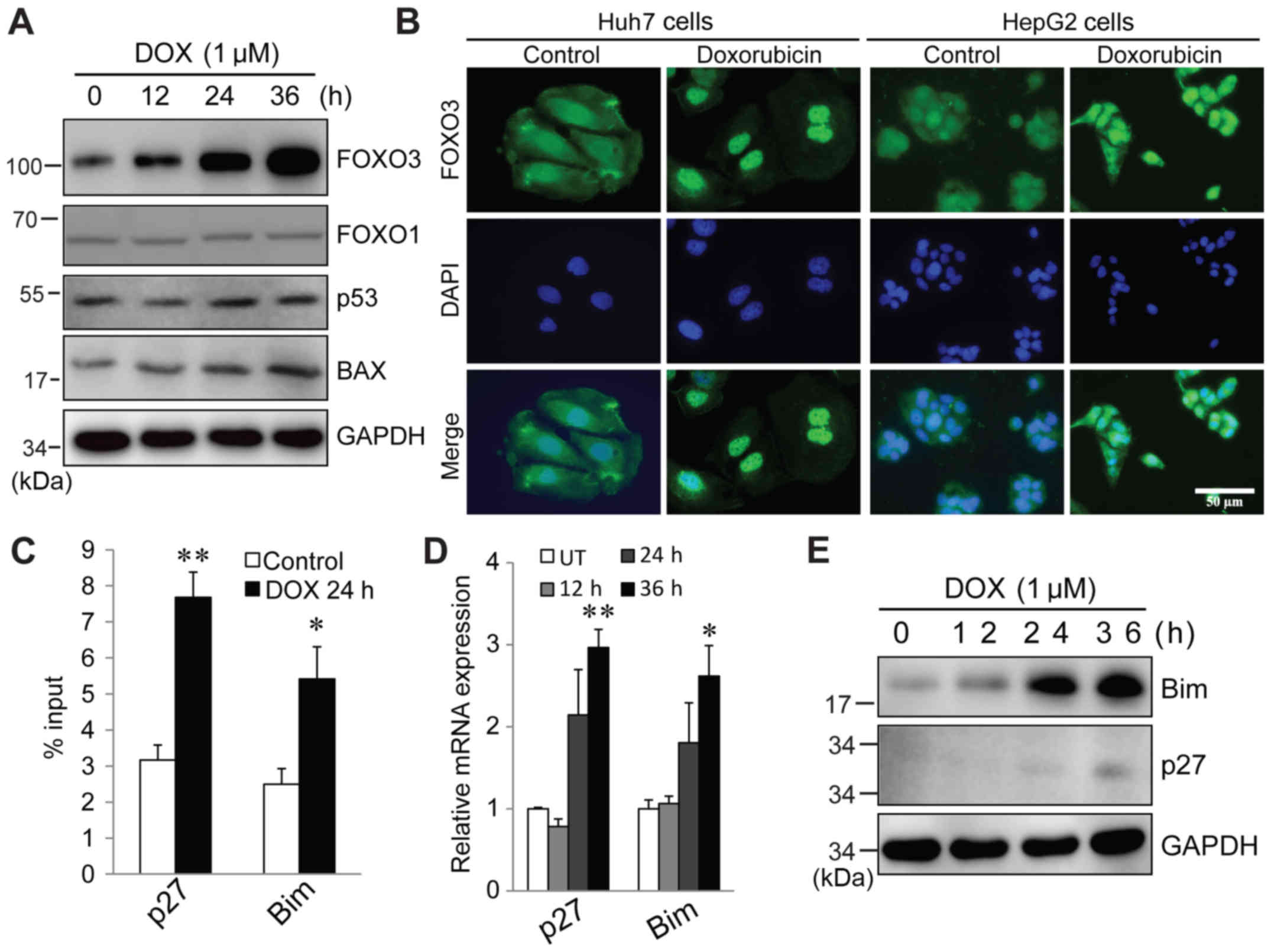
Doxorubicin actives FOXO3, which
triggers pro-apoptotic target gene expression
To determine the potential SIRT6 target that is
responsible for regulating doxorubicin-induced apoptosis, multiple
proteins that are known to induce apoptosis, including p53
(35), FOXO3 (36), FOXO1 (37) and Bax (33), were evaluated. It was identified
that in response to doxorubicin treatment, there were no changes in
FOXO1 and p53 protein expression. Bax expression was increased at
36 h (Fig. 3A). FOXO3 protein was
significantly elevated following doxorubicin treatment (Fig. 3A). Immunofluorescence staining
indicated that in both Huh7 and HepG2 cells, FOXO3 localized to
both the cytosol and nucleus in untreated cells. In response to
doxorubicin, FOXO3 intensity was increased and at 24 h
post-treatment, it was primarily localized in the nucleus (Fig. 3B). In order to investigate whether
nuclear FOXO3 was functionally responsible for apoptosis, a ChIP
assay was performed to assess the promoter binding of FOXO3 to its
target genes, which are responsible for cell cycle arrest and
apoptosis, following doxorubicin treatment. As indicated in
Fig. 3C, at 24 h of doxorubicin
treatment, FOXO3 binding to its target genes p27 and Bim was
significantly increased. Consistent with this, RT-qPCR and western
blotting results indicated that both mRNA and protein levels of p27
and Bim were significantly increased following treatment (Fig. 3D and E). In summary, these data
demonstrated that doxorubicin treatment induces FOXO3 activation,
which triggers expression of its target genes and results in cell
cycle arrest and apoptosis.
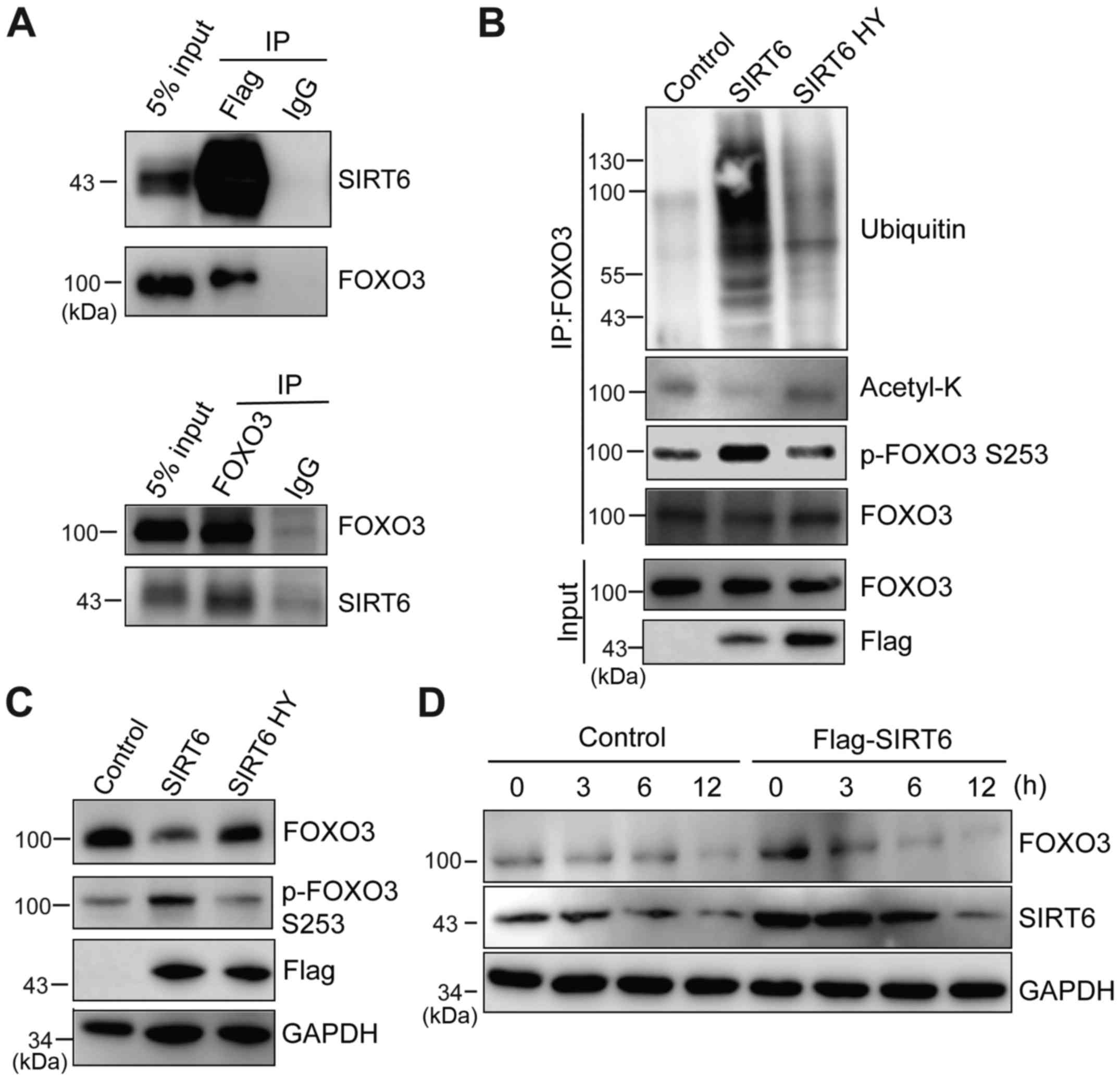
SIRT6 interacts with FOXO3 and
destabilizes FOXO3 by promoting ubiquitination
The aforementioned data indicated that SIRT6 is
downregulated and FOXO3 is upregulated in response to doxorubicin.
Therefore, it was assessed whether SIRT6 regulates FOXO3 protein
stability. SIRT6 was overexpressed in HeLa cells and
immunoprecipitation was performed to examine whether SIRT6
interacts with FOXO3. It was identified that these two proteins
exhibit a direct interaction (Fig.
4A). Previous studies have suggested that multiple SIRT family
members are able to deacetylate FOXO3 (12,14,38).
Thus, it was investigated whether SIRT6 deacetylates FOXO3.
Flag-SIRT6 and SIRT6 H133Y were expressed in HeLa cells and FOXO3
proteins were immunoprecipitated. The acetylation level of FOXO3
was examined using a pan-acetyl Lysine antibody. It was identified
that overexpression of SIRT6, but not the inactive form SIRT6
H133Y, significantly decreased the acetyl-FOXO3 level (Fig. 4B). It was also observed that SIRT6
significantly increased FOXO3 ubiquitination and the p-S253 FOXO3
level, which is critical for FOXO3 degradation (39) (Fig.
4B). These data demonstrated that SIRT6 interacts with FOXO3
and promotes its ubiquitination and degradation. Furthermore, it
was investigated whether SIRT6 regulates FOXO3 protein stability
in vitro. SIRT6 or SIRT6 H133Y was overexpressed in cells,
and then the protein level of FOXO3 was evaluated by western
blotting. SIRT6 overexpression itself was sufficient to increase
p-FOXO3 S253 and reduce FOXO3 protein level, but this effect also
required enzyme activity (Fig. 4C).
The underlying mechanism of this may be that SIRT6 significantly
decreases the FOXO3 protein half-life (Fig. 4D). In summary, these data indicated
that SIRT6 interacts with FOXO3 and regulates FOXO3 protein
stability by promoting its deacetylation and ubiquitination.
SIRT6 regulates doxorubicin-induced
apoptosis through FOXO3
To examine whether FOXO3 is required for
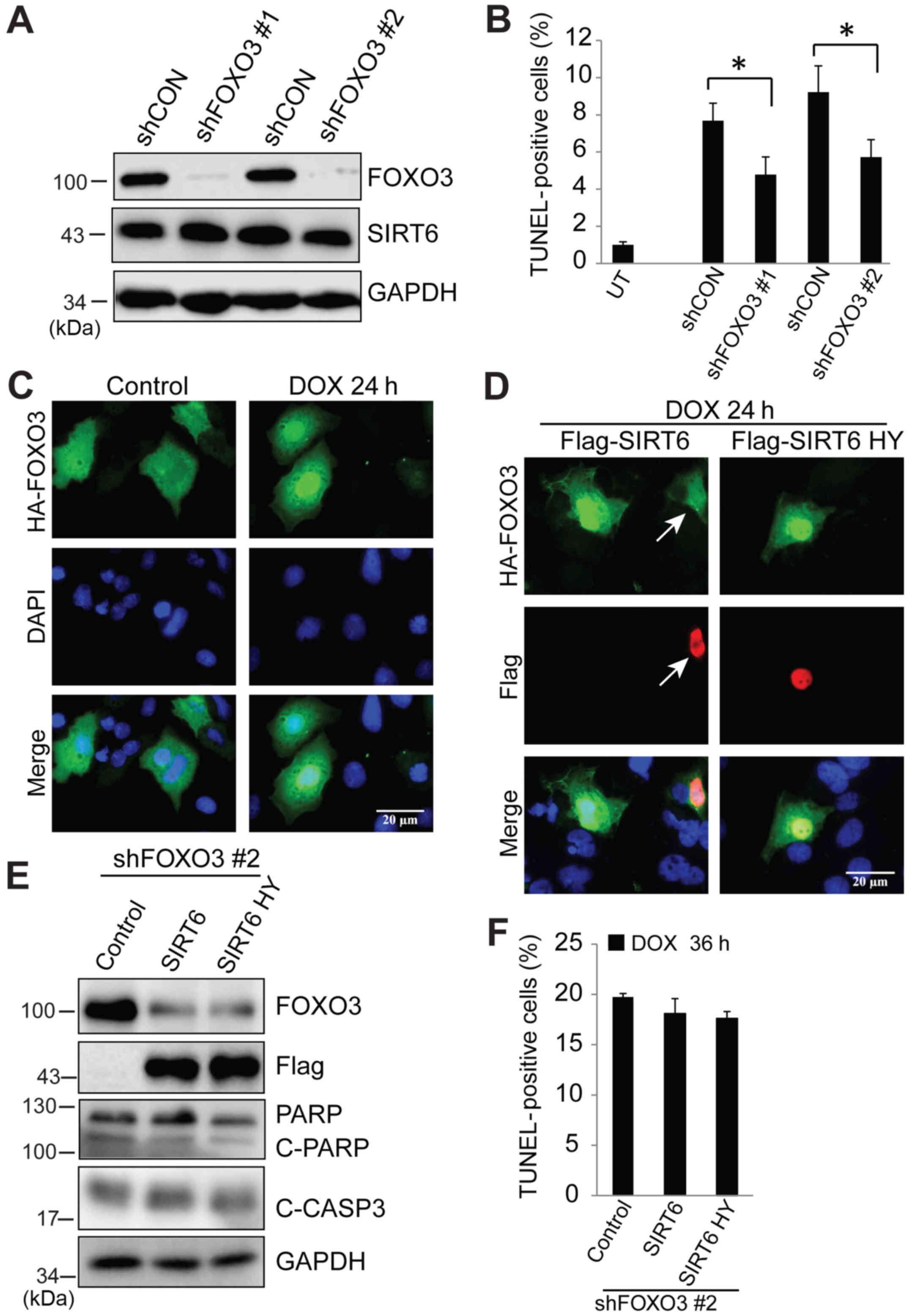
doxorubicin-induced apoptosis; two different lentiviral shFOXO3
transfections were used to knock down FOXO3 protein, and then cells
were treated with doxorubicin. Western blotting results indicated
that shFOXO3 transfection generated >90% knockdown efficiency
and loss of FOXO3 did not have any effects on SIRT6 expression
(Fig. 5A). A TUNEL assay indicated
that absence of FOXO3 significantly abolished doxorubicin-induced
apoptosis (Fig. 5B). In order to
evaluate whether SIRT6 regulates doxorubicin-induced apoptosis
through FOXO3, it was first examined whether SIRT6 blocks nuclear
accumulation of FOXO3 in response to doxorubicin. Cells were
transfected with HA-FOXO3 or co-transfected with Flag-SIRT6.
Following treatment with doxorubicin, cellular localization of
FOXO3 was examined by immunofluorescence. As indicated in Fig. 2B, 24 h of doxorubicin treatment
significantly induced nuclear localization of FOXO3 (Fig. 5C) which could be blocked by SIRT6
(Fig. 5D). Finally, we evaluated
whether the blocking of doxorubicin-induced apoptosis by
overexpression of SIRT6 was via FOXO3. SIRT6 and inactive SIRT6
H133Y were overexpressed in FOXO3-deficient cells followed by
treatment with doxorubicin. SIRT6 and SIRT6 H133Y failed to block
doxorubicin-induced apoptosis, as indicated by similar levels of
cleaved PARP and cleaved caspase-3 (Fig. 5E) and TUNEL-positive cells (Fig. 5F) as compared with control cells. In
summary, these data indicated that in response to doxorubicin
treatment, SIRT6 is a critical factor that regulates FOXO3 nuclear
localization, which is essential for inducing liver cancer cell
death.
Discussion
In the present study, it was demonstrated that
histone deacetylase SIRT6 is a key factor controlling
doxorubicin-induced liver cancer cell death by modulating
transcriptional factor FOXO3. In the absence of doxorubicin
treatment, SIRT6 and FOXO3 interact with each other, and this
interaction promotes FOXO3 deacetylation, ubiquitination and
degradation. Following doxorubicin treatment, SIRT6 is
downregulated, which in turn activates FOXO3 translocation into the
nucleus and binding to its target genes p27 and Bim to cause cell
death. Overexpression of SIRT6 abolishes FOXO3 activation and cell
death following doxorubicin treatment. Finally, it was demonstrated
that SIRT6 regulates doxorubicin-induced cell death through FOXO3;
in the absence of FOXO3, SIRT6 overexpression is not able to
prevent cell death.
SIRT proteins are regulated dynamically by different
stimuli. SIRT6 has long been recognized as an environmental
nutrition sensor; in cases of nutrition restriction, SIRT6 is
rapidly upregulated in a p53-dependent manner (40). Following LPS treatment, SIRTs are
observed to be downregulated (23).
While SIRT1 primarily increases degradation processes via
post-translational mechanisms, SIRT7 decreases both transcription
and protein stability (23). SIRT1
contains several post-translational modification sites that
regulate protein stability and enzyme activity (41). In this study we have demonstrated
that doxorubicin treatment results in a significant decrease in
SIRT6 protein levels, which is critical for cell death. However,
the mechanisms underlying SIRT6 downregulation require further
investigation. It has been indicated that covalent modification of
SIRT1 by oxidants/aldehydes results in a decrease in its enzymatic
activity an increase in its degradation (42). There are multiple serine
phosphorylation sites on SIRT1 (Ser27, Ser47, Ser659 and Ser661),
which are regulated by diverse protein kinases (43–45).
It has been demonstrated that JNK1 phosphorylation of SIRT1 at the
aforementioned sites leads to proteasome-mediated degradation
(46,47). JNK has also been identified to
phosphorylate SIRT6, but it is unclear whether this causes SIRT6
degradation (48). Notably,
doxorubicin treatment induces JNK activation in multiple cancer
types (48,49).
FOXO3 is one of the most well-studied transcription
factors and its activity is tightly regulated by
post-transcriptional modification, including phosphorylation,
ubiquitination, arginine methylation and acetylation (50). It is well documented that
Akt-dependent phosphorylation is the major pathway that controls
FOXO3 activity by promoting nuclear exclusion and degradation
(51). Multiple SIRT proteins have
been reported to deacetylate FOXO3 and regulate its transcriptional
activity, including SIRT1, SIRT2, SIRT3, SIRT6 and SIRT7 (13,14,19,23,51).
In the present study, it was demonstrated that FOXO3 is also an
SIRT6 target in liver cancer cells. SIRT6 interacts with FOXO3;
overexpression of SIRT6, but not the enzyme-inactivated mutant,
reduces FOXO3 protein half-life and redundancy, suggesting that
SIRT6 regulates FOXO3 through deacetylation. Consistent with this,
it was also identified that SIRT6 is able to reduce the acetylation
level of FOXO3. The mechanism by which the FOXO3 acetylation state
controls FOXO3 stability is not clear, but it is reported that
acetylation promotes the binding of FOXO3 to JNK1 (23). Thus, it is speculated that this may
also increase FOXO3 binding to Akt, which triggers its
phosphorylation and degradation. Notably, it was observed that
SIRT6 overexpression increases p-FOXO3 S253 levels, which are
critical for FOXO3 stability. A recent study also indicated that
p38 phosphorylates FOXO3 and promotes its nuclear translocation in
response to doxorubicin; it is not clear whether SIRT6 could block
p38-induced FOXO3 phosphorylation.
The ability of SIRT6 and FOXO3 to regulate
doxorubicin-dependent cell death raises the possibility that
manipulation of this system may have therapeutic implications.
Manipulating SIRT has long been demonstrated to have beneficial
effects in various diseases. Resveratrol, a well-established SIRT
activator, is recognized as an anti-apoptosis and anti-inflammatory
compound that possesses beneficial effects in various diseases,
including rheumatoid arthritis and type I diabetes (52,53).
SIRT inhibitor has been demonstrated to induce cancer cell
apoptosis by increasing p53 acetylation (53,54)
and Myc degradation (55). The
current data suggest that downregulation of SIRT6 is essential for
doxorubicin-induced cancer cell death. Therefore, it is speculated
that high expression of SIRT6 or failure to downregulate SIRT6
following doxorubicin treatment may contribute to TACE resistance.
SIRT6 expression levels in human HCC tissues remain unclear due to
conflicting reports, suggesting that SIRT6 is highly disease stage
or population dependent, but it would also be interesting to
investigate whether tumors with higher SIRT6 expression are also
TACE resistant.
The present study demonstrated that downregulation
of SIRT6 is required for doxorubicin-induced liver cancer cell
death since it promotes FOXO3 nuclear localization and activation,
leading to cell death. The current data also identified that SIRT6
controls FOXO3 by deacetylation, which is a novel mechanism.
Furthermore, acetylation affects kinase binding to FOXO3 and
whether there are apoptotic effects of FOXO3 that are independent
of SIRT6 downregulation. To gain further understanding, these
mechanisms should be investigated in a future study. Nonetheless,
the current findings regarding the interaction between SIRT6 and
FOXO3 elucidate a novel molecular mechanism by which SIRT proteins
regulate cell function and point to SIRT6 as a potentially
important target for liver cancer treatment.
Acknowledgements
We thank Dr Yi-Ming Tao for providing reagents for
this study and his kind help in revising the manuscript.
Funding
The present study was supported in part by grants
from the National Natural Science Foundation of China (grant no.
81473617), the Hunan Natural Science Foundation of China (grant no.
13JJ2032) and The Innovation Platform Open Foundation of Hunan
Educational office (grant no. 16K066).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
The manuscript was written through contributions of
all authors. XFT conceived and designed the study. JQH, FD, XPH, WZ
and XCZ performed the experiments. XFT and JQH reviewed and edited
the manuscript. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Institutional Review Board of the Department of Laboratory Animal
Science of Hunan University of Chinese Medicine (Changsha,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wong MC, Jiang JY, Goggins WB, Liang M,
Fang Y, Fung FD, Leung C, Wang HH, Wong GL, Wong VW, et al:
International incidence and mortality trends of liver cancer: A
global profile. Sci Rep. 7:458462017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Altekruse SF, McGlynn KA and Reichman ME:
Hepatocellular carcinoma incidence, mortality, and survival trends
in the United States from 1975 to 2005. J Clin Oncol. 27:1485–1491.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hadzic N and Finegold MJ: Liver neoplasia
in children. Clin Liver Dis. 15(443–462): vii–x. 2011.
|
|
4
|
Horton JD, Lee S, Brown SR, Bader J and
Meier DE: Survival trends in children with hepatoblastoma. Pediatr
Surg Int. 25:407–412. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Otte JB: Progress in the surgical
treatment of malignant liver tumors in children. Cancer Treat Rev.
36:360–371. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Davis GL, Dempster J, Meler JD, Orr DW,
Walberg MW, Brown B, Berger BD, O'Connor JK and Goldstein RM:
Hepatocellular carcinoma: Management of an increasingly common
problem. Proc. 21:266–280. 2008.
|
|
7
|
Dhanasekaran R, Limaye A and Cabrera R:
Hepatocellular carcinoma: Current trends in worldwide epidemiology,
risk factors, diagnosis, and therapeutics. Hepat Med. 4:19–37.
2012.PubMed/NCBI
|
|
8
|
Cox J and Weinman S: Mechanisms of
doxorubicin resistance in hepatocellular carcinoma. Hepat Oncol.
3:57–59. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
North BJ and Verdin E: Sirtuins:
Sir2-related NAD-dependent protein deacetylases. Genome Biol.
5:2242004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Canto C, Gerhart-Hines Z, Feige JN,
Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P and Auwerx
J: AMPK regulates energy expenditure by modulating NAD+
metabolism and SIRT1 activity. Nature. 458:1056–1060. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Satoh A, Brace CS, Rensing N, Cliften P,
Wozniak DF, Herzog ED, Yamada KA and Imai S: Sirt1 extends life
span and delays aging in mice through the regulation of Nk2
homeobox 1 in the DMH and LH. Cell Metab. 18:416–430. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Zhu Y, Xing S, Ma P and Lin D:
SIRT5 prevents cigarette smoke extract-induced apoptosis in lung
epithelial cells via deacetylation of FOXO3. Cell Stress
Chaperones. 20:805–810. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tseng AH, Shieh SS and Wang DL: SIRT3
deacetylates FOXO3 to protect mitochondria against oxidative
damage. Free Radic Biol Med. 63:222–234. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Brunet A, Sweeney LB, Sturgill JF, Chua
KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et
al: Stress-dependent regulation of FOXO transcription factors by
the SIRT1 deacetylase. Science. 303:2011–2015. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Simic P, Zainabadi K, Bell E, Sykes DB,
Saez B, Lotinun S, Baron R, Scadden D, Schipani E and Guarente L:
SIRT1 regulates differentiation of mesenchymal stem cells by
deacetylating β-catenin. EMBO Mol Med. 5:430–440. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jeong J, Juhn K, Lee H, Kim SH, Min BH,
Lee KM, Cho MH, Park GH and Lee KH: SIRT1 promotes DNA repair
activity and deacetylation of Ku70. Exp Mol Med. 39:8–13. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marquardt JU, Fischer K, Baus K, Kashyap
A, Ma S, Krupp M, Linke M, Teufel A, Zechner U, Strand D, et al:
Sirtuin-6-dependent genetic and epigenetic alterations are
associated with poor clinical outcome in hepatocellular carcinoma
patients. Hepatology. 58:1054–1064. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ponugoti B, Kim DH, Xiao Z, Smith Z, Miao
J, Zang M, Wu SY, Chiang CM, Veenstra TD and Kemper JK: SIRT1
deacetylates and inhibits SREBP-1C activity in regulation of
hepatic lipid metabolism. J Biol Chem. 285:33959–33970. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang F, Nguyen M, Qin FX and Tong Q: SIRT2
deacetylates FOXO3a in response to oxidative stress and caloric
restriction. Aging Cell. 6:505–514. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jin YH, Kim YJ, Kim DW, Baek KH, Kang BY,
Yeo CY and Lee KY: Sirt2 interacts with 14-3-3 beta/gamma and
down-regulates the activity of p53. Biochem Biophys Res Commun.
368:690–695. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rothgiesser KM, Erener S, Waibel S,
Luscher B and Hottiger MO: SIRT2 regulates NF-κB dependent gene
expression through deacetylation of p65 Lys310. J Cell Sci.
123:4251–4258. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vaziri H, Dessain SK, Eaton Ng E, Imai SI,
Frye RA, Pandita TK, Guarente L and Weinberg RA: hSIR2SIRT1
functions as an NAD-dependent p53 deacetylase. Cell. 107:149–159.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li Z, Bridges B, Olson J and Weinman SA:
The interaction between acetylation and serine-574 phosphorylation
regulates the apoptotic function of FOXO3. Oncogene. 36:1887–1898.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Paredes S, Villanova L and Chua KF:
Molecular pathways: Emerging roles of mammalian sirtuin SIRT7 in
cancer. Clin Cancer Res. 20:1741–1746. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kaidi A, Weinert BT, Choudhary C and
Jackson SP: Human SIRT6 promotes DNA end resection through CtIP
deacetylation. Science. 329:1348–1353. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim HS, Xiao C, Wang RH, Lahusen T, Xu X,
Vassilopoulos A, Vazquez-Ortiz G, Jeong WI, Park O, Ki SH, et al:
Hepatic-specific disruption of SIRT6 in mice results in fatty liver
formation due to enhanced glycolysis and triglyceride synthesis.
Cell Metab. 12:224–236. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xiao C, Kim HS, Lahusen T, Wang RH, Xu X,
Gavrilova O, Jou W, Gius D and Deng CX: SIRT6 deficiency results in
severe hypoglycemia by enhancing both basal and insulin-stimulated
glucose uptake in mice. J Biol Chem. 285:36776–36784. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhong L, D'Urso A, Toiber D, Sebastian C,
Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD,
Nir T, et al: The histone deacetylase Sirt6 regulates glucose
homeostasis via Hif1alpha. Cell. 140:280–293. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kugel S, Sebastian C, Fitamant J, Ross KN,
Saha SK, Jain E, Gladden A, Arora KS, Kato Y, Rivera MN, et al:
SIRT6 suppresses pancreatic cancer through control of Lin28b. Cell.
165:1401–1415. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ioris RM, Galie M, Ramadori G, Anderson
JG, Charollais A, Konstantinidou G, Brenachot X, Aras E, Goga A,
Ceglia N, et al: SIRT6 suppresses cancer stem-like capacity in
tumors with PI3K activation independently of its deacetylase
activity. Cell Rep. 18:1858–1868. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sebastian C, Zwaans BM, Silberman DM,
Gymrek M, Goren A, Zhong L, Ram O, Truelove J, Guimaraes AR, Toiber
D, et al: The histone deacetylase SIRT6 is a tumor suppressor that
controls cancer metabolism. Cell. 151:1185–1199. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee N, Ryu HG, Kwon JH, Kim DK, Kim SR,
Wang HJ, Kim KT and Choi KY: SIRT6 depletion suppresses tumor
growth by promoting cellular senescence induced by DNA damage in
HCC. PLoS One. 11:e01658352016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ran LK, Chen Y, Zhang ZZ, Tao NN, Ren JH,
Zhou L, Tang H, Chen X, Chen K, Li WY, et al: SIRT6 overexpression
potentiates apoptosis evasion in hepatocellular carcinoma via
BCL2-associated X protein-dependent apoptotic pathway. Clin Cancer
Res. 22:3372–3382. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Z, Zhao J, Tikkhanovich I, Kuravi S,
Helzberd J, Dorko K, Roberts B, Kumer S and Weinman SA: Serine 574
phosphorylation alters transcriptional programming of FOXO3 by
selectively enhancing apoptotic gene expression. Cell Death Differ.
23:583–595. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang P, Tu B, Wang H, Cao Z, Tang M,
Zhang C, Gu B, Li Z, Wang L, Yang Y, et al: Tumor suppressor p53
cooperates with SIRT6 to regulate gluconeogenesis by promoting
foxO1 nuclear exclusion. Proc Natl Acad Sci U S A. 111:10684–10689.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tao R, Xiong X, DePinho RA, Deng CX and
Dong XC: FoxO3 transcription factor and Sirt6 deacetylase regulate
low density lipoprotein (LDL)-cholesterol homeostasis via control
of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene
expression. J Biol Chem. 288:29252–29259. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Song MY, Wang J, Ka SO, Bae EJ and Park
BH: Insulin secretion impairment in Sirt6 knockout pancreatic beta
cells is mediated by suppression of the FoxO1-Pdx1-Glut2 pathway.
Sci Rep. 6:303212016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang F, Chan CH, Chen K, Guan X, Lin HK
and Tong Q: Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to
Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene.
31:1546–1557. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dobson M, Ramakrishnan G, Ma S, Kaplun L,
Balan V, Fridman R and Tzivion G: Bimodal regulation of FoxO3 by
AKT and 14-3-3. Biochim Biophys Acta. 1813:1453–1464. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kanfi Y, Shalman R, Peshti V, Pilosof SN,
Gozlan YM, Pearson KJ, Lerrer B, Moazed D, Marine JC, de Cabo R, et
al: Regulation of SIRT6 protein levels by nutrient availability.
FEBS Lett. 582:543–548. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Flick F and Luscher B: Regulation of
sirtuin function by posttranslational modifications. Front
Pharmacol. 3:292012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Caito S, Rajendrasozhan S, Cook S, Chung
S, Yao H, Friedman AE, Brookes PS and Rahman I: SIRT1 is a
redox-sensitive deacetylase that is post-translationally modified
by oxidants and carbonyl stress. FASEB J. 24:3145–3159. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sasaki T, Maier B, Koclega KD, Chruszcz M,
Gluba W, Stukenberg PT, Minor W and Scrable H: Phosphorylation
regulates SIRT1 function. PLoS One. 3:e40202008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zschoernig B and Mahlknecht U:
Carboxy-terminal phosphorylation of SIRT1 by protein kinase CK2.
Biochem Biophys Res Commun. 381:372–377. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Olsen JV, Blagoev B, Gnad F, Macek B,
Kumar C, Mortensen P and Mann M: Global, in vivo, and site-specific
phosphorylation dynamics in signaling networks. Cell. 127:635–648.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nasrin N, Kaushik VK, Fortier E, Wall D,
Pearson KJ, de Cabo R and Bordone L: JNK1 phosphorylates SIRT1 and
promotes its enzymatic activity. PLoS One. 4:e84142009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gao Z, Zhang J, Kheterpal I, Kennedy N,
Davis RJ and Ye J: Sirtuin 1 (SIRT1) protein degradation in
response to persistent c-Jun N-terminal kinase 1 (JNK1) activation
contributes to hepatic steatosis in obesity. J Biol Chem.
286:22227–22234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Van Meter M, Simon M, Tombline G, May A,
Morello TD, Hubbard BP, Bredbenner K, Park R, Sinclair DA, Bohr VA,
et al: JNK phosphorylates SIRT6 to stimulate DNA double-strand
break repair in response to oxidative stress by recruiting PARP1 to
DNA breaks. Cell Rep. 16:2641–2650. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kim J and Freeman MR: JNK/SAPK mediates
doxorubicin-induced differentiation and apoptosis in MCF-7 breast
cancer cells. Breast Cancer Res Treat. 79:321–328. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Calnan DR and Brunet A: The foxo code.
Oncogene. 27:2276–2288. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Khongkow M, Olmos Y, Gong C, Gomes AR,
Monteiro LJ, Yagüe E, Cavaco TB, Khongkow P, Man EP, Laohasinnarong
S, et al: SIRT6 modulates paclitaxel and epirubicin resistance and
survival in breast cancer. Carcinogenesis. 34:1476–1486. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Elmali N, Baysal O, Harma A, Esenkaya I
and Mizrak B: Effects of resveratrol in inflammatory arthritis.
Inflammation. 30:1–6. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lee SM, Yang H, Tartar DM, Gao B, Luo X,
Ye SQ, Zaghouani H and Fang D: Prevention and treatment of diabetes
with resveratrol in a non-obese mouse model of type 1 diabetes.
Diabetologia. 54:1136–1146. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Peck B, Chen CY, Ho KK, Di Fruscia P,
Myatt SS, Coombes RC, Fuchter MJ, Hsiao CD and Lam EW: SIRT
inhibitors induce cell death and p53 acetylation through targeting
both SIRT1 and SIRT2. Mol Cancer Ther. 9:844–855. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Jing H, Hu J, He B, Negrón Abril YL,
Stupinski J, Weiser K, Carbonaro M, Chiang YL, Southard T,
Giannakakou P, et al: A SIRT2-selective inhibitor promotes c-Myc
oncoprotein degradation and exhibits broad anticancer activity.
Cancer Cell. 29:297–310. 2016. View Article : Google Scholar : PubMed/NCBI
|