Introduction
Breast cancer is among the most frequently diagnosed
cancer types and the leading cause of cancer mortalities among
females based on the estimates of cancer incidence and mortality
globally for 36 cancer types in 185 countries, which was produced
by the International Agency for Research on Cancer in 2018
(1). In 2012, 1.7 million females
were diagnosed with breast cancer, and there were 6.3 million
females alive who had been diagnosed with breast cancer in the
previous five years globally (2).
Surgery is the most common primary therapy for cancerous lesions of
the breast, followed by radiotherapy or chemotherapy if metastasis
to lymph nodes or other organs is detected (3). Adjuvant hormone therapy, also termed
endocrine therapy, is considered a standard therapy for ~75% of
patients with tumors expressing estrogen receptor (ER) and
progesterone receptor (PR) (4).
However, approximately half of females with ER-positive (ER+ve)
cancer exhibit intrinsic resistance (de novo) to endocrine
therapy and the majority of patients who initially respond develop
acquired resistance during the course of treatment with the
anti-estrogen agents (5). These
refractive patients, together with the patients with ER-negative
(ER-ve) cancer, form a large population, which has poor clinical
outcomes and survival rates, and require other forms of treatment,
primarily chemotherapy.
Chemotherapeutic regimens primarily consist of a
combination of ≥2 cytotoxic drugs (6). Although they have various side
effects, treatment with chemotherapeutic drugs has improved patient
survival and reduced annual relative risk of relapse and mortality
in North America in 2013 (3,7). Drugs
in the taxane class, including paclitaxel, are among the most
frequently used agents in the treatment of various cancer types,
including breast cancer (8).
Paclitaxel (Taxol) exerts its anticancer activity through binding
to specific pockets within β-tubulin, thus stabilizing microtubules
and preventing their depolymerization (7). This results in inhibition of cellular
processes that are dependent on microtubule turnover, including
inhibition of the cellular transition from G0 to
G1, arresting the cell in the G2/M phase,
inhibition of mitosis and eventually induction of apoptosis
(8–10). Paclitaxel also interferes with
mitosis by inducing mitotic block at the metaphase/anaphase
boundary, and forming an incomplete metaphase plate of chromosomes
and an abnormal organization of spindle microtubules (11). Microtubules of different statuses,
including cytoskeletal microtubules and mitotic spindles, may have
different sensitivities to paclitaxel; thus, the concentration of
paclitaxel may be the major determinant of its apoptogenic
mechanisms (12). Paclitaxel may
also exert its killing activity through a gene-directed process,
which is a pathway completely independent of microtubules (11).
The use of paclitaxel is associated with a number of
serious dose-dependent side effects, including paclitaxel-induced
peripheral neuropathy (PIPN), which may necessitate dose reduction
or withdrawal during the course of chemotherapy (13,14).
However, there are no clinically-proven drugs for the prevention of
PIPN, and the best available data support a moderate recommendation
for duloxetine to treat PIPN (14,15).
Therefore, further research is warranted to determine agents that
can relieve PIPN or assist with reducing the doses of paclitaxel
necessary to treat cancer, and thus indirectly reducing the risk of
developing dose-limiting side effects, including PIPN. We
previously observed that co-administration of paclitaxel with
chemically modified tetracycline-3 (COL-3) inhibited the
development of paclitaxel-induced thermal hyperalgesia in mice,
indicating that COL-3 can be used for the prevention of PIPN
(16). COL-3 has matrix
metalloproteinase (MMP) inhibitory properties (17,18),
which resulted in a number of researchers investigating its
anticancer potential in a variety of cancer types, including
melanoma, and lung, breast and prostate cancer (19,20).
Taking this into consideration, it is plausible that the
combination of COL-3 with paclitaxel may have additive or
synergistic activity against a number of cancer types, which would
demonstrate a double advantage of reducing side effects and
increasing antitumor efficacy. Thus, the aim of the present study
was to evaluate the impact of COL-3 on the anticancer activity of
paclitaxel using various ER+ve and ER-ve breast cancer cell lines
in vitro.
Materials and methods
Cell lines
The human breast carcinoma cell lines MCF-7 (ER+ve)
and MDA-MB-231 (ER-ve; de novo resistant form) were obtained
from the American Type Culture Collection (Manassas, VA, USA). The
ER downregulated cell line pII (acquired resistant form) was
established in Professor Yunus Luqmani's and Khajah's lab (Kuwait
University, Safat, Kuwait) by transfection of MCF-7 cells with the
ER directed shRNA plasmid, as described previously (21,22).
Cells were maintained in culture medium containing Advanced
Dulbecco's modified Eagle's medium (Advanced DMEM; Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), supplemented with 5%
fetal bovine serum (FBS, Invitrogen; Thermo Fisher Scientific), 6
ml/500 ml penicillin-streptomycin (10,000 U/ml penicillin and
10,000 µg/ml streptomycin), 6 ml/500 ml (200 mM) L-glutamine and 6
ml/500 ml non-essential amino acids (Invitrogen; Thermo Fisher
Scientific, Inc.). All cells were cultured as monolayers in an
incubator at 37°C in an atmosphere containing 5% CO2 and
95% humidity.
Drugs
COL-3 (purchased from Galderma, Research and
Development SNC, Les Templier, France) was dissolved in dimethyl
sulfoxide (DMSO) to a stock concentration of 1 mM and stored at
−20°C in aliquots. Paclitaxel, purchased from Tocris Bioscience
(Bristol, UK), was dissolved in DMSO to a stock concentration of 1
mM and stored at −20°C in aliquots. All experimental incubation
with drugs was conducted at 37°C, 5% CO2 and 95%
humidity in an incubator. The control vehicle used was 0.01%
DMSO.
Proliferation assay
The effect of various concentrations of paclitaxel
(1, 2.5, 5, 10, 25, 50, 100, and 1,000 nM), COL-3 (50, 100, 1,000,
2,500, 5,000, 10,000, and 20,000 nM) or their combination on cell
proliferation was examined using a colorimetric MTT assay (Promega
Corporation, Madison, WI, USA), as previously described (23,24).
In brief, ~1×104 cells were seeded in triplicate wells
and incubated overnight at 37°C in an atmosphere containing 5%
CO2. Subsequently, the medium was removed and the cells
were treated with the vehicle (control), or various concentrations
of paclitaxel, COL-3 or their combination. The growth was assessed
after 72 h of incubation at 37°C in an atmosphere containing 5%
CO2.
Apoptosis assay
The effect of COL-3, paclitaxel or their combination
on pII cell apoptosis was measured using Annexin
V/7-aminoactinomycin D apoptosis detection kit (BD Biosciences,
Franklin Lakes, NJ, USA), as previously described (24).
Cell cycle assay
Subsequently, ~1×104 pII cells were
seeded in triplicate wells and incubated overnight at 37°C in an
atmosphere containing 5% CO2 and 95% humidity. The cells
were then treated with the vehicle or various concentrations of
paclitaxel, COL-3 or their combination. After 72 h of incubation at
37°C in an atmosphere containing 5% CO2, cells were
trypsinized and washed once with ice-cold PBS. The pellet was then
re-suspended with PBS and fixed by adding ice-cold 70% ethanol
while vortexing at 14,000 × g for 20 sec at 4°C. The samples were
then stored at −20°C overnight. The following day, samples were
centrifuged at 66 × g for 15 min at room temperature and washed
once with PBS. Pellets were treated with RNase, incubated for 15
min at 37°C and 200 µl propidium iodide solution (DNA Prep stain
kit; Beckman Coulter, Inc., Brea, CA, USA) was added. The samples
were analyzed using a Cytomics FC500 flow cytometer with a maximum
emission of 605 nm. The DNA content of cell duplicates during the S
phase of the cell cycle, and the relative amount of cells in the
G0 and G1 phases, in the S phase, and in the
G2 and M phases were determined utilizing the
fluorescence of cells in the G2/M phase, which were
twice as high as that of cells in the G0/G1
phase. This was obtained using CXP Software, version 2 (Beckman
Coulter, Inc.).
Matrigel invasion assay
The degree of pII cell invasion through the basement
membrane matrix was determined using the Cultrex®
24-Well basement membrane extract (BME) Cell Invasion assay kit
(Trevigen, Haithersburg, MD, USA). All procedures and reagent
preparations were conducted according to the manufacturer's
protocols. Briefly, insert membranes were coated with 1X BME and
incubated at 37°C overnight. pII cells, that had been serum-starved
overnight, were re-suspended at 1×106 cells/ml in
Advanced DMEM with or without drugs (100, 1,000, 5,000 and 10,000
nM), and 100 µl (1×105 cells) was loaded into the upper
chambers. To the bottom chamber, 500 µl DMEM (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS was added as a
chemoattractant. After 24 h of incubation at 37°C, the media in the
top chambers as well as the bottom chambers were aspirated followed
by washing with 1X wash buffer. Subsequently, 500 µl Cell
Dissociation Solution/Calcein-acetomethylester (AM) complex was
added to the bottom chamber of each well and incubated at 37°C in
an atmosphere containing CO2 for 60 min. Cells
internalize Calcein-AM and intracellular esterases cleave AM moiety
generating free calcein, which fluoresces. The degree of cell
invasion was determined by recording the fluorescence emission
using a microplate reader (Luminometer) with a filter set of
excitation/emission at 485/535 nm.
Proteome profiler analysis
A total of three different proteome profiler array
kits, Human protease array kit, Human phospho-kinase array kit and
Human apoptosis array kit (R&D Systems, Inc., Minneapolis, MN,
USA), were used to determine the expression levels of a number of
groups of proteins in cell extracts/lysates following the
manufacturer's protocols.
Subsequently, ~1×106 pII cells/well were
cultured for 24 h at 37°C in an atmosphere containing 5%
CO2 under the following treatment conditions: Vehicle
(control); 2.5 nM paclitaxel; 5 µM COL-3; or 2.5 nM paclitaxel and
5 µM COL-3. Upon removal of the advanced DMEM, the cell monolayers
were washed once with ice-cold PBS and then lysed using lysis
buffer supplemented with protease inhibitors [1 µg/ml leupeptin, 1
µg/ml aprotinin and 10 µg/ml phenylmethylsulfonyl fluoride (PMSF);
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany]. Cells were
harvested using a sterile disposable rubber cell scraper and
transferred into Eppendorf® tubes. The cell lysates were
then centrifuged at 14,000 × g for 10 min at 4°C, and the
supernatant was transferred to new Eppendorf tubes for subsequent
protein analysis or stored at −80°C for later analysis. Total
protein concentration in the cell lysates was determined with a
standard Bradford assay.
The relative phosphorylated/expression levels of 45
kinases (cat. no. ARY003B), 35 apoptosis-associated proteins (cat.
no. ARY009) and 35 proteases (cat. no. ARY021B) (R&D Systems,
Inc.) were detected using Proteome Profiler™ Human Protease Array
kits aforementioned, according to the manufacturer's protocols.
MMP activity
The general activity of MMPs was determined using
the MMP activity assay kit from Abcam (cat. no. ab112146;
Cambridge, UK), as previously described (25). pII cells were seeded
(0.1×106 cells) into 6-well plates in culture medium at
37°C and allowed to grow to 80% confluence. Cells were serum
starved overnight at 37°C in an atmosphere containing 5%
CO2, and then left untreated, or treated with 2.5 nM
paclitaxel, 5 µM COL-3 or 2.5 nM paclitaxel and 5 µM COL-3 for 24 h
followed by epidermal growth factor (EGF) stimulation (100 ng/ml
for 30 min) at 37°C in an atmosphere containing 5% CO2.
Subsequently, 25 µl media was removed and added to 25 µl 2 mM APMA
working solution, and then incubated for 15 min at 25°C, followed
by the addition of 50 µl green substrate solution. MMP activity was
measured at 10 min intervals for 1 h at 37°C by recording
fluorescence emission using a microplate reader with a filter set
of excitation/emission at 485/535 nm.
DNA fragmentation assay
Cells were seeded (0.1×106 cells) in
6-well plates to 80% confluence, and then incubated with 2.5 nM
paclitaxel, 5 µM COL-3 or a combination regimen (2.5 nM paclitaxel
+ 5 µM COL-3) for 48 h at 37°C in an atmosphere containing 5%
CO2. Cells were then trypsinized, pelleted at 66 × g for
15 min at room temperature and DNA was isolated from the cells
using a DNA isolation kit (Qiagen, Inc., Gaithersburg, MD, USA),
according to the manufacture's protocol. Subsequently, 1 µg DNA was
run on 2% agarose gel stained with ethidium bromide at 50 V, and
the gel was examined under an UV trans-illuminator. Additionally, a
1 µg DNA ladder was also included in the experiment (λ
DNA/HindIII marker; cat. no. SM0102; Thermo Fisher
Scientific, Inc.). Densitometric analysis of the bands intensity
was calculated using ImageJ software, version k 1.45 (National
Institutes of Health, Bethesda, MD, USA).
Western blotting
Cells were cultured (0.1×106 cells; at
37°C in an atmosphere containing 5% CO2) in 6-well
plates to an 80–90% confluence, and treated with vehicle (control),
2.5 nM paclitaxel, 5 µM COL-3 or combination regimen
poly(ADP-ribose) polymerase (2.5 nM paclitaxel + 5 µM COL-3) for 48
h. The medium was subsequently aspirated off, and cell monolayers
were harvested by scraping and re-suspension into 300 µl lysis
buffer containing 50 mM HEPES, 50 mM NaCl, 5 mM EDTA, 1% Triton X,
100 µg/ml PMSF, 10 µg/ml aprotinin and 10 µg/ml leupeptin, and then
stored at −80°C. Protein concentration was determined with a
Bradford assay using bovine serum albumin (BSA; Sigma-Aldrich;
Merck KGaA) as the standard, and 3 µg protein lysate was mixed with
an equal volume of 2X SDS and incubated at 90°C for 10 min. Samples
were then loaded onto 10% SDS-PAGE and electrophoresed at 150 V for
1 h. Subsequently, proteins were transferred to a nitrocellulose
membrane and blocked with 2% BSA for 1 h at room temperature prior
to being incubated overnight at 4°C with primary antibodies
(prepared in 2% BSA) against β-actin (loading control; 1:1,000
dilution), or phospho- or total- B-cell lymphoma-2
(BCL-2)-associated X (BAX), BCL-2 associated agonist of cell death
(BAD), BCL-2 antagonist/killer (BAK), BCL-2, cytochrome c,
AKT, extracellular signal-regulated kinase (ERK)1/2, p38
mitogen-associated protein kinase (MAPK), apoptotic peptidase
activating factor 1 (APAF-1), and heat shock transcription factor-1
(HSF-1), and un-cleaved or cleaved-caspase-3, −8, −9 and PARP (all
from Cell Signaling Technology, Inc., Danvers, MA, USA; 1:100
dilution). The membrane was then washed 3 times 5 min each with TBS
with Tween-20 (20%) and incubated with anti-rabbit horseradish
peroxidase (HRP)-conjugated secondary antibody (1:500 dilution;
Cell Signaling Technology, Inc.) for 1 h at room temperature and
then developed with Super Signal enhanced chemiluminescent (Pierce;
Thermo Fisher Scientific, Inc.) and visualized with Kodak X-ray
film. The catalogue numbers of the used antibodies are as follows:
β-actin, 4970; phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), 4370;
p44/42 MAPK (Erk1/2), 9102, AKT, 9272; phospho-(Ser/Thr) AKT, 9611;
p38 MAPK, 9212; phospho-p38 MAPK (Thr180/Tyr182), 9215; Apaf-1,
8723; Cytochrome c, 11940; HSF1, 4356, BAK, 12105; BAX,
2772; BAD, 9292; BCL-2, 4223; PARP, 9532; cleaved PARP (Asp214),
5625; caspase-9, 9502; Caspace-8, 4790; cleaved caspase-8 (Asp391),
9496; caspase-3, 9662; cleaved caspase-3 (Asp175), 9579; and
anti-rabbit IgG, HRP-conjugated, 7074.
Caspase-3 activity assay
Cells (1×106) were treated with
paclitaxel (2.5 nM), COL-3 (5 µM) or a combination regimen (2.5 nM
paclitaxel + 5 µM COL-3) for 48 h at 37°C in an atmosphere
containing 5% CO2 and then collected by trypsinization
and washed twice with ice-cold PBS. Subsequently, the cells were
centrifuged at 42 × g for 5 min at 4°C and the pellet was washed
with ice-cold lysis buffer. The protein concentration was
calculated using a Bradford assay. Caspase-3 activity in the lysate
of the different treatment conditions was determined using a
caspase-3 colorimetric assay kit from GenScript (cat. no. L00289;
Piscataway, NJ, USA), and was expressed as caspase-3 activity/mg of
protein.
Data analysis
Statistical analyses were performed using one-way
analysis of variance followed by Dunnett's (for comparing between
treatment conditions) or Bonferroni's (for comparing within
treatment conditions) Multiple Comparison Test using GraphPad Prism
software (version 5.0; GraphPad Software, Inc., La Jolla, CA, USA).
P<0.05 was considered to indicate a statistically significant
difference. Using the GraphPad Prism software, the concentration of
paclitaxel or COL-3 that produced the half-maximal response
(IC50) was calculated using non-linear regression
analysis. The data were fitted to a dose-response-inhibition
equation [log (inhibitor) vs. normalized response curve]. Results
were expressed as the mean ± standard error of the mean.
To test for synergism, summation or antagonism, the
combination index (CI) was calculated by adapting the Chou and
Talalay equation for mutually non-exclusive drugs: CI =
(D)1(Dx)1+(D)2(Dx)2+(D)1 (D)2(Dx)1 (Dx)2, where CI<1 indicates
synergism, CI=1 indicates summation and CI>1 indicates
antagonism (25,26). (D)1 and (D)2 represent the
concentrations of paclitaxel and COL-3 in the combination that also
inhibits cell growth by x%, respectively. (Dx)1 and (Dx)2 represent
the corresponding individual concentrations of paclitaxel and COL-3
that produced the same x% growth inhibition as the combination
regimen, respectively. Since a portion of the obtained data were
negative, such as slightly enhanced growth instead of inhibiting it
at reduced concentrations, the CompuSyn software developed by Chou
and Talalay (26,27), which only accepts values between
0–1, could not be used. Thus, the individual concentration of
paclitaxel or COL-3 that would produce the equivalent effect as the
combined concentrations were calculated using the identical
dose-response-inhibition equation used to calculate the
IC50 using GraphPad Prism software (version 5.0).
Results
Effects of monotherapy with paclitaxel
or COL-3 on breast cancer cell proliferation
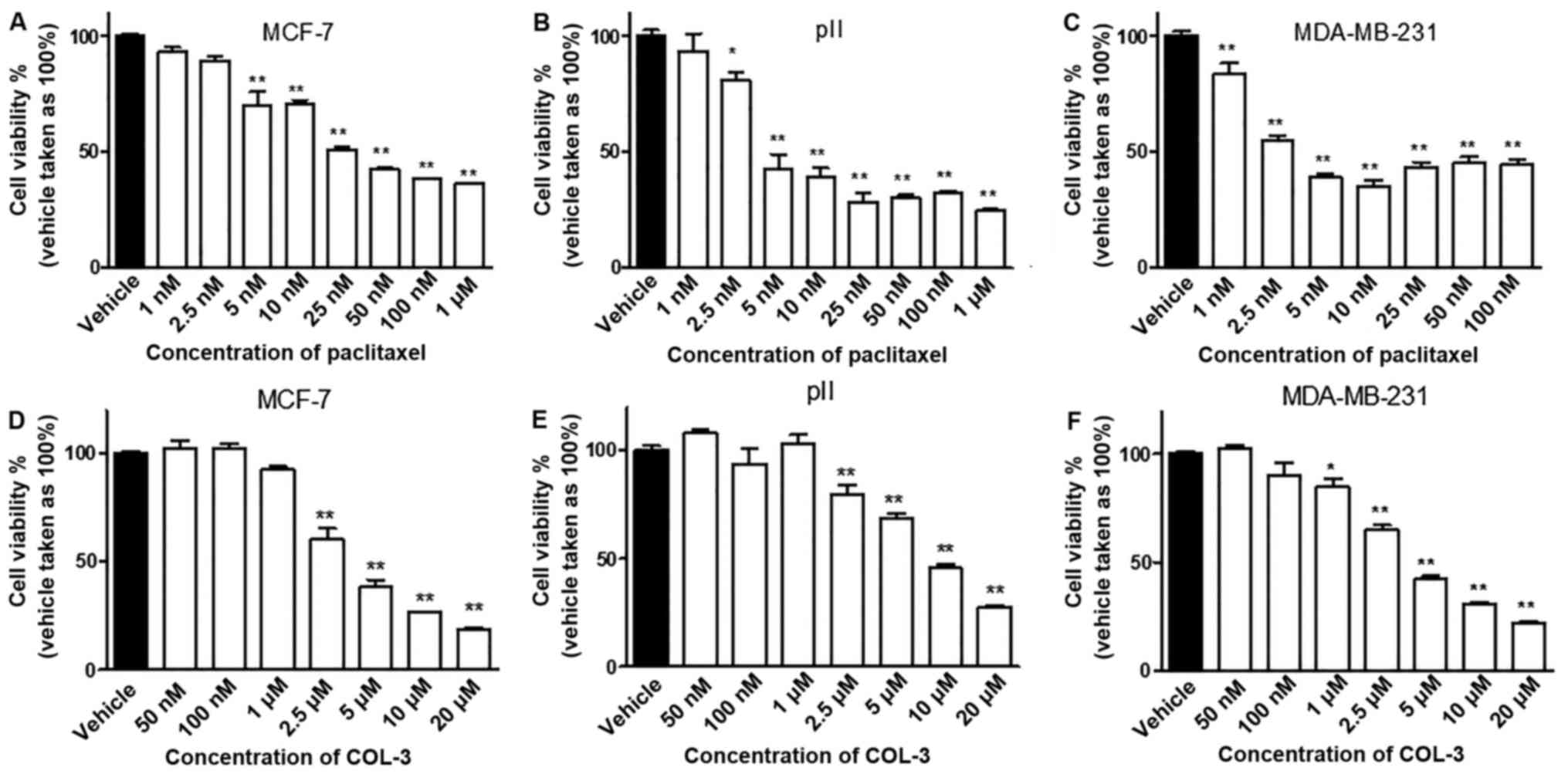
Paclitaxel inhibited cell proliferation in all of
the tested cell lines in a concentration-dependent manner (Fig. 1A-C). Additionally, paclitaxel had
similar inhibitory effects (60–70%) in all cell lines at the
highest concentrations (50 nM-1 µM); however, the degree of
sensitivity of the cell lines to paclitaxel varied. The
IC50 of paclitaxel for MCF-7 cells was 36 nM (95%
confidence interval, 22.96–56.39 nM), for pII cells it was 8.6 nM
(95% confidence interval, 6.188–11.97 nM) and for MDA-MB-231 cells
it was 6.5 nM (95% confidence interval, 4.777–8.930 nM). The slight
increase in cell viability observed at the highest doses of
paclitaxel (25–100 nM) in MDA-MB-231 cells was not statistically
significant (P>0.05), compared with the 10 nM dose, and the
effects of paclitaxel may have reached a plateau phase at a
concentration of 10 nM. Notably, the IC50 of the
ER+ cell line MCF-7 was 4–5.5 times increased, compared
with the ER− cell lines (pII and MDA-MB-231). Thus, the
order of sensitivity of the cell lines to paclitaxel from the most
to the least sensitive was MDA-MB-231, pII and then MCF-7.
COL-3 inhibited cell proliferation in all of the
tested cell lines in a concentration-dependent manner (Fig. 1D-F). It also demonstrated similar
anti-proliferative effects (60–70%) for all the cell lines at the
highest concentrations (10–20 µM). However, the degree of
sensitivity of the cell lines to COL-3 varied. The IC50
of COL-3 for MCF-7 cells was 4 µM (95% confidence interval,
3.456–4.755 µM), for pII cells it was 10 µM (95% confidence
interval, 8.196–12.428 µM) and for MDA-MB-231 cells it was 4.4 µM
(95% confidence interval, 3.854–5.052 µM). Notably, the
IC50 of pII cells was ~2 times increased, compared with
MDA-MB-231 and MCF-7 cells. Thus, the order of sensitivity of the
cell lines to COL-3 from the most to the least sensitive was MCF-7,
MDA-MB-231 and then pII. Additionally, it is notable that the
IC50 for COL-3 was 100–1,100 times increased, compared
with paclitaxel, thus indicating that the latter drug is more
potent at inhibiting cell proliferation.
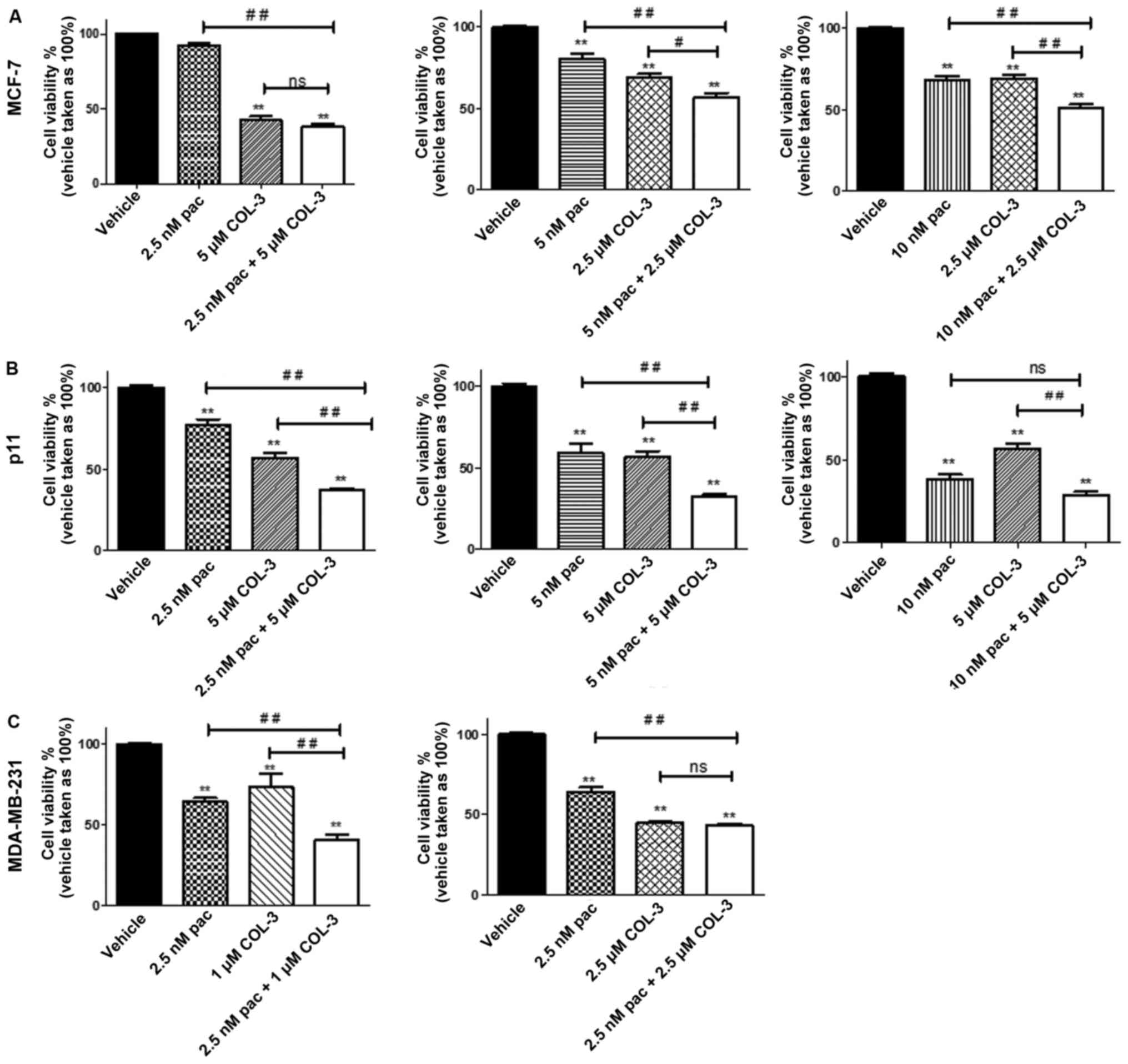
Effects of combination regimens of
COL-3 and paclitaxel on cell proliferation
The effects of various combination regimens on MCF-7
cell proliferation varied from notably additive to a slight
antagonism. Combining 2.5 nM paclitaxel with 5 µM COL-3 resulted in
a notably additive effect (CI=0.99) although the combination
regimen did not significantly inhibit proliferation, compared with
treatment with COL-3 alone (62 vs. 58%, respectively; P>0.05;
Fig. 2A). The combination of 5 nM
paclitaxel with 2.5 µM COL-3 resulted in a slight antagonistic
effect (CI=1.17; Table I). However,
combination of 10 nM paclitaxel with 2.5 µM COL-3 resulted in a
notably additive inhibitory effect (CI=1.09). This combination
regimen resulted in 49% inhibition, which is significant
(P<0.01), compared with treatment with each drug individually,
which achieved 30–32% inhibition (Fig.
2A). Additionally, the calculated individual concentrations of
paclitaxel and COL-3 that would produce the identical inhibitory
effect as the combination regimen were 34.6 nM and 4 µM,
respectively. Thus, by combining the drugs, the concentration of
paclitaxel and COL-3 would be reduced by 3.5- and 1.6-fold,
respectively (Table I).
 | Table I.Comparison of the anti-proliferative
effects of combined concentrations of pac and COL-3 with the
calculated individual concentrations of pac or COL-3 that would
produce equivalent effect as combination for all the cell
lines. |
Table I.
Comparison of the anti-proliferative
effects of combined concentrations of pac and COL-3 with the
calculated individual concentrations of pac or COL-3 that would
produce equivalent effect as combination for all the cell
lines.
|
|
|
| Calculated
individual concentration that produces equivalent % inhibition of
cell viability as the combination regimen | Folds of reduction
of the drug concentration in the combination regimen vs. individual
drug treatment |
|---|
|
|
|
|
|
|
|---|
| Cell lines | Combined
concentrations | % inhibition of
cell viability of the combination regimen | Pac (nM) | COL-3 (µM) | CI | Pac (nM) | COL-3 (µM) |
|---|
| MCF-7 | 2.5 nM pac + 5 µM
COL-3 | 62.1 | 58.8 |
6.6 | 0.99 | 23.5 | 1.32 |
|
| 5 nM pac + 2.5 µM
COL-3 | 43 | 27.2 | 3 | 1.17 | 5.44 |
1.2 |
|
| 10 nM pac + 2.5 µM
COL-3 | 49 | 34.6 | 4 | 1.09 |
3.5 |
1.6 |
| pII | 2.5 nM pac + 5 µM
COL-3 | 63.1 | 14.7 | 17.2 | 0.51 | 6 |
3.4 |
|
| 5 nM pac + 5 µM
COL-3 | 67.8 | 18.1 | 21.3 | 0.58 | 4 |
4.3 |
|
| 10 nM pac + 5 µM
COL-3 | 71 | 21.2 | 24.9 | 0.767 |
2.1 | 4.98 |
| MDA-MB-231 | 2.5 nM pac + 1 µM
COL-3 | 59.5 | 9.6 |
6.5 | 0.45 |
3.8 |
6.5 |
|
| 2.5 nM pac + 2.5 µM
COL-3 | 56.7 | 8.5 |
5.8 | 0.85 |
3.4 |
2.3 |
For pII cells, the combination regimen resulted in
synergistic inhibitory effects on cell proliferation. Combination
of 2.5 nM paclitaxel with 5 µM COL-3 resulted in a synergistic
effect (CI=0.51; Table I). It also
significantly inhibited cell proliferation by 63%, compared with
treatment with individual drugs, which achieved 30–40% inhibition
(P<0.01; Fig. 2B). Additionally,
the calculated individual concentrations of paclitaxel and COL-3
that would produce the identical inhibitory effect as the
combination regimen were 14.7 nM and 17.2 µM, respectively. Thus,
by combining the drugs, the concentration of paclitaxel and COL-3
would be reduced by 5.9- and 3.4-fold, respectively (Table I). Similarly, combination of the
highest concentration of paclitaxel (5 nM) with the highest
concentration of COL-3 (5 µM) resulted in a synergistic inhibitory
effect on cell proliferation (CI=0.58). It also significantly
inhibited cell proliferation by 67.8%, compared with treatment with
individual drugs, which achieved 40–50% inhibition (P<0.01;
Fig. 2B). Furthermore, the
calculated individual concentrations of paclitaxel and COL-3 that
would produce the identical inhibitory effect as the combination
regimen were 18.1 nM and 21.3 µM, respectively. Thus, by combining
an increased concentration of paclitaxel with an increased
concentration of COL-3, the concentration of paclitaxel and COL-3
would be reduced by 3.6- and 4.3-fold, respectively (Table I). However, combining 10 nM
paclitaxel, which caused >50% inhibition, with the identical
concentration of COL-3 (5 µM) resulted in moderate synergism due to
it having a slightly increased inhibitory effect, compared with
paclitaxel alone (70 vs. 60% reduction, respectively; P>0.05;
Fig. 2B).
With regards to MDA-MB-231 cells, the combination
regimen also resulted in synergistic inhibitory effects on cell
proliferation. Synergism was achieved when combining 2.5 nM
paclitaxel with 1 µM COL-3 (CI=0.45; Table I), which also significantly
inhibited cell proliferation by 60%, compared with treatment with
individual drugs, which achieved 30–40% inhibition (P<0.01,
Fig. 2C). The calculated individual
concentrations of paclitaxel and COL-3 that would produce the
identical inhibitory effect as the combination regimen were 9.6 nM
and 6.5 µM, respectively. Thus, by combining the drugs, the
concentration of paclitaxel and COL-3 would be reduced by 3.8- and
6.5-fold, respectively (Table I).
Combining 2.5 nM paclitaxel with 2.5 µM COL-3 resulted in a slight
synergistic inhibitory effects on cell proliferation, although the
combination did not have a significant inhibitory effect, compared
with COL-3 alone (57 vs. 55%, respectively; P>0.05. Fig. 2C).
Notably, the ER-ve cell lines were more responsive
to the combination regimens, which resulted in synergistic
inhibitory effects on cell proliferation, compared with the ER+ve
cell line.
Effects of paclitaxel, COL-3 or their
combination on pII cell apoptosis and cell cycle
The rational for using different concentrations of
COL-3 or paclitaxel was due to the different sensitivities of the
cell lines to the effects of the drugs. A concentration, which
inhibited cell proliferation by ~50% in the combination regiments,
was selected for the subsequent experiments.
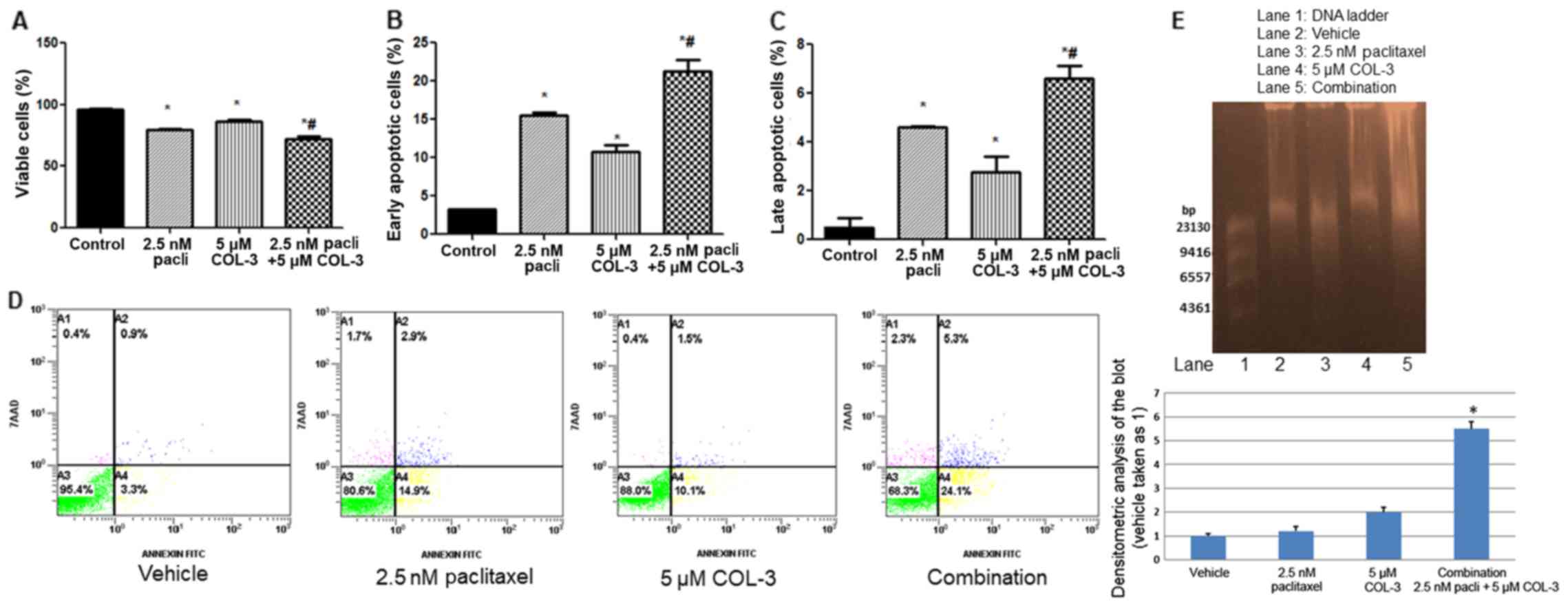
At the tested concentrations, treatment with 2.5 nM
paclitaxel or 5 µM COL-3 alone significantly decreased viable cells
and increased early/late apoptotic cells, compared with treatment
with vehicle (P<0.05; Fig.
3A-C). The combination regimen resulted in an enhanced effect,
a significantly decreased number of viable cells and an increased
percentage of early/late apoptotic cells, compared with monotherapy
(P<0.05; Fig. 3A-C). Fig. 3D depicts representative examples of
the dot-blot for the FACS data presented in Fig. 3A-C. In the DNA fragmentation
analysis, slight DNA fragmentation was observed in vehicle treated
cells (48 h) and with monotherapy with COL-3 or paclitaxel in
agreement with Annexin-V/7AAD data (Fig. 3D). The combination regimen resulted
in a significantly increased percentage of apoptotic cells and DNA
fragmentation, compared with vehicle or monotherapy with COL-3 or
paclitaxel (P<0.05), as depicted in Fig. 3D and E.
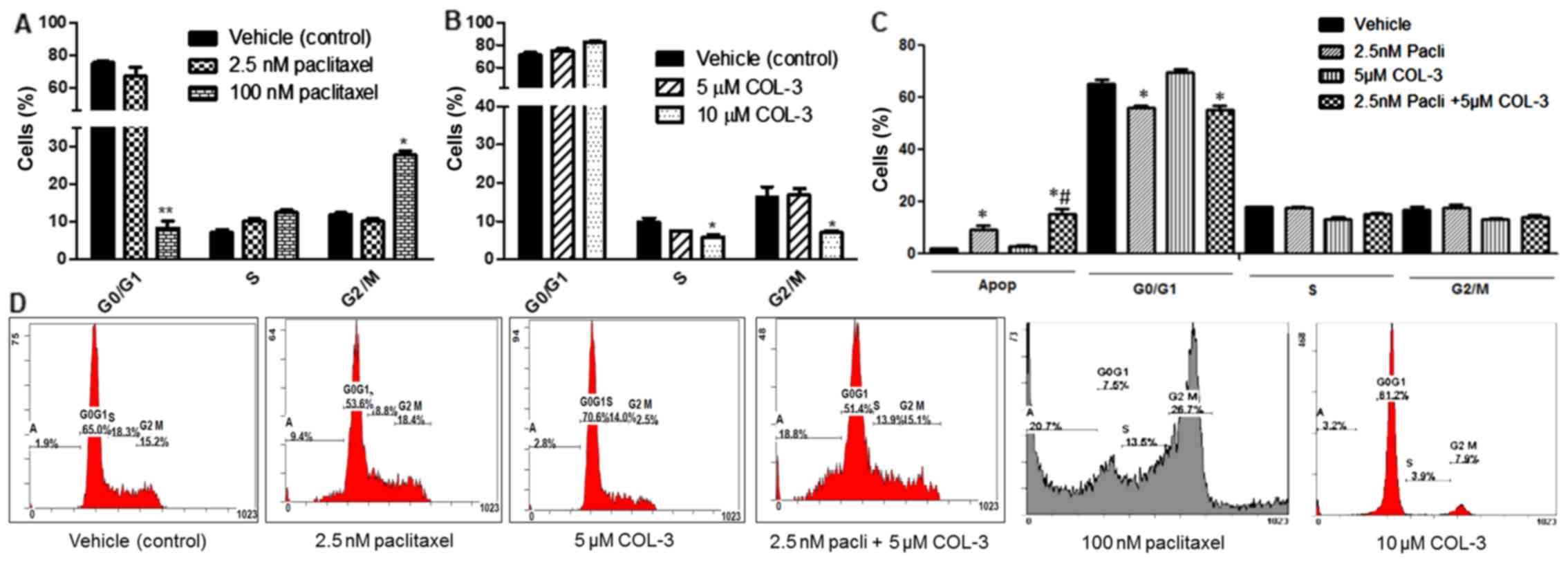
Treatment with paclitaxel at a concentration of 100
nM, significantly decreased (P<0.01) the cells in the
G0/G1 phase, from 75.3±1.2% to 8.4±1.9%, and
increased (P<0.05) cells in the G2/M phase, from
12.0±0.5% to 27.8±1.1%, compared with the vehicle treatment; thus,
it arrested the cell cycle at the G2/M phase (Fig. 4A). Treatment with COL-3 at a
concentration of 10 µM, but not 5 µM, significantly decreased cells
in the S and G2/M phases, from 9.9±1.2 to 5.8±0.7% and
from 16.3±2.7 to 6.9±0.6%, respectively, while it increased cells
in the G0/G1 phase, from 71±3 to 82±1%,
compared with the vehicle treatment; thus, it arrested the cell
cycle at the G0/G1 phase (P<0.05; Fig. 4B). Treatment with a reduced
concentration of paclitaxel 2.5 nM also decreased the cells in
G0/G1 phase, but to a lesser extent, compared
with the 100 nM concentration (Fig.
4C). Paclitaxel 2.5 nM, but not COL-3 5 µM, significantly
decreased the cells in the G0/G1 phase,
compared with vehicle (P<0.05; Fig.
4C). The combination of paclitaxel and COL-3 had similar
effects on the cell cycle to paclitaxel alone but resulted in a
synergistic effect in enhancing the number of cells in the
apoptotic phase, confirming the data presented in Fig. 3. Fig.
4D depicts the flow cytometry graphs for each treatment
condition.
Effects of paclitaxel, COL-3 or their
combination on the expression level of phosphokinases and
apoptosis-associated proteins
No statistically significant effects on the
expression levels of phosphokinases or apoptosis-associated
proteins were observed following treatment with paclitaxel, COL-3
or their combination, which may be due to variation in the
expression of phosphokinases or proteins between the samples
analyzed (Fig. 5A and B). However,
there was an increased phosphorylation level of p53 at S392 and S46
in cells treated with monotherapy or with the combination regimen,
compared with the vehicle-treated cells (Fig. 5B). Treatment with paclitaxel or
COL-3 alone increased phosphorylated p53 at S392 by 10% and at S46
by 20%, while the combination regimen increased it by 15.5 and 17%,
respectively. However, a trend of decreased phosphorylation level
of heat shock protein 27 (S78/S82), p70S6 kinase (T389), epithelial
nitric oxide synthase (S1177) and EGFR (Y1086) was observed in all
of the treatment regimens, compared with the vehicle-treated cells
(Fig. 5A).
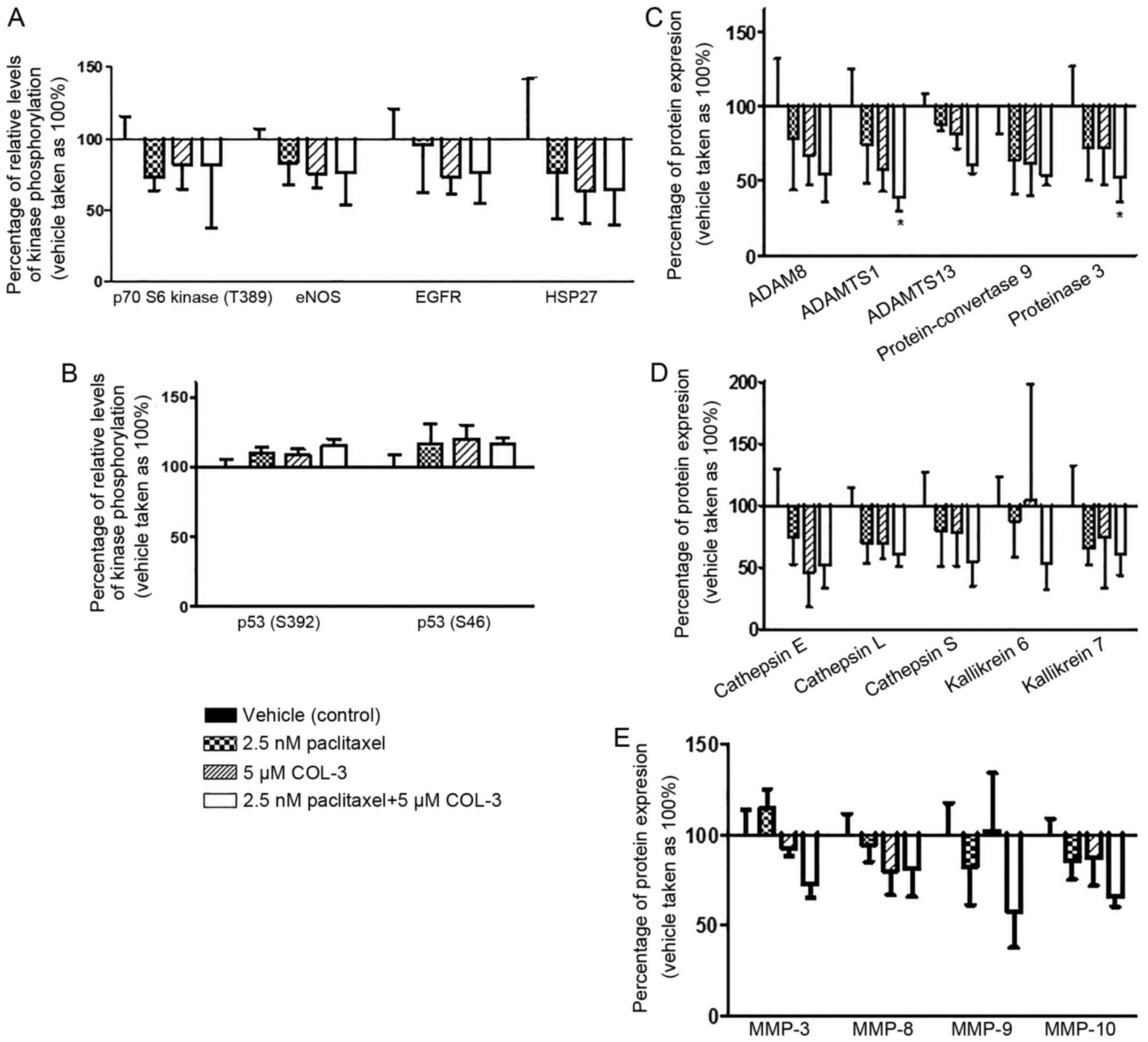 | Figure 5.Effect of combination regimens on the
phosphorylation levels of tested kinases and apoptosis-associated
proteins in pII cells. Lysates of cells were collected after 24 h
of incubation with the tested drugs. Densitometric values are means
normalized to the mean of the reference spots on each blot, and the
percentage of relative expression levels of selected phosphokinases
and apoptosis-associated proteins was determined (vehicle set as
100%) (A) Expression levels of p70S6 kinase (T389), eNOS, EGFR and
HSP27. (B) Expression levels of p53 at S392 and S46. (C) Expression
levels of ADAM8, ADAMTS1, ADAMTS13, protein-convertase 9 and
proteinase 3. (D) Expression levels of cathepsins E, L and S, and
kallikreins 6 and 7. (E) Expression levels of MMPs −3, −8, −9 and
−10. Each bar represents the mean ± standard error of the mean of 4
independent experiments. *P<0.05, compared with the vehicle
treated group. eNOS, epithelial nitric oxide synthase; EGFR,
epidermal growth factor receptor; HSP27, heat shock protein 27;
COL-3, chemically modified tetracycline-3; MMP, matrix
metalloproteinase; ADAMTS1, ADAM metallopeptidase with
thrombospondin type 1 motif 1. |
Among the 35 tested proteases, the expression of the
proteases ADAM metallopeptidase with thrombospondin type 1 motif 1
(ADAMTS1) and proteinase 3 (PR3) was significantly reduced by the
combination regimen, compared with the vehicle-treated cells (61
and 48% respectively; P<0.05; Fig.
5C), but this difference was not significant with paclitaxel or
COL-3 monotherapy. However, a trend towards decreased expression
was observed among 12 other proteases in cells treated with
monotherapy and/or the combination regimen, compared with the
vehicle-treated cells. These proteases include ADAM8, ADAMTS13,
cathepsin E, cathepsin L, cathepsin S, kallikrein 6, kallikrein 7,
MMP-3, MMP-8, MMP-9, MMP-10 and protein-convertase 9 (Fig. 5C-E).
Effect of paclitaxel, COL-3 or their
combination on the expression/phosphorylated levels of various
signaling molecules
Fig. 6A and B
depicts western blot analysis for the
expression/phosphorylated/cleaved levels of various downstream
signaling molecules important for cell survival, proliferation and
motility/invasion. The combination regimen enhanced the expression
of caspase-3, cleaved caspase-3 and phosphorylated P70S6K, which is
a downstream target of mammalian target of rapamycin (mTOR)
(28). However, COL-3 treatment
(alone or in combination with paclitaxel) enhanced the expression
levels of BCL-2 and BAK, which are important in cell apoptosis
(29), as well as the
phosphorylated AKT, the downstream target of phosphoinositide
3-kinase (28). The expression
levels of cytochrome c and APAF-1 were decreased with COL-3
treatment, compared with paclitaxel monotherapy. The expression
levels of BAD and phosphorylated p38 MAPK were increased with all
treatment regiments, compared with untreated cells. However, the
expression/phosphorylated/cleaved levels of caspase-8 and −9, BAX,
ERK1/2, and HSF-1 were not modulated in all the tested groups. The
expression profile of the total and the cleaved form of PARP was
enhanced by paclitaxel, but not COL-3, treatment as well as with
the combination regimen. Notably, the absence of detection of the
total levels of P70S6K and p38 MAPK is a limitation of the present
study. However, the total levels of ERK1/2 and AKT were not
modulated, although the phosphorylated AKT level was modulated, and
also the β-actin bands indicate equal loading. This indicated that
the phosphorylated levels of a number of the tested molecules are
modulated without affecting their total protein levels. These data
demonstrated that the observed synergistic effect of the
combination regimen is partially through increased total and
cleaved caspase-3 levels, and P70S6K phosphorylation in pII
cells.
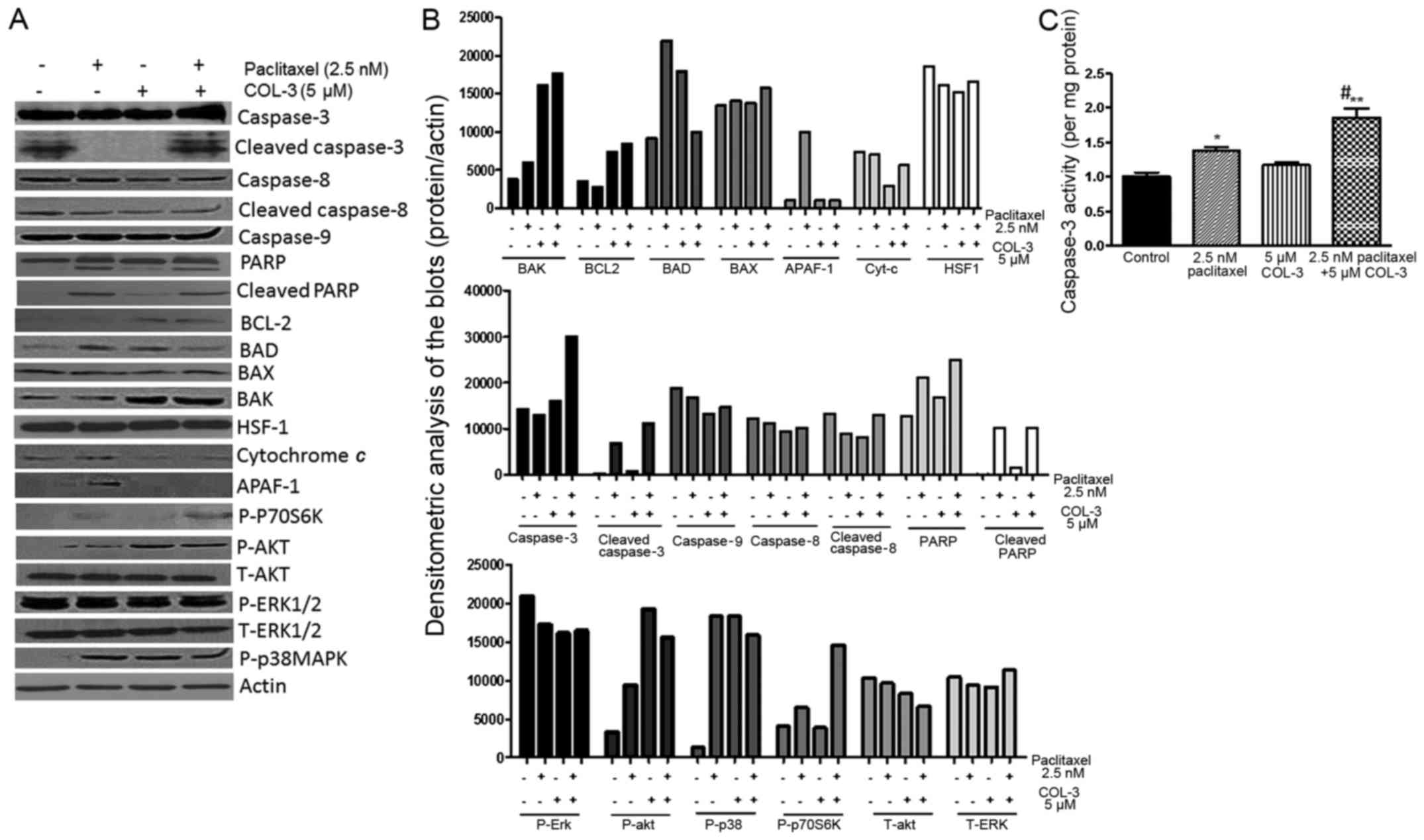 | Figure 6.Effect of drug treatment on the
expression and phosphorylated levels of various signaling molecules
and caspase-3 activity. (A) Cells were treated with paclitaxel,
COL-3 or combination regimen for 48 h. Total protein lysate (3 µg)
was electrophoresed on 10% SDS-PAGE, blotted onto nitrocellulose
membrane and probed with antisera to P- or T-BAX, BAD, BAK, BCL-2,
cytochrome c, AKT, ERK1/2, p38 MAPK, APAF-1, HSF-1, and
un-cleaved or cleaved-caspase-3, −8, −9, and PARP, and β-actin
(loading control). This blot represents 1/3 experiments. (B)
Densitometric analysis of the tested markers (normalized to
β-actin). (C) Caspase-3 activity for pII cells treated with
paclitaxel, COL-3 or combination regimen for 48 h. Histobars
represent the mean ± standard error of the mean of 3 independent
determinations, *P<0.05 and **P<0.01, compared with the UT
group. #P<0.05, compared with the paclitaxel-treated
group. p-, phospho-; t-, total-; UT, untreated; PARP,
poly(ADP-ribose) polymerase; BCL-2, B-cell lymphoma-2; BAD, BCL-2
associated agonist of cell death; BAX, BCL-2-associated X; BAK,
BCL-2 antagonist/killer; HSF-1, heat shock transcription factor-1;
APAF-1, apoptotic peptidase activating factor 1; ERK, extracellular
signal-regulated kinase; MAPK, mitogen-associated protein kinase;
COL-3, chemically modified tetracycline-3; pac, paclitaxel. |
Effect of paclitaxel, COL-3 or their
combination on caspase-3 activity
Treatment with paclitaxel, but not COL-3,
significantly increased caspase-3 activity in pII cells, compared
with vehicle treatment (P<0.05). The combination regimen
resulted in an enhanced effect and significantly increased
caspase-3 activity, compared with monotherapy (P<0.05; Fig. 6C).
Effect of paclitaxel, COL-3 or their
combination on total MMP activity
EGF stimulation of pII cells significantly increased
the total MMP activity, compared with vehicle treatment (P<0.05;
Fig. 7A). COL-3, but not
paclitaxel, treatment blocked the EGF-induced MMP activity to
baseline levels, and the combination regimen had similar effects to
COL-3 monotherapy.
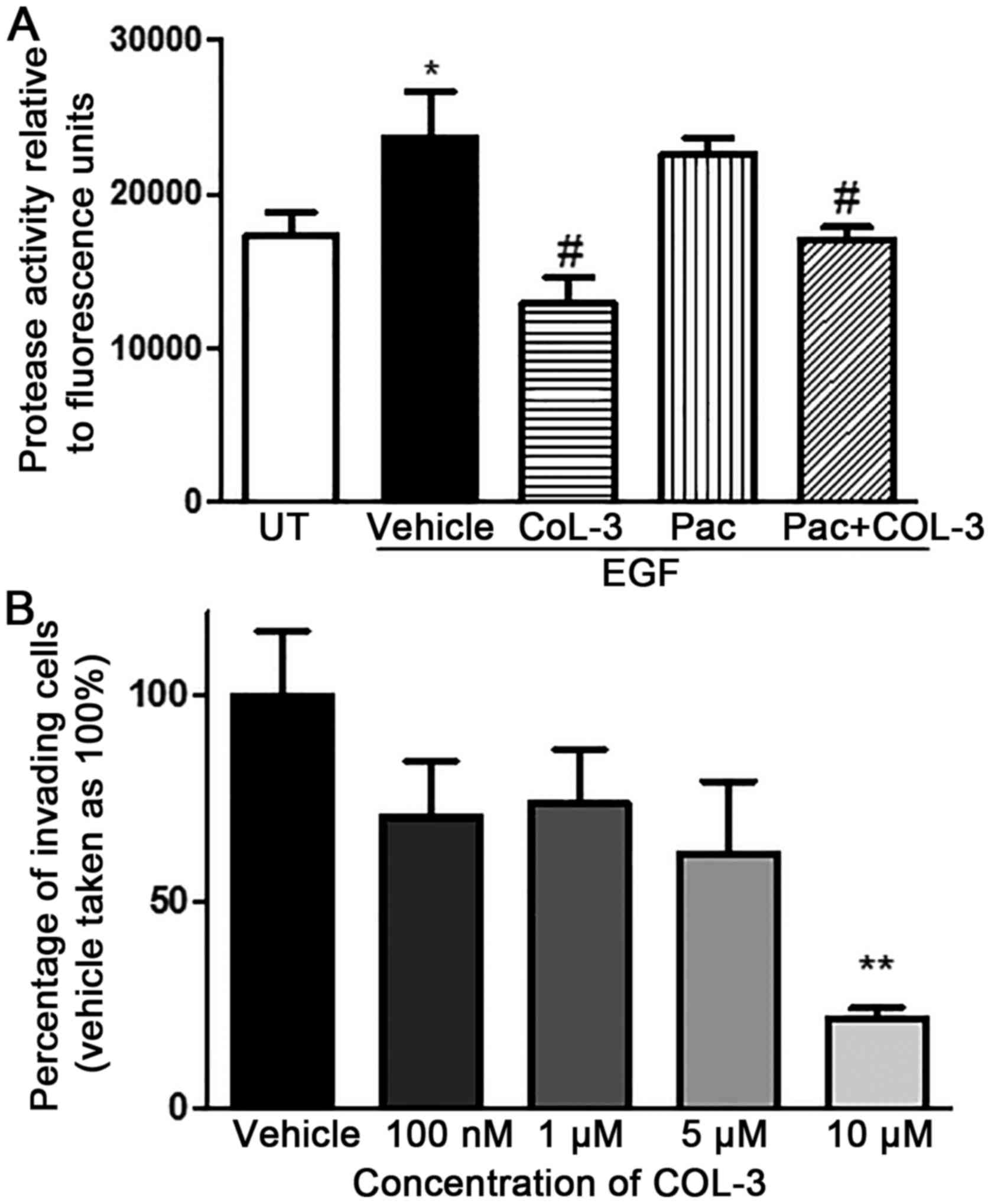 | Figure 7.Effect of drug treatment in total MMP
activity and invasion of pII cells. (A) Cells were left untreated
(UT) or stimulated with EGF (100 ng/ml) with or without
pre-treatment with individual drugs or combination regiment, and
total MMP activity was determined in the supernatant. Histobars
represent the mean ± SEM of 6 independent determinations,
*P<0.05, compared with the UT group. #P<0.05,
compared with the EGF/vehicle treated group. (B) Cells were left
untreated (vehicle) or exposed to various concentrations of COL-3.
The total number of pII cells penetrating through a basement
membrane extract towards serum components was determined by
fluorometric analysis. Histobars represent the mean ± SEM of 6
independent determinations. **P<0.01, compared with the
vehicle-treated group. SEM, standard error of the mean; COL-3,
chemically modified tetracycline-3; pac, paclitaxel; MMP, matrix
metalloproteinase; UT, untreated; EGF, epidermal growth factor. |
Effects of COL-3 on pII cell
invasion
COL-3 produced a concentration-dependent inhibitory
effect on pII cell invasion in the Cultrex® 24-well
invasion assay and reached statistical significance at 10 µM,
compared with vehicle (78% inhibition; P<0.01; Fig. 7B).
Discussion
The present study demonstrated that the chemically
modified tetracycline COL-3 enhances the anticancer effects of
paclitaxel against ER+ve and ER-ve breast cancer cell lines. COL-3
on its own had anti-proliferative effects against ER+ve and ER-ve
breast cancer cell lines, although it was less potent than
paclitaxel. There was synergistic anti-proliferative activity
between COL-3 and paclitaxel against ER-ve breast cancer cell
lines, and the anti-proliferative effects varied from notably
additive to slight antagonism against the ER+ve cell line. Reduced
concentrations of the drugs in combination produced similar
anti-proliferative activity, compared with high concentrations of
paclitaxel or COL-3 alone. The synergistic anti-proliferative
activity of the drug combination against the highly invasive and
proliferative ER-ve breast cancer cell line pII may be due to the
enhanced anti-apoptotic activity, but not cell cycle arrest.
Additionally, COL-3 inhibited pII cell invasion and MMP activity
in vitro. The combination of paclitaxel and COL-3 also
significantly increased the expression and activity of caspase-3
and phosphorylated P70S6K, a downstream target of the mTOR pathway
(28). The combination inhibited
the expression of two proteases, ADAMTS1 and PR3.
Although paclitaxel is considered an integral agent
in the treatment of breast cancer and other solid tumor types, the
dose-dependent side effects associated with the development of
peripheral neuropathy (PIPN) limits its use and may render the
treatment ineffective (14,30). Identification of agents that prevent
PIPN while preserving the anticancer effectiveness of paclitaxel
would allow for the treatment of aggressive cancer cases that
require high chemotherapeutic doses, and improve the quality of
life for patients with cancer and survivors of cancer who are
affected by chemotherapy-induced neuropathy. Taking into
consideration the indicated role of MMPs and proteinase-activated
receptors in the development of paclitaxel-induced neuropathy
(31,32), as well as the previously observed
protective effect of the potent MMP inhibitor COL-3 against
paclitaxel-induced thermal hyperalgesia in mice (16), and its indicated potential as an
anticancer drug (20,33), the present study evaluated whether
COL-3 affects the anticancer activity of paclitaxel using various
breast cancer cell lines.
Paclitaxel significantly inhibited cell
proliferation in all the tested cell lines in a
concentration-dependent manner, which is consistent with previous
in vitro reports, where it was reported to have
concentration-dependent cytotoxicity on a variety of human tumor
cell lines, including those derived from the breast carcinoma, lung
adenocarcinoma, cervical carcinoma and colon carcinoma (34–37).
Additionally, no further increase in cell killing was observed at
paclitaxel concentrations >50 nM, similar to what has been
reported previously (34).
Furthermore, the degree of sensitivity of the cell lines to
paclitaxel was variable. Paclitaxel was more effective against the
ER-ve cell lines, compared with the ER+ve cell line, which may be
due to the increased proliferative rate of the ER-ve cells,
compared with the ER+ve cells. The present data are in agreement
with previous studies that demonstrated that MDA-MB-231 and other
ERα-silenced cells (derived from MCF-7 cells) were more sensitive
to the anti-proliferative effects of paclitaxel, compared with the
ER+ve cells (38,39). Similarly, COL-3 significantly
inhibited cell proliferation in all the tested cell lines in a
concentration-dependent manner. Previous studies with COL-3 have
also indicated that it has a strong ability to inhibit the
proliferation of various cancer cells, including those from the
prostate, colon, cervical and breast, in a concentration-dependent
manner (40–44). However, the degree of sensitivity of
the tested cell lines to COL-3 was variable. Unlike the case for
paclitaxel, the ER status may not affect the sensitivity to COL-3.
The present data are in contrast with previous studies that
determined that COL-3 was not cytotoxic at concentrations up to 50
µM or 20 µg/ml against the breast cancer cell lines MDA-MB-468 and
MMP-9-overexpressing sub-clone (E10) of MDA-MB-231, respectively
(40,43). This difference could be due to the
different cell lines used, the reduced period of incubation (48 h)
used in the previous studies (40,43) or
different experimental designs, including cells being plated onto
extracellular matrix (ECM)-coated wells (40).
The combination regimens of paclitaxel and COL-3 had
synergistic anti-proliferative effects against ER-ve cell lines and
the anti-proliferative effects varied from notably additive to
slightly antagonism on the ER+ve cell line. The synergistic
anti-proliferative effect observed against the ER-ve pII cells was
partially via the induction of apoptosis rather than inducing cell
cycle arrest. Additionally, it was possible to reduce the
concentrations of paclitaxel as well as COL-3 when the drugs were
used in combination, while producing the identical degree of
inhibition in cell proliferation as increased concentrations of
each drug alone. Thus, the combination could assist in reducing the
risk of developing dose-dependent side effects, including PIPN. It
may also be possible to reduce the risk of developing cutaneous
photo-toxicity, which is the dose-limiting side effect of COL-3
(34). A previous study
demonstrated that the combination of paclitaxel with the novel
epigenetic agent phenethyl isothiocyanate (PEITC) had a synergistic
effect on the inhibition of growth of breast cancer cells (MCF-7
and MDA-MB-231), as well as increasing apoptosis and cell cycle
arrest at the G2/M phase (35). This indicates that it is possible to
reduce the dosage of paclitaxel when it is used in combination with
other drugs, which could reduce the dose-dependent side effects,
while maintaining its clinical efficacy against breast cancer cells
and other solid tumor types. In that study, they also observed an
antagonistic effect when combining a number of doses, including 1
nM paclitaxel with 5 or 10 µM PEITC (35). Another previous study demonstrated
that paclitaxel combined with a cannabidiol produced an additive to
a synergistic inhibition of breast cancer cell viability (45). Cannabidiol was also indicated by the
identical research group to prevent the onset of paclitaxel-induced
mechanical and thermal sensitivity in rodents (46). However, the concentrations of
paclitaxel that were used ranged from 2.5–10 µM, which are
1,000-fold increased, compared with the concentrations used in the
present study (2.5–10 nM).
Numerous studies demonstrated that paclitaxel kills
cancer cells through the induction of apoptosis (11,47–50).
Previous studies have also indicated that COL-3 induces apoptosis
as a mechanism of its cytotoxicity. It was also reported that COL-3
causes caspase-dependent and -independent apoptosis (42,44,51).
In the present study, it was determined that the tested
concentration of paclitaxel (2.5 nM) had pro-apoptotic effects on
pII cells, which differs from what was reported by Jordan et
al (11) on cervical cancer
HeLa cells using a concentration of 3 nM. COL-3 treatment at
increased concentration (5 µM) also significantly decreased viable
cells and increased early/late apoptotic cells similar to what was
reported in prostate, colon and cervical cancer cells using the
identical concentration range (42,44,51).
Notably, the combination regimen enhanced the percentage of
early/late apoptotic cells. This indicated that the synergistic
inhibitory effect on cell proliferation is partially due to the
induction of cell apoptosis.
It is considered that stabilization of microtubules
of paclitaxel induces mitotic arrest at the G2/M phase
of the cell cycle (47,52). However, it was reported that at low
concentrations, paclitaxel increases cells in the sub-G1
and G1 phases, indicating G1 arrest (52). Additionally, studies using
bromodeoxyuridine for DNA labeling indicated that cells were not
arrested in the G1 phase directly (47,52).
Instead, G1 cells went through the S phase and then
divided to generate 2C DNA cells, indicating that G1
arrest does not occur in the first cell cycle, and did not prevent
cells from passing through S phase and entering mitosis, but rather
in the second or third cell cycle (52). However, COL-3 was determined to
cause cell cycle arrest at the G1/S phase transition in
prostate and cervical cancer cells, indicating that COL-3 inhibits
mitogenic signaling (42,44). The exact mechanism by which it
arrests the cell cycle remains unclear due to limited studies
investigating its cytotoxic effects. In agreement with previous
reports, the cell cycle arrest at the G2/M phase was
observed in cells treated with increased concentration of
paclitaxel (100 nM) with a significant decrease in
G0/G1 cells, whereas the cell cycle arrest at
G0/G1 was observed at the increased
concentration of COL-3 (10 µM). The reduced concentration of COL-3
(5 µM) did not have a significant effect on the cell cycle in the
G1 or G2/M phases. However, the reduced
concentration of paclitaxel (2.5 nM) and the combination of these
concentrations, which induced apoptosis as aforementioned,
similarly reduced cells in the G0/G1 phases
of the cell cycle. Previously, it was reported that mitotic arrest
is not only responsible for the efficacy of paclitaxel, but also
for chromosome mis-segregation on highly abnormal, multi-polar
spindles that results in cell death, which may be also the case in
the present study (37). Thus,
performing DNA labeling should be considered in future studies to
examine the effect of the reduced concentrations of paclitaxel,
COL-3 or their combination on the cell cycle.
COL-3 treatment significantly reduced cell invasion
partially through inhibiting total MMP activity. CMTs have been
demonstrated to inhibit the enzymatic activities of gelatinases
(MMP-2 and MMP-9), stromelysins (MMP-3, MMP-10 and MMP-11),
collagenases (MMP-1, MMP-8 and MMP-13) as well as
non-collagenolytic proteases (53–55).
COL-3 has been demonstrated to significantly reduce cancer cell
invasion and migration in vitro by inhibiting MMP activity
and/or expression and upregulating the expression of E-cadherins
(41–43,56).
The majority studies have reported its specificity towards
gelatinases not only by inhibiting their activity but also their
expression levels (41–43). In addition to its anti-MMP function,
COL-3 directly inhibits the amidolytic (proteolytic) activity of
human leukocyte elastase (HLE), a serine proteinase also termed
neutrophil elastase, and the extracellular matrix degradation
mediated by HLE (19). Thus, the
pleiotropic properties of CMT-3, which includes, but is not limited
to, the inhibition of serine proteinases (19), MMPs (19,20)
and cytokines (57), provide
therapeutic potential to reduce excessive connective tissue
breakdown during various pathologic processes, including
inflammatory diseases, cancer metastasis and metabolic bone
diseases. Previous studies using the MDA-MB-468 cells demonstrated
a concentration-dependent inhibition of invasion, with 67%
inhibition observed with 10 µg/ml (30 µM) COL-3 (43,47,52).
Additionally, culturing E-10 cells, a transfected subclone of
MDA-MB-231 cells, in 10 or 20 µM COL-3 diminished secreted MMP-9
levels by 45 or 60%, respectively, as well as inhibited their ECM
degradative ability by 20–30% (43). Consistent with the previous studies,
COL-3 demonstrated a concentration-dependent inhibition of pII cell
invasion with a significant inhibition observed at 10 µM.
Consequently, the effects of COL-3 as well as paclitaxel and their
combination on the expression level of various proteases were
further evaluated. Among the 35 tested proteases, the expression of
ADAMTS1 and PR3 were significantly decreased in cells treated with
the combination of COL-3 and paclitaxel.
In conclusion, the present study indicates that
COL-3 potentiates the anticancer activity of paclitaxel by
enhancing its inhibitory effects on cell proliferation, inducing
apoptosis as well as inhibiting invasiveness. The molecular
mechanism may involve the modulation of the expression of
proteases, including ADAMTS1 and PR3, caspase-3 expression/activity
and P70S6K phosphorylation. Furthermore, the combination regimen
would also offer opportunities for the reduction in the effective
dose of paclitaxel, which in turn would have further beneficial
effects for the reduction of the dose-dependent side effects,
including PIPN. This indicates that the combination of paclitaxel
and COL-3 in addition to reducing the development of PIPN, as
recently reported (16), would also
have enhanced anticancer activity against breast cancer. Therefore,
the potential of the combination against breast cancer growth and
metastasis in vivo warrants further research.
Acknowledgements
The authors would like to acknowledge the Core
facility in the Health Science Center (Kuwait University, Safat,
Kuwait) for the technical assistance.
Funding
This work was supported by grants from Kuwait
University Research Sector (grant nos. YP01/14 and SRUL02/13).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MAK and WM conceived and designed the experiments.
RMET, PM and MAK performed the experiments. RMET, MAK, PM and WM
analyzed the data. WM and MAK contributed reagents, materials and
analysis tools. RMET, MAK and WM wrote the paper. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 12:214922018.
|
|
2
|
Ferlay J, Steliarova-Foucher E,
Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D and
Bray F: Cancer incidence and mortality patterns in Europe:
Estimates for 40 countries in 2012. Eur J Cancer. 49:1374–1403.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hernandez-Aya LF and Gonzalez-Angulo AM:
Adjuvant systemic therapies in breast cancer. Surg Clin North Am.
93:473–491. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Clark GM, Osborne CK and McGuire WL:
Correlations between estrogen receptor, progesterone receptor, and
patient characteristics in human breast cancer. J Clin Oncol.
2:1102–1109. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dorssers LC, Van der Flier S, Brinkman A,
van Agthoven T, Veldscholte J, Berns EM, Klijn JG, Beex LV and
Foekens JA: Tamoxifen resistance in breast cancer: Elucidating
mechanisms. Drugs. 61:1721–1733. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hassan MS, Ansari J, Spooner D and Hussain
SA: Chemotherapy for breast cancer (Review). Oncol Rep.
24:1121–1131. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mincey BA, Palmieri FM and Perez EA:
Adjuvant therapy for breast cancer: Recommendations for management
based on consensus review and recent clinical trials. Oncologist.
7:246–250. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rowinsky EK: The development and clinical
utility of the taxane class of antimicrotubule chemotherapy agents.
Annu Rev Med. 48:353–374. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schiff PB and Horwitz SB: Taxol stabilizes
microtubules in mouse fibroblast cells. Proc Natl Acad Sci USA.
77:1561–1565. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Woods CM, Zhu J, McQueney PA, Bollag D and
Lazarides E: Taxol-induced mitotic block triggers rapid onset of a
p53-independent apoptotic pathway. Mol Med. 1:506–526. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jordan MA, Wendell K, Gardiner S, Derry
WB, Copp H and Wilson L: Mitotic block induced in HeLa cells by low
concentrations of paclitaxel (Taxol) results in abnormal mitotic
exit and apoptotic cell death. Cancer Res. 56:816–825.
1996.PubMed/NCBI
|
|
12
|
Wang TH, Wang HS and Soong YK:
Paclitaxel-induced cell death: Where the cell cycle and apoptosis
come together. Cancer. 88:2619–2628. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Crown J and O'Leary M: The taxanes: An
update. Lancet. 355:1176–1178. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Esin E and Yalcin S: Neuropathic cancer
pain: What we are dealing with? How to manage it? Onco Targets
Ther. 7:599–618. 2014.PubMed/NCBI
|
|
15
|
Hershman DL, Lacchetti C, Dworkin RH,
Lavoie Smith EM, Bleeker J, Cavaletti G, Chauhan C, Gavin P, Lavino
A, Lustberg MB, et al: ; American Society of Clinical Oncology:
Prevention and management of chemotherapy-induced peripheral
neuropathy in survivors of adult cancers: American Society of
Clinical Oncology clinical practice guideline. J Clin Oncol.
32:1941–1967. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Parvathy SS and Masocha W: Matrix
metalloproteinase inhibitor COL-3 prevents the development of
paclitaxel-induced hyperalgesia in mice. Medical principles and
practice: International journal of the Kuwait University. Health
Sci Cent. 22:35–41. 2013.
|
|
17
|
Golub LM, Ramamurthy NS, McNamara TF,
Greenwald RA and Rifkin BR: Tetracyclines inhibit connective tissue
breakdown: new therapeutic Implications for an old family of drugs.
Crit Rev Oral Biol Med. 2:297–321. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Golub LM, Suomalainen K and Sorsa T: Host
modulation with tetracyclines and their chemically modified
analogues. Curr Opin Dent. 2:80–90. 1992.PubMed/NCBI
|
|
19
|
Gu Y, Lee HM, Simon SR and Golub LM:
Chemically modified tetracycline-3 (CMT-3): A novel inhibitor of
the serine proteinase, elastase. Pharmacol Res. 64:595–601. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lokeshwar BL: Chemically modified
non-antimicrobial tetracyclines are multifunctional drugs against
advanced cancers. Pharmacol Res. 63:146–150. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Al Saleh S, Al Mulla F and Luqmani YA:
Estrogen receptor silencing induces epithelial to mesenchymal
transition in human breast cancer cells. PLoS One. 6:e206102011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Luqmani YA, Al Azmi A, Al Bader M, Abraham
G and El Zawahri M: Modification of gene expression induced by
siRNA targeting of estrogen receptor alpha in MCF7 human breast
cancer cells. Int J Oncol. 34:231–242. 2009.PubMed/NCBI
|
|
23
|
Khajah MA, Al Saleh S, Mathew PM and
Luqmani YA: Differential effect of growth factors on invasion and
proliferation of endocrine resistant breast cancer cells. PLoS One.
7:e418472012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Khajah MA, Mathew PM and Luqmani YA:
Inhibitors of PI3K/ERK1/2/p38 MAPK show preferential activity
against endocrine-resistant breast cancer cells. Oncol Res.
25:1283–1295. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Khajah MA, Mathew PM, Alam-Eldin NS and
Luqmani YA: Bleb formation is induced by alkaline but not acidic pH
in estrogen receptor silenced breast cancer cells. Int J Oncol.
46:1685–1698. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chou TC, Tan QH and Sirotnak FM:
Quantitation of the synergistic interaction of edatrexate and
cisplatin in vitro. Cancer Chemother Pharmacol. 31:259–264. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Al Saleh S, Sharaf LH and Luqmani YA:
Signalling pathways involved in endocrine resistance in breast
cancer and associations with epithelial to mesenchymal transition
(Review). Int J Oncol. 38:1197–1217. 2011.PubMed/NCBI
|
|
29
|
Krajewski S, Krajewska M, Turner BC, Pratt
C, Howard B, Zapata JM, Frenkel V, Robertson S, Ionov Y, Yamamoto
H, et al: Prognostic significance of apoptosis regulators in breast
cancer. Endocr Relat Cancer. 6:29–40. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee JJ and Swain SM: Peripheral neuropathy
induced by microtubule-stabilizing agents. J Clin Oncol.
24:1633–1642. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen Y, Yang C and Wang ZJ:
Proteinase-activated receptor 2 sensitizes transient receptor
potential vanilloid 1, transient receptor potential vanilloid 4,
and transient receptor potential ankyrin 1 in paclitaxel-induced
neuropathic pain. Neuroscience. 193:440–451. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nishida K, Kuchiiwa S, Oiso S, Futagawa T,
Masuda S, Takeda Y and Yamada K: Up-regulation of matrix
metalloproteinase-3 in the dorsal root ganglion of rats with
paclitaxel-induced neuropathy. Cancer Sci. 99:1618–1625. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
D'Agostino P, Ferlazzo V, Milano S, La
Rosa M, Di Bella G, Caruso R, Barbera C, Grimaudo S, Tolomeo M, Feo
S, et al: Chemically modified tetracyclines induce cytotoxic
effects against J774 tumour cell line by activating the apoptotic
pathway. Int Immunopharmacol. 3:63–73. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liebmann JE, Cook JA, Lipschultz C, Teague
D, Fisher J and Mitchell JB: Cytotoxic studies of paclitaxel
(Taxol) in human tumour cell lines. Br J Cancer. 68:1104–1109.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu K, Cang S, Ma Y and Chiao JW:
Synergistic effect of paclitaxel and epigenetic agent phenethyl
isothiocyanate on growth inhibition, cell cycle arrest and
apoptosis in breast cancer cells. Cancer Cell Int. 13:102013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tommasi S, Mangia A, Lacalamita R,
Bellizzi A, Fedele V, Chiriatti A, Thomssen C, Kendzierski N,
Latorre A, Lorusso V, et al: Cytoskeleton and paclitaxel
sensitivity in breast cancer: The role of beta-tubulins. Int J
Cancer. 120:2078–2085. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zasadil LM, Andersen KA, Yeum D, Rocque
GB, Wilke LG, Tevaarwerk AJ, Raines RT, Burkard ME and Weaver BA:
Cytotoxicity of paclitaxel in breast cancer is due to chromosome
missegregation on multipolar spindles. Sci Transl Med.
6:229ra432014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Izbicka E, Campos D, Carrizales G and
Patnaik A: Biomarkers of anticancer activity of R115777
(Tipifarnib, Zarnestra) in human breast cancer models in vitro.
Anticancer Res. 25:3215–3223. 2005.PubMed/NCBI
|
|
39
|
Tokuda E, Seino Y, Arakawa A, Saito M,
Kasumi F, Hayashi S and Yamaguchi Y: Estrogen receptor-α directly
regulates sensitivity to paclitaxel in neoadjuvant chemotherapy for
breast cancer. Breast Cancer Res Treat. 133:427–436. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gu Y, Lee HM, Roemer EJ, Musacchia L,
Golub LM and Simon SR: Inhibition of tumor cell invasiveness by
chemically modified tetracyclines. Curr Med Chem. 8:261–270. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lokeshwar BL, Escatel E and Zhu B:
Cytotoxic activity and inhibition of tumor cell invasion by
derivatives of a chemically modified tetracycline CMT-3 (COL-3).
Curr Med Chem. 8:271–279. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lokeshwar BL, Selzer MG, Zhu BQ, Block NL
and Golub LM: Inhibition of cell proliferation, invasion, tumor
growth and metastasis by an oral non-antimicrobial tetracycline
analog (COL-3) in a metastatic prostate cancer model. Int J Cancer.
98:297–309. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Meng Q, Xu J, Goldberg ID, Rosen EM,
Greenwald RA and Fan S: Influence of chemically modified
tetracyclines on proliferation, invasion and migration properties
of MDA-MB-468 human breast cancer cells. Clin Exp. 18:139–146.
2000. View Article : Google Scholar
|
|
44
|
Zhao L, Xu J, Yang Y, Chong Y, Liu C, Jiao
Y and Fan S: Inhibitory impacts of chemically modified
tetracycline-3 and underlying mechanism in human cervical cancer
cells. Anticancer Drugs. 24:799–809. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ward SJ, McAllister SD, Kawamura R, Murase
R, Neelakantan H and Walker EA: Cannabidiol inhibits
paclitaxel-induced neuropathic pain through 5-HT(1A) receptors
without diminishing nervous system function or chemotherapy
efficacy. Br J Pharmacol. 171:636–645. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ward SJ, Ramirez MD, Neelakantan H and
Walker EA: Cannabidiol prevents the development of cold and
mechanical allodynia in paclitaxel-treated female C57Bl6 mice.
Anesth Analg. 113:947–950. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bhalla K, Ibrado AM, Tourkina E, Tang C,
Mahoney ME and Huang Y: Taxol induces internucleosomal DNA
fragmentation associated with programmed cell death in human
myeloid leukemia cells. Leukemia. 7:563–568. 1993.PubMed/NCBI
|
|
48
|
Fan W: Possible mechanisms of
paclitaxel-induced apoptosis. Biochem Pharmacol. 57:1215–1221.
1999.PubMed/NCBI
|
|
49
|
Lieu CH, Chang YN and Lai YK: Dual
cytotoxic mechanisms of submicromolar taxol on human leukemia HL-60
cells. Biochem Pharmacol. 53:1587–1596. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Torres K and Horwitz SB: Mechanisms of
Taxol-induced cell death are concentration dependent. Cancer Res.
58:3620–3626. 1998.PubMed/NCBI
|
|
51
|
Onoda T, Ono T, Dhar DK, Yamanoi A and
Nagasue N: Tetracycline analogues (doxycycline and COL-3) induce
caspase-dependent and -independent apoptosis in human colon cancer
cells. Int J Cancer. 118:1309–1315. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Demidenko ZN, Kalurupalle S, Hanko C, Lim
CU, Broude E and Blagosklonny MV: Mechanism of G1-like arrest by
low concentrations of paclitaxel: Next cell cycle p53-dependent
arrest with sub G1 DNA content mediated by prolonged mitosis.
Oncogene. 27:4402–4410. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mäkelä M, Sorsa T, Uitto VJ, Salo T,
Teronen O and Larjava H: The effects of chemically modified
tetracyclines (CMTs) on human keratinocyte proliferation and
migration. Adv Dent Res. 12:131–135. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Myers SA and Wolowacz RG:
Tetracycline-based MMP inhibitors can prevent fibroblast-mediated
collagen gel contraction in vitro. Adv Dent Res. 12:86–93. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ryan ME, Ramamurthy NS and Golub LM:
Tetracyclines inhibit protein glycation in experimental diabetes.
Adv Dent Res. 12:152–158. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gu Y, Lee HM, Golub LM, Sorsa T, Konttinen
YT and Simon SR: Inhibition of breast cancer cell extracellular
matrix degradative activity by chemically modified tetracyclines.
Ann Med. 37:450–460. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sandler C, Ekokoski E, Lindstedt KA,
Vainio PJ, Finel M, Sorsa T, Kovanen PT, Golub LM and Eklund KK:
Chemically modified tetracycline (CMT)-3 inhibits histamine release
and cytokine production in mast cells: Possible involvement of
protein kinase C. Inflamm Res. 54:304–312. 2005. View Article : Google Scholar : PubMed/NCBI
|