Introduction
Cancer is one of the leading causes of mortality
globally, causing ~8 million instances of mortality and 13 million
new cases annually (1). Colorectal
cancer (CRC) is the third most common non-cutaneous malignancy, and
is responsible for a marked proportion of cancer morbidity and
mortality, particularly in developed countries (2). In recent years, the mortality rate of
CRC has increased rapidly in China and ranks fourth among mortality
caused by malignant tumors (3). CRC
is typically diagnosed at advanced stages; surgery combined with
chemotherapy and radiotherapy is currently the most effective
approach for treatment, but the 5-year survival rate of patients
with CRC has decreased to ~8% with poor prognosis (4). Thus, understanding the molecular
mechanisms underlying CRC development may provide insights into a
novel clinical therapy strategy.
Evidence from experimental, epidemiological and
clinical studies indicate that aspirin or non-steroidal
anti-inflammatory drugs (NSAIDs) may decrease the risk of CRC
markedly through cyclooxygenase (COX) inhibition. Two isoforms of
COX, identified as COX-1 and COX-2, have ~60% amino acid sequence
homology and catalyze the same reactions. However, they are encoded
by distinct genes and differ in their tissue distribution (5). COX-2 is overexpressed in sporadic
colorectal adenomas and carcinomas, compared with normal mucosa
(6). It has been identified that
targeting COX-2 successfully prevented CRC. That COX-2 is the only
COX isoform involved in colorectal tumorigenesis has been
challenged in a number of studies. It is widely accepted that a low
dose of aspirin (between 80 and 100 mg) decreases the incidence and
mortality of CRC primarily by targeting COX-1 rather than COX-2
(7). Genetic knockout of COX-1 in
mice markedly decreased intestinal polyposis (8). Upregulation of COX-1 expression
protected intestinal stem cells from DNA damage in the early phase
of intestinal tumorigenesis (9).
The chemopreventive activity of 6-C-(E-phenylethenyl)-naringenin
suppresses CRC growth by inhibiting COX-1 expression (10). Thus, these studies indicate that
COX-1 serves an important function in colorectal
carcinogenesis.
Apoptosis is a highly regulated and controlled
process of programmed cell death in organism development and tissue
homeostasis to remove infected cells, and thus maintain internal
environment stability. Under pathological conditions, normal cells
may lose their ability to undergo apoptosis, leading to
uncontrolled cell growth and autoimmune diseases. Insufficient
apoptosis may also lead to uncontrolled cell proliferation followed
by tumorigenesis. One of the characteristics of tumor cells is the
capability to evade apoptosis regulated by the balance of pro- and
anti-apoptotic factors through the extrinsic or mitochondrial
pathway (11). A complex interplay
between mitochondrial proteins, such as the B-cell lymphoma 2
(Bcl-2) family proteins, serve essential functions in apoptosis.
Anti-apoptotic Bcl-2 and pro-apoptotic proteins Bcl-2-associated X
protein (Bax) dimerized to control mitochondrial membrane
permeabilization and mitochondrial membrane potential (MMP or
ΔΨm). The equilibrium between Bcl-2 and Bax determines
the cell's fate to undergo apoptosis or cell survival (12). Therefore, understanding the function
of apoptosis in tumors and exploiting the targets for anticancer
drugs by controlling apoptosis in cancer cells has value in cancer
therapy.
Nuclear factor κB (NF-κB) is a major transcription
factor involved in a myriad of physiological and pathological
scenarios, including apoptosis, proliferation, invasion, and
angiogenesis (13). In mammals,
there are five homologous subunits in the NF-κB family: RelA (p65),
c-Rel, RelB, NF-κB1 (p105-p50) and NF-κB (p100-p52). All NF-κB
family members share a conserved Rel homology domain in their
N-terminus that is required for DNA binding, homo- and
heterodimerization formation, nuclear localization and interaction
with inhibitor of NF-κB (IκB). A number of divergent stimuli
activate two NF-κB pathways: The canonical and non-canonical
pathways (otherwise referred to as the ‘classical’ and
‘non-classical’). Under inactive conditions, NF-κB is localized in
the cytoplasm with three heterotrimeric complexes, including p50,
p65, and IκBα in the classical NF-κB pathway. Upon stimulation,
such as by cytokines, bacterial products and tumor necrosis factor
α, the signaling pathway is activated, followed by the
phosphorylation, ubiquitination and degradation of IκBα and
phosphorylation of p65. Subsequently, p105-RelA is processed to
p50-RelA (NF-κB), which translates into the nucleus and binds
promoters of target genes to switch on the expression of diverse
target genes involved in cell proliferation (such as c-Myc) and
anti-apoptosis (such as Bcl-2) (14). The non-classical NF-κB pathway is
activated through IκB kinase α, and p100-RelB is processed into
p52-RelB heterodimers, which translates into the nucleus to bind
the promoter of NF-κB-responsive elements in NF-κB target genes
regulating lymphoid organogenesis and maintaining the malignant
phenotype in certain types of cancer (15). The activation of the two pathways
relies on the inducible phosphorylation-inhibitory proteins IκB
(IκBs and p100), and regulates cell survival, death and
carcinogenesis. It has been identified that COX-2 promotes
angiogenesis in aggressive forms of CRC induced by the
non-classical NF-κB pathway (16),
and overexpression of COX-2 increases the expression of Bcl-2 to
inhibit apoptosis, which appears to be a key pathway in the
survival of cancer cells. To the best of our knowledge, the
association between COX-1 and NF-κB in colorectal carcinogenesis
has not been comprehensively investigated, and the underlying
molecular mechanism requires elucidation.
Thus, the aim of the present study was to
investigate the function of COX-1 in colorectal carcinogenesis
using a short hairpin RNA (shRNA) knockdown strategy in CRC cells.
The results of the present study indicated that COX-1 is required
for maintaining malignant characteristics of CRC cells. Knockdown
of COX-1 suppressed cell proliferation and survival, involved in
mitochondrial dysfunction and triggered caspase-dependent
mitochondrial apoptosis by inhibiting the p65 subunit
phosphorylation of NF-κB in HCT116 and HT29 CRC cells, which
indicated a potential molecular target in developing prevention or
treatment of CRC.
Materials and methods
Patients and tissue samples
All colorectal tumor samples and matching normal
tissues were from 14 patients with CRC (9 males and 5 females; age
range, 33–68 years) at the Department of Surgery, Pu'er City
People's Hospital (Pu'er, Yunnan, China), between September 2016
and November 2017. The fresh tumor tissue specimens were excised
from the tumors, and their corresponding adjacent normal tissues
were taken ≥3 cm from the tumor edge. All specimens were stabilized
by snap-freezing immediately in cryovials, immersed in liquid
nitrogen and stored at −80°C until analysis.
Cell lines and cell culture
The human embryonic kidney epithelial cell line
293T, the human CRC cell lines Caco2, Colo205, SW480, HT29 and
HCT116, and the normal intestinal epithelial cell FHC were obtained
from the Biochemistry and Molecular Biology Laboratory of Yunnan
University (Yunnan, China). Cells were cultured in high-glucose
Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% (v/v)
fetal bovine serum (FBS; BI Biological Industries, Wentworthville,
Australia) and 1% (v/v) antibiotics (100 mg/l streptomycin, 100
U/ml penicillin; HyClone; GE Healthcare Life Sciences, Logan UT,
USA) in a cell incubator at 37°C with an humidified atmosphere
containing 5% CO2. Cell numbers were determined using a
cell counting chamber.
Virus packaging and cellular stable
transfection with shRNAs
A total of four shRNAs were used to target the COX-1
gene and were cloned into pLKO.1-puro-based lentiviral vectors
(pLKO.1-puro-COX-1) synthesized by Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany). The COX-1 target sequences are listed in
Table I. The vector pLKO.1-puro
(pLKO.1-puro) was used as a negative control (mock). Lentiviruses
were created in 293T cells by transfection of packaging plasmid and
envelope plasmid with X-tremeGENE™ HP DNA transfection reagent
(Roche Diagnostics, Indianapolis, IN, USA), according to the
manufacturer's protocol. Virus-containing supernatants were
harvested at 24 and 48 h, respectively, post-transfection and
passed through a 0.45 µm filter (Sigma-Aldrich; Merck KGaA) and
used immediately or stored at −80°C for future use. HCT116 and HT29
cells were infected twice with lentiviruses within 48 h, and the
stable silenced cells were selected using puromycin (Thermo Fisher
Scientific, Inc.) at a final concentration of 6 µg/ml. The protein
expression level of COX-1 was determined using western blot
analysis.
 | Table I.shRNA targets used in the present
study. |
Table I.
shRNA targets used in the present
study.
| shRNA | Clone accession
number | Sequence
(5′-3′) |
|---|
| shCOX-1-1 |
NM_000962.2–1266s1c1 |
CCGGCATGGAGTTCAACCATCTCTACTCGAGTAGAGATGGTTGAACTCCATGTTTTTG |
| shCOX-1–2 |
NM_000962.2–822s1c1 |
CCGGCGGCCACATTTATGGAGACAACTCGAGTTGTCTCCATAAATGTGGCCGTTTTTG |
| shCOX-1–3 |
NM_000962.2–225s1c1 |
CCGGGCCAGTGAATCCCTGTTGTTACTCGAGTAACAACAGGGATTCACTGGCTTTTTG |
| shCOX-1–4 |
NM_000962.2–564s1c1 |
CCGGCGTGAGCTATTACACTCGTATCTCGAGATACGAGTGTAATAGCTCACGTTTTTG |
Western blot analysis
Cells or tissues were washed twice with ice-cold PBS
and incubated on ice for 30 min in radioimmunoprecipitation buffer
(50 mM Tris/HCl, pH 7.4, 150 mM NaCl, 0.5% sodium deoxycholate,
0.1% SDS, 1% NP-40, 2 mM sodium EDTA; Beyotime Institute of
Biotechnology, Haimen, China), supplemented with Complete™ protease
inhibitor cocktail (Roche Diagnostics). The protein concentration
was determined using a Bradford protein assay kit (Beijing Dingguo
Changsheng Biotechnology Co., Ltd., Beijing, China). Equal amounts
(20 µg) of protein samples were separated by SDS-PAGE (12.5% gel)
using a standard protocol, and then transferred onto 0.22 µm
Immun-Blot® polyvinylidene difluoride membranes (Merck
KGaA) with wet transfer electrophoresis (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). The membranes was blocked using 10% (w/v)
non-fat milk in PBS containing 1% (w/v) Tween-20 (pH 7.4) at room
temperature for 1 h. Membranes were incubated with specific primary
antibodies overnight on a rocker at 4°C. Following washing with PBS
containing 1% Tween-20, the membranes were incubated again for 2 h
at room temperature with corresponding secondary antibodies. All
primary antibodies were used at the dilutions specified by the
manufacturers' protocols and secondary antibodies were used at
1:2,000 dilutions. The protein-antibody complexes were visualized
using an Enhanced Chemiluminescence detection kit (Thermo Fisher
Scientific, Inc.) with a chemiluminescence instrument (Tanon
Science and Technology Co., Ltd., Shanghai, China). Each immunoblot
depicted is representative of at least three similar independent
experiments. The protein expression of each band was quantified by
densitometric analysis using ImageJ software (version 1.52a;
National Institutes of Health, Bethesda, MD, USA).
Antibodies
The primary antibodies for the detection of COX-1
(69 kDa; cat. no. C0159) and COX-2 (72 kDa; cat. no. C0160) were
purchased from Anbo Biotechnology Co., Ltd. (San Francisco, CA,
USA). Anti-phospho-IκBα (36 kDa; cat. no. ab133462), anti-Bax (20
kDa; cat. no. ab32503), anti-voltage-dependent anion channel 2 (32
kDa; cat. no. ab47104) and anti-cleaved caspase-3 (17 kDa; cat. no.
ab2302) were purchased from Abcam (Cambridge, UK). Anti-phospho-p65
(65 kDa; cat. no. 3033) and anti-c-Myc (63 kDa; cat. no. 5605) were
purchased from Cell Signaling Technology (Danvers, MA, USA).
Anti-Bcl-2 (26 kDa; cat. no. 551097) and anti-cyclin D1
(36 kDa; cat. no. 554180) were purchased from BD Biosciences (San
Jose, CA, USA). Other primary antibodies against IκBα (34 kDa; cat.
no. AF1282), p65 (65 kDa; cat. no. AF1234), cytochrome c (15
kDa; cat. no. AC908), caspase-9 (47 kDa; cat. no. AC062), cleaved
caspase-9 (37 kDa; cat. no. AC062), caspase-3 (35 kDa; cat. no.
AC030), poly(ADP-ribose) polymerase (PARP; 116 kDa; cat. no.
AF1657), cleaved PARP (89 kDa; cat. no. AF1567), cyclin
E2 (47 kDa; cat. no. AF2494) and GAPDH (36 kDa; cat. no.
AF0006) were purchased from Beyotime Institute of Biotechnology.
The secondary antibodies used were horseradish peroxidase
(HRP)-labeled goat anti-rabbit immunoglobulin G (IgG) [heavy and
light chains (H+L)] (cat. no. A0208) or HRP-labeled goat anti-mouse
IgG (H+L) (cat. no. A0216) (both Beyotime Institute of
Biotechnology).
Analysis of apoptotic cells using
Annexin V/propidium iodide (PI) dual staining
The analysis of apoptosis-mediated cell death of
tumor cells was performed using a double-staining method with a
fluorescein isothiocyanate (FITC)-labeled Annexin V and PI
Apoptosis Detection kit (Beyotime Institute of Biotechnology),
according to the manufacturer's protocol. Briefly, cell pellets
were resuspended in 400 µl binding buffer [10 mM
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid/NaOH, 140 mM
NaCl and 2.5 mM CaCl2 at pH 7.4] at room temperature for
5 min, stained with FITC-conjugated Annexin V and PI, and incubated
at 4°C for 20 min in the dark. Fluorescence intensities were
analyzed using an epifluorescence microscope at a magnification of
×20 (Leica DMI8; Leica Microsystems GmbH, Wetzlar, Germany). The
Annexin V-positive/PI-negative cells were recognized as early
apoptotic cells, the Annexin V-positive/PI-positive cells were
recognized as late apoptotic cells and the Annexin
V-negative/PI-negative cells were recognized as viable cells. Three
independent experiments were performed with three replicates per
experiment.
Cell cycle analysis by flow
cytometry
The distribution of various phases of the cell cycle
(G0/G1, S and G2/M) was determined
by quantification of the DNA content of cells stained with PI
solution (containing 50 µg/ml PI, 0.2% Triton X-100 and 100 µg/ml
RNase A). Briefly, cells were harvested and fixed with ice-cold 70%
ethanol for 30 min at 4°C, then incubated with PI solution at 37°C
for 45 min. The cell cycle profiles were analyzed by flow cytometry
(BD Biosciences).
Cell viability
The cell viability was measured by the CellTiter
96® AQueous One Solution Cell Proliferation assay kit
(Promega Corporation, Madison, WI, USA), according to the
manufacturer's protocol, for various durations (0, 24, 48, 96 and
144 h). In brief, 40 µl MTT solution was added to each well
(containing 800 cells), followed by incubation at 37°C for 4 h. The
optical density was determined at a wavelength of 490 nm using a
microplate reader (Bio-Rad Laboratories, Inc.). Experiments were
performed in triplicate.
Determination of adenosine
triphosphate (ATP)
The production of ATP was determined using a
luciferin/luciferase assay with an ATP assay kit (Beyotime
Institute of Biotechnology), according to the manufacturer's
protocol. Briefly, the cells were collected and permeabilized prior
to the addition of ATP lysis buffer, and incubated on ice for 30
min to complete lysis. The cell lysates were centrifuged at 12,000
× g for 10 min at 4°C, and luminescence was measured using a Pi-102
fluorescence luminometer (Hygiena, Camarillo, CA, USA) immediately
following mixing. The content of the cellular ATP level was
calculated using the ATP standard curve. ATP content was normalized
to protein concentration.
MMP assay
Breakdown of the MMP was monitored using a JC-1
Mitochondrial Membrane Potential assay kit (Beyotime Institute of
Biotechnology). The cells were incubated with freshly prepared JC-1
solution (10 mg/ml in culture medium) for 20 min at 37°C under 5%
CO2 in the dark, and fluorescence was quantified by flow
cytometry (BD Biosciences). In healthy mitochondria with normal
MMP, JC-1 forms aggregates that emit red fluorescence
(excitation/emission, 490/530 nm). However, in the apoptotic and
necrotic cells with the loss of MMP, JC-1 does not aggregate and
remains in the monomeric form that emits green fluorescence.
Therefore, the increase in green florescence was due to the loss of
MMP.
Detection of intracellular ROS
levels
The Reactive Oxygen Species assay kit (Beyotime
Institute of Biotechnology) was used to determine the accumulation
of intracellular ROS, according to the manufacturer's protocol.
Briefly, cells were loaded with 10 µm
2′,7′-dichlorodihydrofluorescein diacetate at 37°C for 30 min in
the dark, followed by three washes in PBS, and were resuspended in
300 µl PBS and filtered. The fluorescence intensity of the
resulting 2′,7′-dichlorofluorescein was determined using flow
cytometry (BD Biosciences).
Pyrrolidinedithiocarbamate (PDTC)
assay
To validate the association between NF-κB and COX-1,
the specific inhibitor of NF-κB, PDTC, was dissolved in PBS. Cells
(1.5×105) were incubated with 50 or 100 µM PDTC for 48
h, and the protein expression of IκBα, phospho-IκBα, total p65,
phospho-p65 and COX-1 was determined by western blot analysis.
Co-immunoprecipitation (co-IP)
assay
The co-IP assay was performed using Protein
A+G-agarose beads (Beyotime Institute of Biotechnology), according
to the manufacturer's protocol. Briefly, HCT116 or HT29 cells were
collected and incubated on ice for 30 min in 300 µl native lysis
buffer and centrifuged at 12,000 × g for 15 min at 4°C. A 40 µl
volume of supernatant was used as input and the remainder of the
supernatant was precleared by mixing with 1 µg normal IgG and 30 µl
Protein A+G-agarose beads at 4°C for 4 h. The beads were pelleted
and the supernatant was retained for co-IP by incubation with 3 µg
anti-COX-1 antibody (1.5 mg/ml; cat. no. ab695; Abcam) at 4°C
overnight, or with 3 µg IgG (1 mg/ml; cat. no. A7028; Beyotime
Institute of Biotechnology) as the negative control. Then, Protein
A+G-agarose beads were added to above mixture for 4 h at 4°C. The
beads were collected and washed with ice-cold PBS five times, and
boiled at 95°C for 5 min in 60 µl 1X loading buffer. Western
blotting was performed to detect p65, phospho-p65 and COX-1.
Statistical analysis
Results are presented as the mean ± standard error
of the mean obtained from at least three independent experiments.
Statistical significance was determined by one-way analysis of
variance with Dunnett's analysis using SPSS software (version 19.0;
IBM Corp., Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
COX-1 is required for maintaining CRC
malignant characteristics
To investigate whether human COX-1 is associated
with the tumorigenic properties of CRC, the expression of COX-1 in
colorectal tumor samples and matching normal tissues was
investigated. The results indicated that COX-1 was more markedly
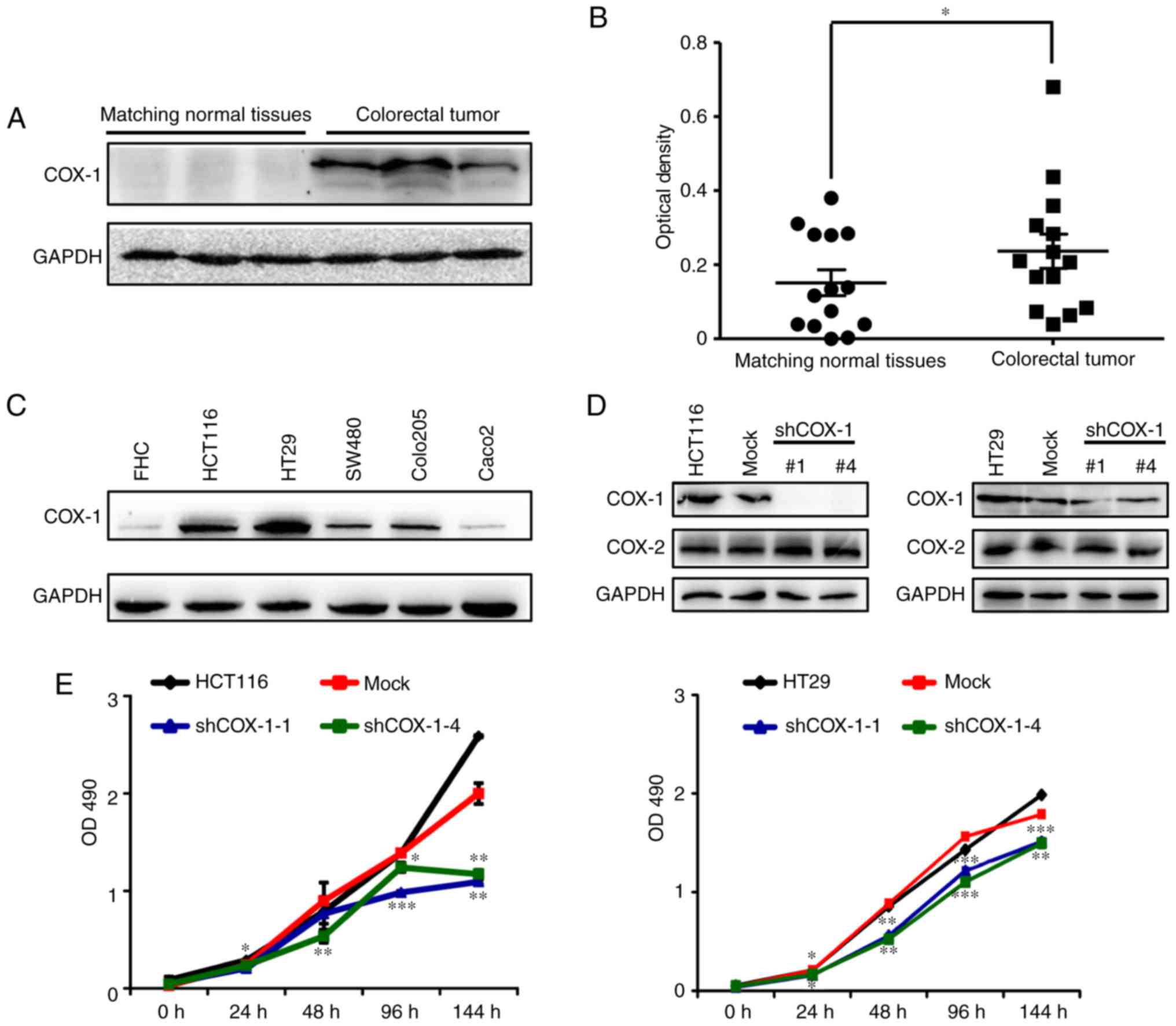
expressed in CRC tissues compared with in normal tissues (Fig. 1A). COX-1 expression was determined
to be significantly increased in the 14 colorectal tumor tissues
(Fig. 1B). Next, the expression of
COX-1 in five CRC cell lines (Caco2, Colo205, SW480, HT29 and
HCT116) and a normal intestinal epithelial cell FHC was
investigated. Consistent with the results in tissues, the CRC cells
expressed increased COX-1 levels compared with FHC cells (Fig. 1C), particularly in HT29 and HCT116
cells, which were therefore selected for shRNA interference for
subsequent assays. Together, these results indicate that COX-1
serves an important function in maintaining the malignant
characteristics in CRC.
COX-1 knockdown suppresses
proliferation of HCT116 and HT29 CRC cells
To investigate whether the decreased expression of
COX-1 affects the development of CRC tumors, COX-1 was depleted in
HCT116 and HT29 CRC cells using four specific shRNAs (termed
shCOX-1-1, shCOX-1-2, shCOX-1-3 and shCOX-1-4, respectively) and
negative control (plasmid without COX-1 shRNA, termed mock) to
establish the stable shCOX-1-knockdown HCT116 and HT29 cell lines
with lentivirus particles. It was identified that COX-1 was
markedly downregulated by shCOX-1-1 and shCOX-1-4 as determined by
western blotting in HCT116 and HT29 cells. However, the expression
of COX-2 protein was unchanged (Fig.
1D). It confirmed that the shRNAs specifically targeted the
COX-1 in HCT116 and HT29 CRC cells. It was investigated whether
COX-1 inhibition was able to decrease the viability of HCT116 and
HT29 CRC cells. An MTT assay was used to quantify cell
proliferation at 0, 24, 48, 96 and 144 h after shRNA knockdown. The
results indicated that COX-1 downregulation significantly inhibited
the cell proliferation in the two cell lines (Fig. 1E). These observations indicated that
COX-1 is required for the proliferation of HCT116 and HT29 CRC
cells.
COX-1 downregulation depolarizes MMP,
eliminates ATP production and increases intracellular ROS
The significant decrease in cell proliferation
raised the hypothesis that depletion of COX-1 alters mitochondrial
function in CRC cells, as mitochondria provide energy to support
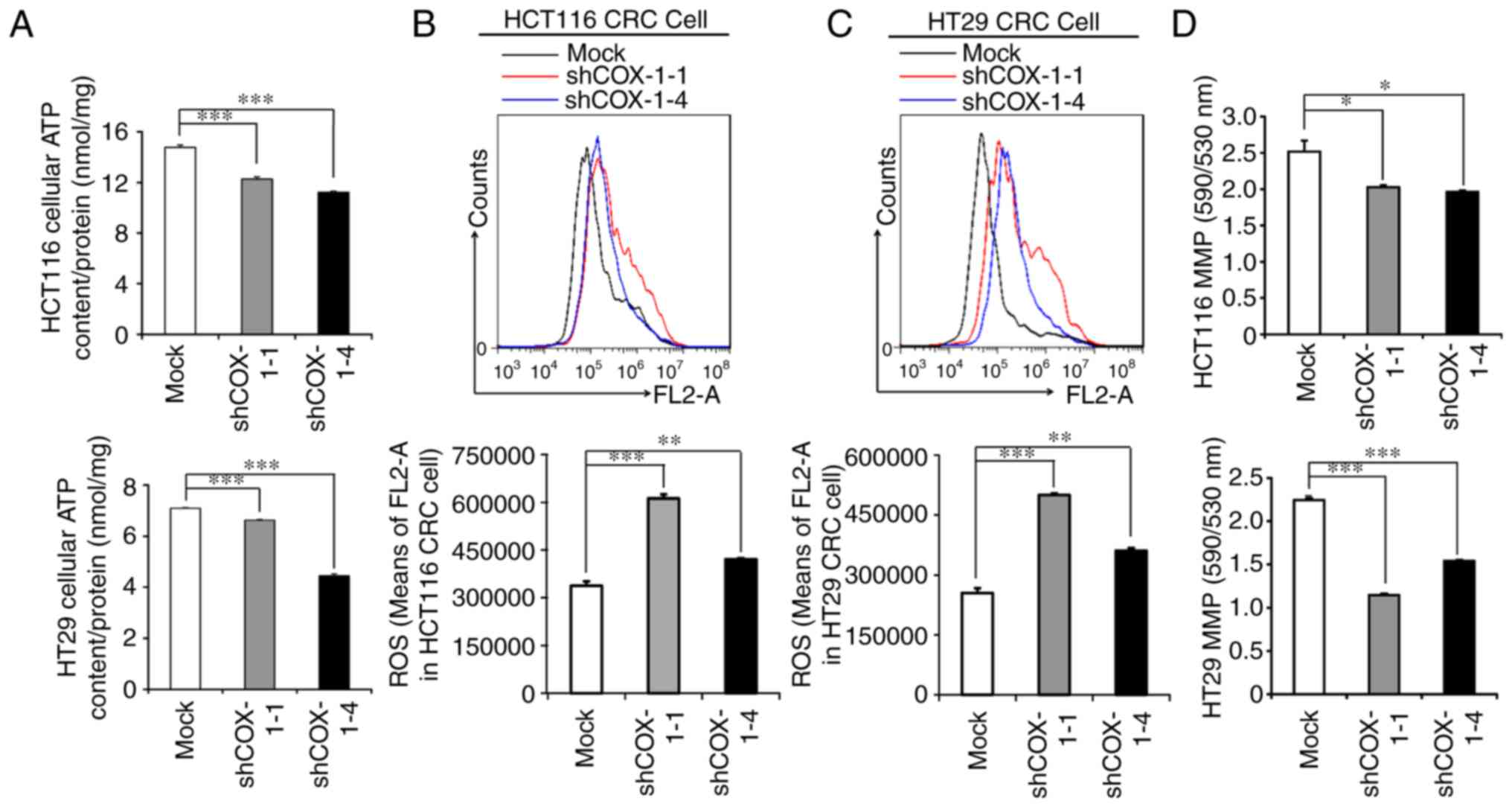
the cell proliferation. It was identified that the generation of
ATP was significantly inhibited in HCT116 and HT29 CRC cells
treated with shCOX-1-1 and shCOX-1-4 compared with the mock
(Fig. 2A). In addition, ROS
production was significantly induced in COX-1-knockdown cells
(Fig. 2B and C). Mitochondrial
membrane integrity by MMP was investigated using the cationic
mitochondrial dye JC-1; the COX-1-knockdown CRC cells exhibited
depolarization of mitochondria and loss of MMP (Fig. 2D). These results suggested that
depletion of COX-1 led to mitochondrial dysfunction in CRC
cells.
COX-1 downregulation halts cells in
G0/G1 phase and triggers apoptosis
A significant accumulation in
G0/G1 phase and a reciprocal decrease in S
phase were observed in shCOX-1-treated HCT116 and HT29 CRC cells,
compared with the control. In addition, markedly decreased
expression of cyclin E2 in the COX-1-knockdown HCT116
and HT29 cells was observed (Fig. 3A
and B). These results indicated that the inhibitory effect of
COX-1 knockdown on cell proliferation was partially due to
G0/G1 phase arrest caused by the decreased
expression of cyclin E2. Induction of apoptosis in
COX-1-downregulated cells was investigated using Annexin V/PI dual
staining using an epifluorescence microscope; the early apoptotic
cells (green color) and the late apoptotic cells (green and red
color) were increased in shCOX-1-treated HCT116 and HT29 cells
(Fig. 3C and D). Collectively,
these results indicated that cell cycle and apoptosis were involved
in the proliferation mediated by shCOX-1 depletion in HCT116 and
HT29 cells.
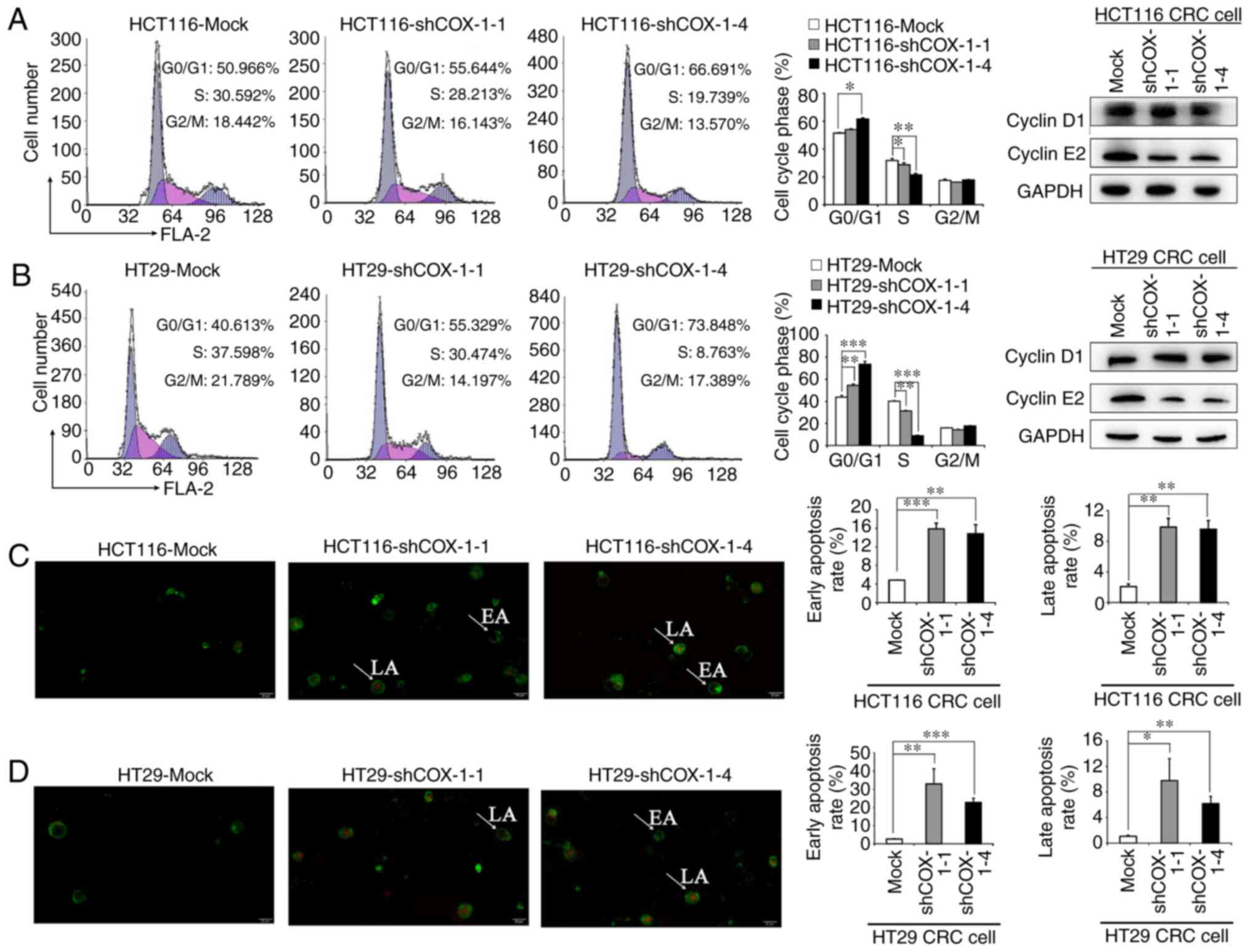 | Figure 3.Depletion of COX-1 induces cell cycle
arrest and triggers apoptosis in HCT116 and HT29 CRC cells.
Downregulation of COX-1 induced G0/G1 arrest
in (A) HCT116 and (B) HT29 CRC cells, as determined by flow
cytometry, and expression of cell cycle-associated proteins cyclin
D1 and cyclin E2 as determined by western
blotting. Apoptosis assay of (C) HCT116 and (D) HT29 CRC cells with
fluorescein isothiocyanate-Annexin V/propidium iodide double
staining visualized under a fluorescence microscope. Representative
fluorescence images analysis are presented. *P<0.05,
**P<0.01, ***P<0.001. Original magnification, ×20. COX-1,
cyclooxygenase 1; CRC, colorectal cancer; sh, short hairpin RNA;
EA, early apoptosis; LA, late apoptosis. |
COX-1 downregulation activates
caspase-dependent mitochondrial apoptosis
The aforementioned results indicated that the
apoptosis triggered by downregulation of COX-1 serves an important
function in HCT116 and HT29 CRC cell proliferation. Thus, the
apoptotic pathway involved in COX-1-regulated cell death was
investigated, focusing on initiator pro-caspase-9 and effector
pro-caspase-3, as well as their substrate PARP. The western blot
results indicated that depletion of COX-1 in HCT116 and HT29 cells
promoted the accumulation of cleaved executioner caspases-9 and −3,
and PARP fragment, and decreased the precursor proteins
pro-caspase-9 and −3, and full-length PARP (Fig. 4A and B). It suggested that a marked
proportion of the precursors were cleaved to execute the apoptotic
process following stimulation by COX-1 knockdown. In addition, it
was revealed that the expression of anti-apoptotic Bcl-2 was
markedly downregulated and a marked increase in pro-apoptotic Bax
was observed upon COX-1 depletion which usually regulated the
intrinsic apoptosis through the mitochondrial pathway (Fig. 4A and B). Thus, the Bax/Bcl-2 ratio
was increased, indicating that mitochondrial apoptosis was induced
upon COX-1 depletion. Bcl-2 antagonizes the dimerization of Bax to
prevent the loss of MMP and block the release of cytochrome
c from the mitochondria to the cytosol. The release of
cytochrome c initiates the caspase cascade cleavage as a
hallmark of the mitochondrial apoptotic pathway. Consistent with
this, an increase in cytosolic cytochrome c and a decrease
in mitochondria cytochrome c were observed (Fig. 4C and D). Taken together, the results
revealed that depletion of COX-1 in HCT116 and HT29 cells led to
mitochondrial dysfunction with the depolarized MMP, less ATP and
excessive ROS stress. At the molecular level, the Bax/Bcl-2 ratio
was increased, the release of cytochrome c was initiated and
the caspase cascade was activated, which ultimately resulted in
apoptotic cell death.
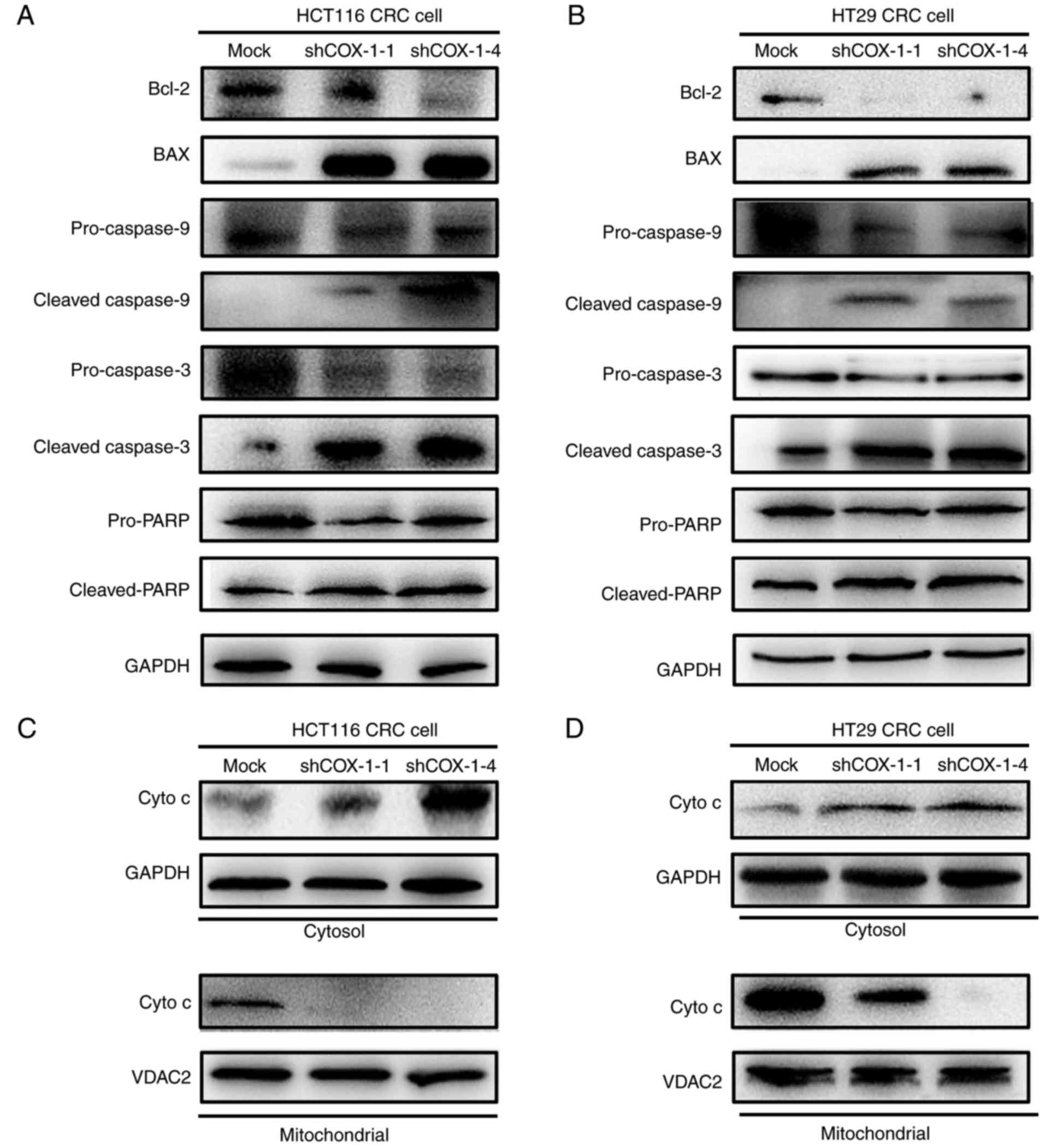 | Figure 4.Downregulation of COX-1 induces
apoptotic cell death via the intrinsic mitochondrial and
caspase-dependent pathway. Cells were harvested and lysed for
determination of the expression of apoptosis-associated proteins
Bcl-2, Bax, procasapse-9, cleaved caspase-9, procasapse-3, cleaved
caspase-3, pro-PARP, cleaved PARP in (A) HCT116 and (B) HT29 CRC
cells, and cytosolic cytochrome c and mitochondrial
cytochrome c in (C) HCT116 and (D) HT29 CRC cells, by
western blotting. GAPDH was used as a loading control for cytosolic
proteins and VDAC2 was used as a loading control for mitochondrial
proteins. COX-1, cyclooxygenase 1; Bcl-2, B-cell lymphoma 2; Bax,
Bcl-2-associated X protein; PARP, poly(ADP-ribose) polymerase; CRC,
colorectal cancer; VDAC2, voltage-dependent anion channel 2; sh,
short hairpin RNA; cyto c, cytochrome c. |
Depletion of COX-1 in CRC cells
induces apoptosis via the NF-κB signaling pathway
Considering that the NF-κB signaling pathway is
involved in a variety of cellular processes including apoptosis,
proliferation, cell survival and invasion, combined with the fact
that the anti-apoptotic gene Bcl-2 is the downstream target of the
NF-κB signaling pathway, we hypothesized that the effects of COX-1
knockdown on cell proliferation may be associated with the
activation of the NF-κB signaling pathway. It is well-known that
the p65 subunit of NF-κB is phosphorylated and translocates to
nucleus when the NF-κB signaling pathway was activated. In the
present study, the phosphorylation of the p65 subunit was
suppressed in shCOX-1-transfected HCT116 and HT29 CRC cells
compared with the control, whereas the total p65 level was stable
following COX-1 depletion (Fig.
5A). By contrast, the expression of the oncogene c-Myc
(Fig. 5A) and Bcl-2 were
downregulated (Fig. 4A and B) in
COX-1-knockdown cells compared with the control. These results
indicated that COX-1 downregulation induced apoptosis and decreased
cell proliferation, in part, via inhibiting the phosphorylation of
the p65 subunit of NF-κB. Furthermore, a specific inhibitor of
NF-κB, PDTC, was used to validate the association between NF-κB and
COX-1. It was identified that the phosphorylated IκBα subunit,
total p65 and phosphorylated p65 were markedly inhibited by PDTC;
however, the expression of total IκBα was not altered with 48 h of
treatment with either 50 or 100 µM PDTC. The expression of COX-1
was also markedly decreased following PDTC treatment (Fig. 5B). These results suggested that
knockdown of COX-1 suppressed the phosphorylation of p65 subunit of
NF-κB, which in turn downregulated the downstream Bcl-2 and c-Myc.
However, the results of the co-immunoprecipitation assay indicated
that there was no direct interaction between COX-1 and the
phospho-p65/p65 subunit of NF-κB (data not shown). We therefore
hypothesized that there are intermediate signaling proteins between
COX-1 and NF-κB.
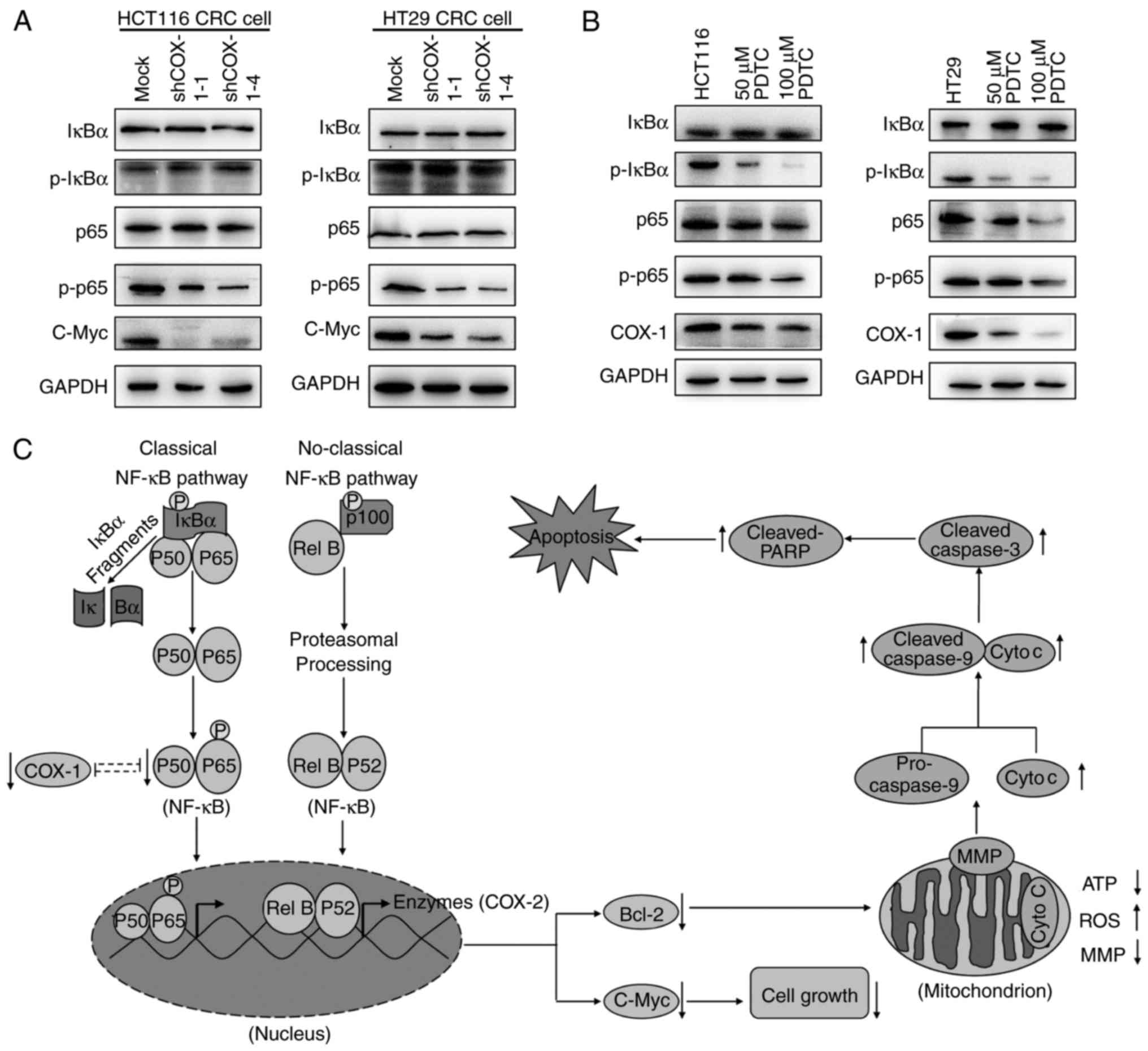 | Figure 5.Depletion of COX-1 suppresses the
activation of the NF-κB signaling pathway. (A) Immunoblot analysis
of NF-κB signaling pathway-associated genes in COX-1-knockdown
HCT116 and HT29 CRC cells. (B) HCT116 and HT29 CRC cells were
treated with PDTC, a p65 (NF-κB)-specific inhibitor. (C) Schematic
diagram indicating potential molecular mechanisms by which
knockdown of COX-1 induces apoptosis in HCT116 and HT29 CRC cells.
COX-1, cyclooxygenase 1; NF-κB, nuclear factor κB; PDTC,
pyrrolidinedithiocarbamate; IκBα, inhibitor of NF-κB α; p-,
phospho-; sh, short hairpin RNA; Cyto c, cytochrome c; ROS,
reactive oxygen species; ATP, adenosine triphosphate; MMP,
mitochondrial membrane potential; Bcl-2, B-cell lymphoma 2; PARP,
poly(ADP-ribose) polymerase. |
Discussion
The incidence of CRC has increased as a result of
the change in diets and lifestyles in society. Surgery combined
with chemotherapy and radiotherapy is a useful treatment for
advanced-stage (III and IV) CRC. However, chemoresistance is the
major obstacle hindering effective therapy of patients with
advanced CRC, with poor survival of patients and frequent relapse.
Therefore, searching for highly effective antitumor targets and
novel therapeutic anticancer agents is an urgent requirement. COX-2
has been intensively investigated as an essential factor involved
in the development of various types of cancer; however, COX-1 has
emerged as exhibiting an important function in tumor progression,
particularly in colon carcinogenesis. Wu et al (17) indicted that treatment of colon
cancer cells with a COX-1 inhibitor decreased cell proliferation,
and induced alterations in morphological and biochemical
characteristics associated with cell cycle arrest and
macroautophagy. Li et al (10) identified that COX-1 is required for
the maintenance of malignant characteristics of colon cancer cells
or tumor promoter-induced transformation of pre-neoplastic cells,
and the specific COX-1 inhibitor as a potential preventive agent
against CRC. Chulada et al (8) observed that knockout of COX-1 markedly
decreased the incidence of polyposis in Min mice to suppress
colorectal carcinogenesis. Clinical studies indicated that
inhibiting COX-1 and COX-2 with conventional NSAIDs to decrease the
number of intestinal polyps in patients with familial adenomatous
polyposis is more effective than with COX-2-selective inhibitors
only (18). However, the precise
molecular mechanism underlying COX-1 regulation in CRC remains to
be determined. The results of the present study indicated that
COX-1 is upregulated in CRC clinical tissues and cell lines, which
indicated COX-1 may be required for maintaining malignant
characteristics of CRC cells. To investigate this hypothesis, a DNA
vector-based COX-1-specific shRNA approach was used to knock down
COX-1 to determine the biological consequences in HCT116 and HT29
CRC cells. An MTT assay was used to determine the inhibition of
proliferation in COX-1-knockdown CRC cells, and flow cytometric
analysis revealed G0/G1 arrest, which
indicated COX-1-mediated cell proliferation in CRC.
Mitochondrial metabolism was identified to
participate in various biological processes. Mitochondrial DNA has
been used as a promising strategy for cancer therapy and a number
of physical and chemical stimuli causing mitochondrial dysfunction
have been reported (19). In
healthy cells, the inner mitochondrial membrane is nearly
impermeable to all ions, which contribute to the robust formation
of an electrochemical gradient resulting in the MMP required for
ATP synthesis. However, the long-lasting opening of the
mitochondrial permeability transition pore will lead to permanent
MMP dissipation and cell death (20). In the present study, it was
demonstrated that depletion of COX-1 depolarized the MMP, increased
the accumulation of ROS and diminished ATP. ROS typically promote
mitochondrial dysfunction and serve an upstream function in the
inhibition of proliferation and mitochondria-mediated apoptosis
(21). Accumulation of
intracellular ROS altered the mitochondrial membrane integrity and
depolarized MMP, which caused mitochondrial swelling and increased
permeability of the mitochondrial membrane, led to cytochrome
c leaks from the perforated mitochondria and induced
mitochondrial intrinsic apoptosis (also called the
mitochondria-mediated pathway of apoptosis). The leaked cytochrome
c leads to formation of an apoptosome which activates
pro-caspase-9 and initiates the chain reaction of the classical
caspase cascade, defined as caspase dependent-apoptosis (22). COX-1 knockdown in the present study
induced caspase-dependent apoptosis with upregulation of cytosolic
cytochrome c, caspase-9 and −3 activation, and PARP
cleavage, along with downregulation of mitochondria cytochrome
c, which indicated that cytochrome c leaks from the
mitochondria to the cytosol. In addition, Bcl-2 family members are
key factors in mitochondrial function, cell cycle and apoptosis
(23,24). In the present study, it was also
revealed that downregulation of COX-1 in CRC cells significantly
decreased anti-apoptotic Bcl-2 expression and increased
pro-apoptotic Bax expression, with deleterious mitochondrial
function. It is generally accepted that the imbalance between the
expression of Bcl-2 and Bax proteins leads to permeabilization of
the mitochondrial outer membrane, followed by release of cytochrome
c and subsequent cell death (25). These alterations indicated that
COX-1 induced mitochondrial apoptosis by mediating Bcl-2 family
members in CRC cells. Taken together, the results of the present
study shed new light on the association between COX-1 and cell
death in CRC.
In CRC, NF-κB was constitutively activated; aberrant
NF-κB activation is involved in enhanced proliferation (regulating
c-Myc), evasion of apoptosis (regulating Bcl-2 and Bax), invasion
(regulating vascular endothelial growth factor) and angiogenesis
(regulating cytokines and chemokines) by regulating the expression
of diverse target genes (14). It
is suggested that a series of pharmacological NF-κB inhibitors may
be potential anticancer agents in cancer therapy (26). Communication between COX-2 and
upstream NF-κB has been suggested previously, with results
indicating that COX-2 is increased in CRC to promote angiogenesis
induced by NF-κB (16). The
interaction between COX-2 and NF-κB occurs directly; the COX-2
promoter contains a consensus binding sequence (−223/-214,
GGGACTACCC) for NF-κB (27). COX-2
is one of the target genes for non-classical NF-κB pathway that
primarily targets p52-RelB dimers translocated into the nucleus and
activated the target genes for regulating lymphoid organogenesis
and maintaining the malignant phenotype in certain types of cancer
(14). However, COX-1 lacks the
corresponding promoter elements for NF-κB (28). In the present study, the specific
inhibition of COX-1 by shRNA led to downregulation of
phosphorylation of the p65 subunit of NF-κB. Furthermore,
phosphorylation of p65, as well as COX-1, was markedly inhibited by
the NF-κB inhibitor PDTC. These results indicated that COX-1
activated NF-κB through phosphorylation of its subunit p65, and
could also respond to NF-κB signaling as a feedback loop. However,
co-immunoprecipitation assays were performed, and the results
indicated that there was no direct interaction between COX-1 and
the phospho-p65/p65 subunit of NF-κB. We therefore hypothesized
that certain intermediate signaling proteins must exist between
COX-1 and NF-κB. A previous study indicated that epidermal growth
factor (EGF) receptor (EGFR) signaling pathways were partial
inhibitors in EGF-induced neoplastic transformation by
downregulation of COX-1 in CRC (10). In addition, constitutive EGFR
signaling activates NF-κB through the IκBα phosphorylation on
Ser32/Ser36, thereby affecting the
translocation of p65 subunit signal to the nucleus in prostate
cancer cells (29). Therefore, we
hypothesized that the EGFR/NF-κB signaling pathway may be the
intermediate signaling pathway between COX-1 and NF-κB; however,
this requires verification. We also hypothesize that the different
responses to NF-κB of COX-1 and COX-2 are based on their
differences in the promoter region; COX-2 is the target gene of
non-classical NF-κB pathway through p52, and COX-1 may be the
target gene of the classical NF-κB pathway involved in regulating
cell survival and carcinogenesis through p65.
In summary, the results of the present study
identified an association between COX-1 and CRC. COX-1 was
upregulated in CRC tissues and cells, and deletion of COX-1 from
CRC cells inhibited proliferation with G0/G1
arrest and triggered the caspase-dependent apoptotic response to
mitochondrial dysfunction. The cell proliferation assay, flow
cytometry analysis of cell cycle and apoptotic analysis indicated
that COX-1 inhibition could significantly decrease the viability of
HCT116 and HT29 CRC cells. Therefore, we hypothesize that depletion
of COX-1 in HCT116 and HT29 CRC cells will suppress tumor growth in
mice, which requires validation using xenograft mice models. In
addition, the results of the present study revealed that COX-1
downregulation could deactivate the NF-κB signaling pathway by
inhibiting the phosphorylation of the p65 subunit, that in turn
suppresses the expression of its downstream anti-apoptosis gene
Bcl-2, and decreases the Bcl-2/Bax ratio to promote the release of
cytochrome c from the intermembrane space of mitochondria
into the cytosol, together with the cleavage of caspase-9 and-3,
and PARP. Finally, all these responses induced by COX-1 knockdown
contributed to the inhibition of cell proliferation. The present
study provides an improved understanding of the diverse pathways
including apoptosis and mitochondrial metabolism for the
involvement of COX-1 in CRC, indicating the potential for COX-1 as
a target for CRC therapy.
Acknowledgements
Not applicable.
Funding
The present study was financially supported by the
National Natural Science Foundation of China (grant nos. 81760507,
81360310, 81360175 and 81660583), Key Project of Yunnan Education
Department (grant no. ZD2015002) and Natural Scientific Research
Foundation of Yunnan Education Department (grant no. 2015Z019).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
LD, HG, ZL and QC conceived and designed the
research, collected and assembled the data, and wrote the
manuscript. LD, HG and ZL performed the experiments. LD and QC
reviewed and edited the manuscript. QL, WW, JR, MY and JL analyzed
the data. All authors have read and approved the content of the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Written informed consent for the present study use
of tissue specimens was obtained from all patients, and ethical
approval was approved from the Ethics Committees of Pu'er City
People's Hospital (Pu'er, China) and School of Medicine in Yunnan
University (Kunming China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lloyd SC, Harvey NR, Hebert JR, Daguise V,
Williams D and Scott DB: Racial disparities in colon cancer.
Primary care endoscopy as a tool to increase screening rates among
minority patients. Cancer. 109:378–385. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xiang L, Wang S, Jin X, Duan W, Ding X and
Zheng C: Expression of BMP2, TLR3, TLR4 and COX2 in colorectal
polyps, adenoma and adenocarcinoma. Mol Med Rep. 6:973–976. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
O'Connell JB, Maggard MA and Ko CY: Colon
cancer survival rates with the new American Joint Committee on
Cancer sixth edition staging. J Natl Cancer Inst. 96:1420–1425.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ricciotti E and FitzGerald GA:
Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol.
31:986–1000. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chapple KS, Cartwright EJ, Hawcroft G,
Tisbury A, Bonifer C, Scott N, Windsor AC, Guillou PJ, Markham AF,
Coletta PL, et al: Localization of cyclooxygenase-2 in human
sporadic colorectal adenomas. Am J Pathol. 156:545–553. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tsikas D, Tewes KS, Gutzki FM, Schwedhelm
E, Greipel J and Frolich JC: Gas chromatographic-tandem mass
spectrometric determination of acetylsalicylic acid in human plasma
after oral administration of low-dose aspirin and guaimesal. J
Chromatogr B Biomed Sci Appl. 709:79–88. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chulada PC, Thompson MB, Mahler JF, Doyle
CM, Gaul BW, Lee C, Tiano HF, Morham SG, Smithies O and Langenbach
R: Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces
intestinal tumorigenesis in Min mice. Cancer Res. 60:4705–4708.
2000.PubMed/NCBI
|
|
9
|
Riehl TE, George RJ, Sturmoski MA, May R,
Dieckgraefe B, Anant S and Houchen CW: Azoxymethane protects
intestinal stem cells and reduces crypt epithelial mitosis through
a COX-1-dependent mechanism. Am J Physiol Gastrointest Liver
Physiol. 291:G1062–G1070. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li H, Zhu F, Chen H, Cheng KW, Zykova T,
Oi N, Lubet RA, Bode AM, Wang M and Dong Z:
6-C-(E-phenylethenyl)-naringenin suppresses colorectal cancer
growth by inhibiting cyclooxygenase-1. Cancer Res. 74:243–252.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zahedifard M, Faraj FL, Paydar M, Yeng
Looi C, Hajrezaei M, Hasanpourghadi M, Kamalidehghan B, Abdul Majid
N, Mohd Ali H and Ameen Abdulla M: Synthesis, characterization and
apoptotic activity of quinazolinone Schiff base derivatives toward
MCF-7 cells via intrinsic and extrinsic apoptosis pathways. Sci
Rep. 5:pp. 115442015, View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Martinou JC and Youle RJ: Mitochondria in
apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang S, Liu Z, Wang L and Zhang X:
NF-kappaB signaling pathway, inflammation and colorectal cancer.
Cell Mol Immunol. 6:327–334. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Southern SL, Collard TJ, Urban BC, Skeen
VR, Smartt HJ, Hague A, Oakley F, Townsend PA, Perkins ND,
Paraskeva C and Williams AC: BAG-1 interacts with the p50-p50
homodimeric NF-κB complex: Implications for colorectal
carcinogenesis. Oncogene. 31:2761–2772. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tsujii M, Kawano S, Tsuji S, Sawaoka H,
Hori M and DuBois RN: Cyclooxygenase regulates angiogenesis induced
by colon cancer cells. Cell. 93:705–716. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu WK, Sung JJ, Wu YC, Li HT, Yu L, Li ZJ
and Cho CH: Inhibition of cyclooxygenase-1 lowers proliferation and
induces macroautophagy in colon cancer cells. Biochem Biophys Res
Commun. 382:79–84. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dolara P, Caderni G and Tonelli F:
Nimesulide, a selective anti-inflammatory cyclooxygenase-2
inhibitor, does not affect polyp number and mucosal proliferation
in familial adenomatous polyposis. Scand J Gastroenterol.
34:11681999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yusnita Y, Norsiah MD and Rahman AJ:
Mutations in mitochondrial NADH dehydrogenase subunit 1 (mtND1)
gene in colorectal carcinoma. Malays J Pathol. 32:103–110.
2010.PubMed/NCBI
|
|
20
|
Kroemer G, Galluzzi L and Brenner C:
Mitochondrial membrane permeabilization in cell death. Physiol Rev.
87:99–163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liang W, Cai A, Chen G, Xi H, Wu X, Cui J,
Zhang K, Zhao X, Yu J, Wei B, et al: Shikonin induces
mitochondria-mediated apoptosis and enhances chemotherapeutic
sensitivity of gastric cancer through reactive oxygen species. Sci
Rep. 6:382672016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cregan SP, Dawson VL and Slack RS: Role of
AIF in caspase-dependent and caspase-independent cell death.
Oncogene. 23:2785–2796. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fu XF, Yao K, Du X, Li Y, Yang XY, Yu M,
Li MZ and Cui QH: PGC-1α regulates the cell cycle through ATP and
ROS in CH1 cells. J Zhejiang Univ Sci B. 17:136–146. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Du X, Fu X, Yao K, Lan Z, Xu H, Cui Q and
Yang E: Bcl-2 delays cell cycle through mitochondrial ATP and ROS.
Cell Cycle. 16:707–713. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Goldar S, Khaniani MS, Derakhshan SM and
Baradaran B: Molecular mechanisms of apoptosis and roles in cancer
development and treatment. Asian Pac J Cancer Prev. 16:2129–2144.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Baud V and Karin M: Is NF-kappaB a good
target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov.
8:33–40. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Reddy ST, Wadleigh DJ and Herschman HR:
Transcriptional regulation of the cyclooxygenase-2 gene in
activated mast cells. J Biol Chem. 275:3107–3113. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Griswold DE and Adams JL: Constitutive
cyclooxygenase (COX-1) and inducible cyclooxygenase (COX-2):
Rationale for selective inhibition and progress to date. Med Res
Rev. 16:181–206. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shostak K and Chariot A: EG FR N F-κB
Partners in cancer. Trends Mol Med. 21:385–393. 2015. View Article : Google Scholar : PubMed/NCBI
|